Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Whole Exome Sequencing
3. Results
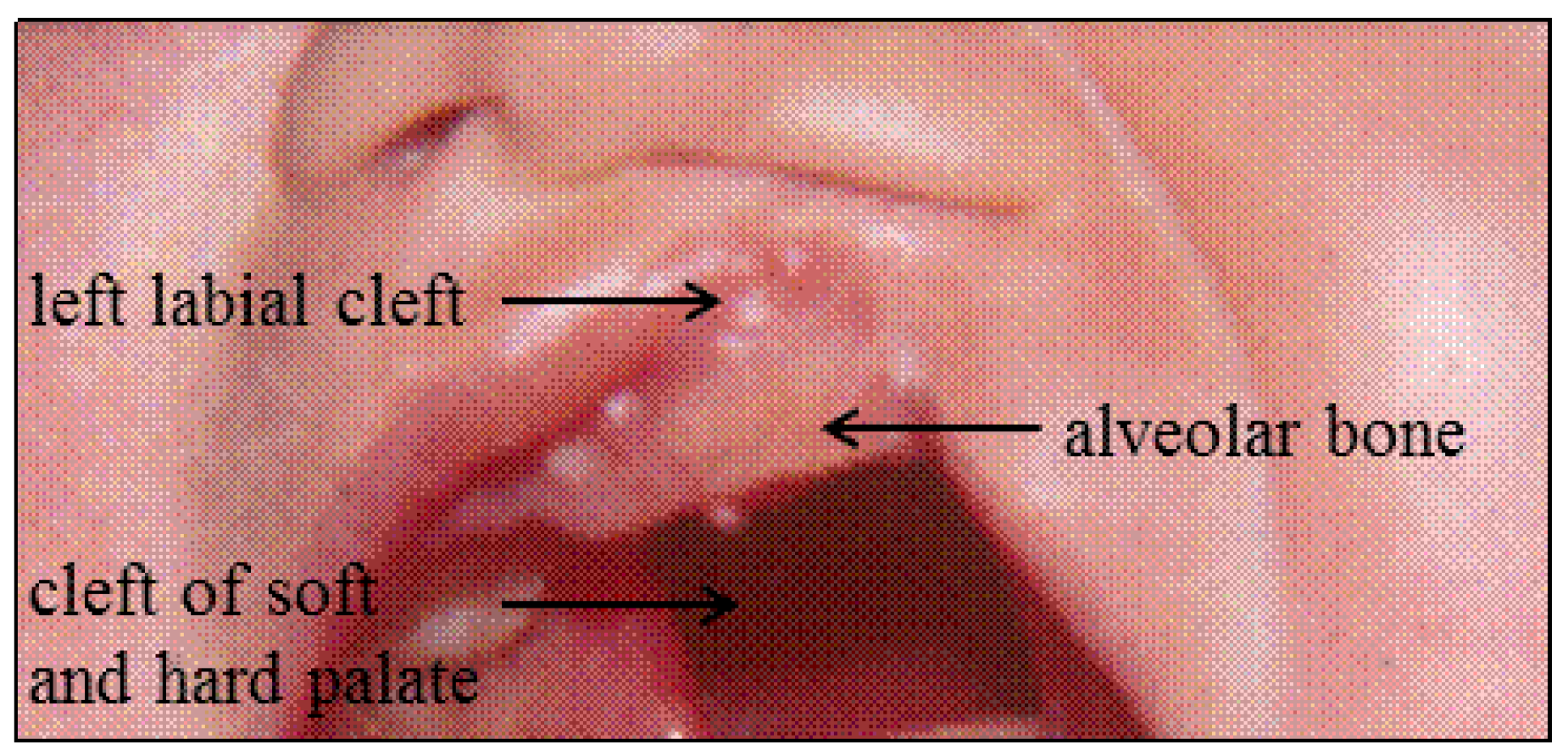
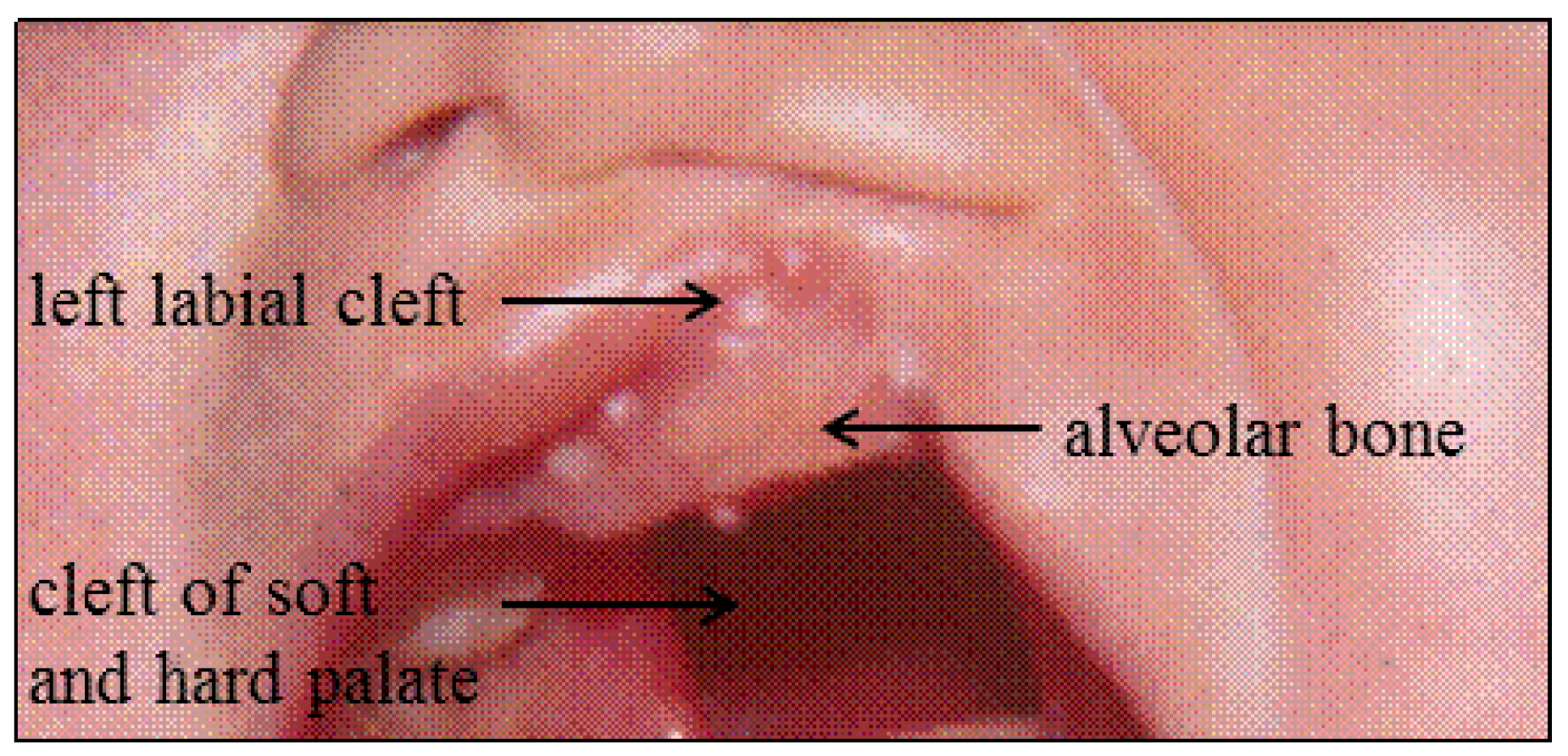
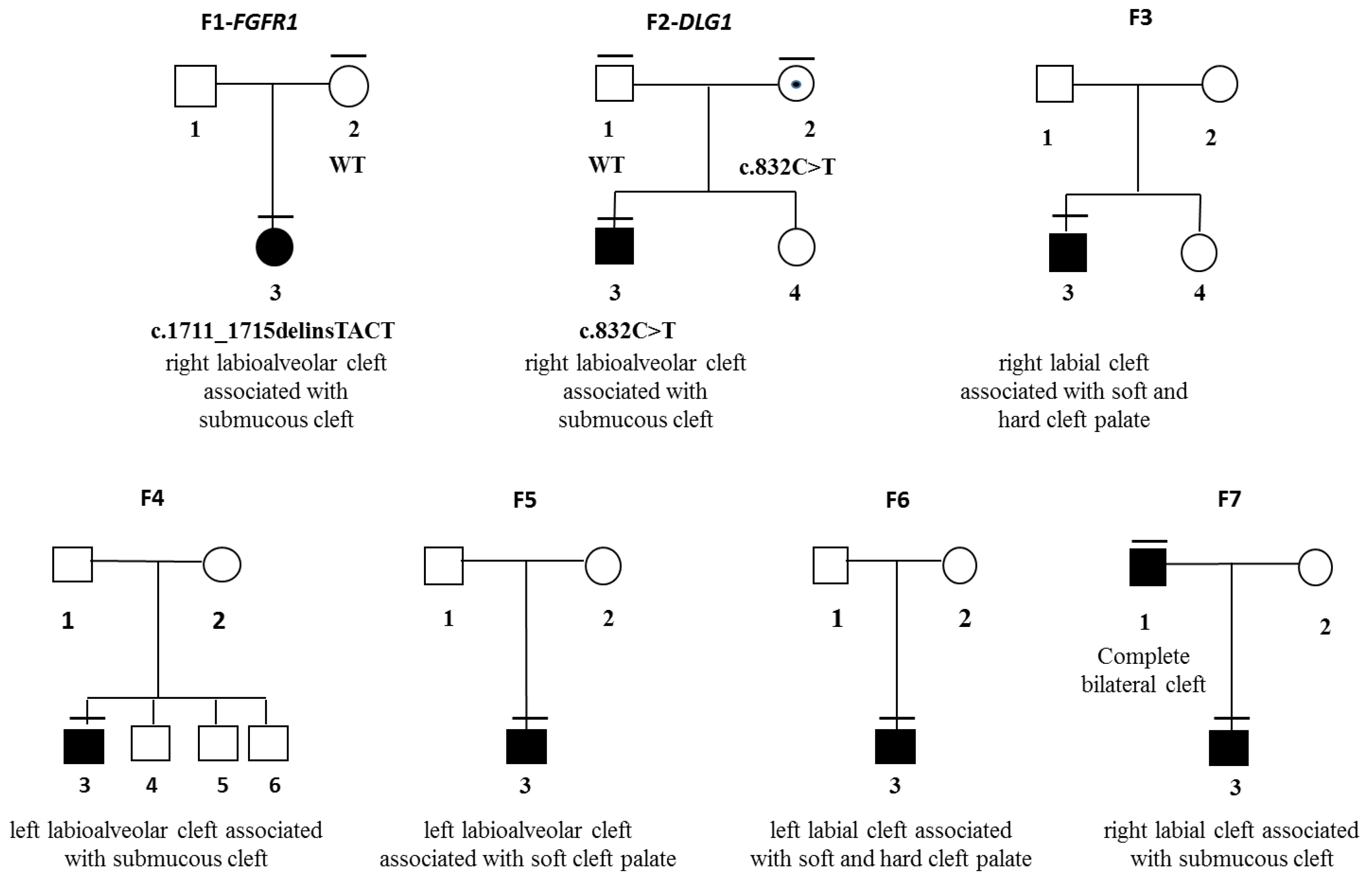
3.1. Patient F1-3
3.2. Patient F2-3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Unlocking mendelian disease using exome sequencing. Genome Biol. 2011, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Bureau, A.; Parker, M.M.; Ruczinski, I.; Taub, M.A.; Marazita, M.L.; Murray, J.C.; Mangold, E.; Noethen, M.M.; Ludwig, K.U.; Hetmanski, J.B.; et al. Whole exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics 2014, 197, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.; Demeer, B.; Revencu, N.; Helaers, R.; Theys, S.; Bou Saba, S.; Boute, O.; Devauchelle, B.; Francois, G.; Bayet, B.; et al. Whole exome sequencing identifies mutations in 10% of patients with familial non-syndromic cleft lip and/or palate in genes mutated in well-known syndromes. J. Med. Genet. 2018, 55, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Neiswanger, K.; Walker, K.; Klotz, C.M.; Cooper, M.E.; Bardi, K.M.; Brandon, C.A.; Weinberg, S.M.; Vieira, A.R.; Martin, R.A.; Czeizel, A.E.; et al. Whorl patterns on the lower lip are associated with nonsyndromic cleft lip with or without cleft palate. Am. J. Med. Genet. A 2009, 149, 2673–2679. [Google Scholar] [CrossRef]
- Jiang, R.; Bush, J.O.; Lidral, A.C. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev. Dyn. 2006, 235, 1152–1166. [Google Scholar] [CrossRef]
- Alvarez, C.W.; Guion-Almeida, M.L.; Richieri-Costa, A. Clinical and genetic study on 356 brazilian patients with a distinct phenotype of cleft lip and palate without alveolar ridge involvement. J. Cranio-Maxillofac. Surg. 2014, 42, 1952–1957. [Google Scholar] [CrossRef]
- Cuddapah, S.R.; Kominek, S.; Grant, J.H., 3rd; Robin, N.H. Irf6 sequencing in interrupted clefting. Cleft Palate Craniofac. J. 2016, 53, 373–376. [Google Scholar] [CrossRef]
- Highlander. Available online: http://sites.uclouvain.be/highlander/ (accessed on 23 May 2019).
- Genome Aggregation Database (gnomAD). Available online: http://gnomad.broadinstitute.org/ (accessed on 23 May 2019).
- Exome Aggregation Consortium (ExAC). Available online: http://exac.broadinstitute.org/ (accessed on 23 May 2019).
- 1000 Genomes. Available online: http://www.1000genomes.org/ (accessed on 23 May 2019).
- Genome of the Netherlands (GoNL). Available online: http://www.nlgenome.nl/ (accessed on 23 May 2019).
- Stanier, P.; Moore, G.E. Genetics of cleft lip and palate: Syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 2004, 13 (Suppl. 1), R73–R81. [Google Scholar] [CrossRef]
- Leslie, E.J.; Murray, J.C. Evaluating rare coding variants as contributing causes to non-syndromic cleft lip and palate. Clin. Genet. 2013, 84, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Jugessur, A.; Farlie, P.G.; Kilpatrick, N. The genetics of isolated orofacial clefts: From genotypes to subphenotypes. Oral Dis. 2009, 15, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Kousa, Y.A.; Mansour, T.A.; Seada, H.; Matoo, S.; Schutte, B.C. Shared molecular networks in orofacial and neural tube development. Birth Defects Res. 2017, 109, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Conte, F.; Oti, M.; Dixon, J.; Carels, C.E.; Rubini, M.; Zhou, H. Systematic analysis of copy number variants of a large cohort of orofacial cleft patients identifies candidate genes for orofacial clefts. Hum. Genet. 2016, 135, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Lough, K.J.; Byrd, K.M.; Spitzer, D.C.; Williams, S.E. Closing the gap: Mouse models to study adhesion in secondary palatogenesis. J. Dent. Res. 2017, 96, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Mansilla, M.A.; Biggs, L.C.; Schuette, K.; Bullard, S.; Cooper, M.; Dunnwald, M.; Lidral, A.C.; Marazita, M.L.; Beaty, T.H.; et al. Expression and mutation analyses implicate arhgap29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 934–942. [Google Scholar] [CrossRef]
- Peyrard-Janvid, M.; Leslie, E.J.; Kousa, Y.A.; Smith, T.L.; Dunnwald, M.; Magnusson, M.; Lentz, B.A.; Unneberg, P.; Fransson, I.; Koillinen, H.K.; et al. Dominant mutations in grhl3 cause van der woude syndrome and disrupt oral periderm development. Am. J. Hum. Genet. 2014, 94, 23–32. [Google Scholar] [CrossRef]
- Baek, J.A.; Lan, Y.; Liu, H.; Maltby, K.M.; Mishina, Y.; Jiang, R. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev. Biol. 2011, 350, 520–531. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, K.U.; Ahmed, S.T.; Bohmer, A.C.; Sangani, N.B.; Varghese, S.; Klamt, J.; Schuenke, H.; Gultepe, P.; Hofmann, A.; Rubini, M.; et al. Meta-analysis reveals genome-wide significance at 15q13 for nonsyndromic clefting of both the lip and the palate, and functional analyses implicate grem1 as a plausible causative gene. PLoS Genet. 2016, 12, e1005914. [Google Scholar] [CrossRef]
- Jugessur, A.; Shi, M.; Gjessing, H.K.; Lie, R.T.; Wilcox, A.J.; Weinberg, C.R.; Christensen, K.; Boyles, A.L.; Daack-Hirsch, S.; Trung, T.N.; et al. Genetic determinants of facial clefting: Analysis of 357 candidate genes using two national cleft studies from scandinavia. PLoS ONE 2009, 4, e5385. [Google Scholar] [CrossRef]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in fgfr1 cause autosomal dominant kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Acierno, J.S., Jr.; Meysing, A.; Eliseenkova, A.V.; Ma, J.; Ibrahimi, O.A.; Metzger, D.L.; Hayes, F.J.; Dwyer, A.A.; Hughes, V.A.; et al. Mutations in fibroblast growth factor receptor 1 cause both kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2006, 103, 6281–6286. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.M.; Mansilla, M.A.; Ma, J.; Daack-Hirsch, S.; Maher, B.S.; Raffensperger, L.M.; Russo, E.T.; Vieira, A.R.; Dode, C.; Mohammadi, M.; et al. Impaired fgf signaling contributes to cleft lip and palate. Proc. Natl. Acad. Sci. USA 2007, 104, 4512–4517. [Google Scholar] [CrossRef] [PubMed]
- Mostowska, A.; Gaczkowska, A.; Zukowski, K.; Ludwig, K.U.; Hozyasz, K.K.; Wojcicki, P.; Mangold, E.; Bohmer, A.C.; Heilmann-Heimbach, S.; Knapp, M.; et al. Common variants in dlg1 locus are associated with non-syndromic cleft lip with or without cleft palate. Clin. Genet. 2018, 93, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Caruana, G.; Bernstein, A. Craniofacial dysmorphogenesis including cleft palate in mice with an insertional mutation in the discs large gene. Mol. Cell. Biol. 2001, 21, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Rivera, C.; Simonson, S.J.; Yamben, I.F.; Shatadal, S.; Nguyen, M.M.; Beurg, M.; Lambert, P.F.; Griep, A.E. Requirement for dlgh-1 in planar cell polarity and skeletogenesis during vertebrate development. PLoS ONE 2013, 8, e54410. [Google Scholar] [CrossRef]
- Willatt, L.; Cox, J.; Barber, J.; Cabanas, E.D.; Collins, A.; Donnai, D.; FitzPatrick, D.R.; Maher, E.; Martin, H.; Parnau, J.; et al. 3q29 microdeletion syndrome: Clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet. 2005, 77, 154–160. [Google Scholar] [CrossRef]
- Genotype to Mendelian Phenotype (Geno2MP). Available online: https://geno2mp.gs.washington.edu (accessed on 23 May 2019).
- De Lima, R.L.; Hoper, S.A.; Ghassibe, M.; Cooper, M.E.; Rorick, N.K.; Kondo, S.; Katz, L.; Marazita, M.L.; Compton, J.; Bale, S.; et al. Prevalence and nonrandom distribution of exonic mutations in interferon regulatory factor 6 in 307 families with van der woude syndrome and 37 families with popliteal pterygium syndrome. Genet. Med. 2009, 11, 241–247. [Google Scholar] [CrossRef]
- Ghassibe, M.; Bayet, B.; Revencu, N.; Verellen-Dumoulin, C.; Gillerot, Y.; Vanwijck, R.; Vikkula, M. Interferon regulatory factor-6: A gene predisposing to isolated cleft lip with or without cleft palate in the belgian population. Eur. J. Hum. Genet. 2005, 13, 1239–1242. [Google Scholar] [CrossRef]
- Desmyter, L.; Ghassibe, M.; Revencu, N.; Boute, O.; Lees, M.; Francois, G.; Verellen-Dumoulin, C.; Sznajer, Y.; Moncla, A.; Benateau, H.; et al. Irf6 screening of syndromic and a priori non-syndromic cleft lip and palate patients: Identification of a new type of minor vws sign. Mol. Syndromol. 2010, 1, 67–74. [Google Scholar] [CrossRef]
- Grosen, D.; Chevrier, C.; Skytthe, A.; Bille, C.; Molsted, K.; Sivertsen, A.; Murray, J.C.; Christensen, K. A cohort study of recurrence patterns among more than 54,000 relatives of oral cleft cases in denmark: Support for the multifactorial threshold model of inheritance. J. Med. Genet. 2010, 47, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Busch, T.; Eliason, S.; Anand, D.; Bullard, S.; Gowans, L.J.J.; Nidey, N.; Petrin, A.; Augustine-Akpan, E.A.; Saadi, I.; et al. Exome sequencing provides additional evidence for the involvement of arhgap29 in mendelian orofacial clefting and extends the phenotypic spectrum to isolated cleft palate. Birth Defects Res. 2017, 109, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Pengelly, R.J.; Arias, L.; Martinez, J.; Upstill-Goddard, R.; Seaby, E.G.; Gibson, J.; Ennis, S.; Collins, A.; Briceno, I. Deleterious coding variants in multi-case families with non-syndromic cleft lip and/or palate phenotypes. Sci. Rep. 2016, 6, 30457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aylward, A.; Cai, Y.; Lee, A.; Blue, E.; Rabinowitz, D.; Haddad, J., Jr.; University of Washington Center for Mendelian Genomics. Using whole exome sequencing to identify candidate genes with rare variants in nonsyndromic cleft lip and palate. Genet. Epidemiol. 2016, 40, 432–441. [Google Scholar] [CrossRef]
- Hoebel, A.K.; Drichel, D.; van de Vorst, M.; Bohmer, A.C.; Sivalingam, S.; Ishorst, N.; Klamt, J.; Golz, L.; Alblas, M.; Maaser, A.; et al. Candidate genes for nonsyndromic cleft palate detected by exome sequencing. J. Dent. Res. 2017, 96, 1314–1321. [Google Scholar] [CrossRef]
- Holzinger, E.R.; Li, Q.; Parker, M.M.; Hetmanski, J.B.; Marazita, M.L.; Mangold, E.; Ludwig, K.U.; Taub, M.A.; Begum, F.; Murray, J.C.; et al. Analysis of sequence data to identify potential risk variants for oral clefts in multiplex families. Mol. Genet. Genom. Med. 2017, 5, 570–579. [Google Scholar] [CrossRef]
- Cox, L.L.; Cox, T.C.; Moreno Uribe, L.M.; Zhu, Y.; Richter, C.T.; Nidey, N.; Standley, J.M.; Deng, M.; Blue, E.; Chong, J.X.; et al. Mutations in the epithelial cadherin-p120-catenin complex cause mendelian non-syndromic cleft lip with or without cleft palate. Am. J. Hum. Genet. 2018, 102, 1143–1157. [Google Scholar] [CrossRef]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies mll2 mutations as a cause of kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef]
- Leslie, E.J.; Koboldt, D.C.; Kang, C.J.; Ma, L.; Hecht, J.T.; Wehby, G.L.; Christensen, K.; Czeizel, A.E.; Deleyiannis, F.W.; Fulton, R.S.; et al. Irf6 mutation screening in non-syndromic orofacial clefting: Analysis of 1521 families. Clin. Genet. 2016, 90, 28–34. [Google Scholar] [CrossRef]
- Demeer, B.; Revencu, N.; Helaers, R.; Devauchelle, B.; Francois, G.; Bayet, B.; Vikkula, M. Unmasking familial cpx by wes and identification of novel clinical signs. Am. J. Med. Genet. A 2018, 176, 2661–2667. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Patient | Gender | CLP Familial History | Discontinuous CLP Description | Gene, Variant and Protein Change | |
|---|---|---|---|---|---|
| Cleft of the Primary Palate | Cleft of the Secondary Palate | ||||
| 1 | F | − | right labioalveolar | submucous | FGFR1, c.1809_1810insTC; p.Glu571Tyrfs*61 |
| 2 | M | − | left labioalveolar | submucous | DLG1, c.832C>T; p.Arg278* |
| 3 | M | − | right labioalveolar | submucous | |
| 4 | M | − | left labioalveolar | submucous | |
| 5 | M | − | right labial | soft and hard palate | |
| 6 | M | − | left labial | soft and hard palate | |
| 7 | M | + | left labial | submucous | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demeer, B.; Revencu, N.; Helaers, R.; Gbaguidi, C.; Dakpe, S.; François, G.; Devauchelle, B.; Bayet, B.; Vikkula, M. Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients. Genes 2019, 10, 833. https://doi.org/10.3390/genes10100833
Demeer B, Revencu N, Helaers R, Gbaguidi C, Dakpe S, François G, Devauchelle B, Bayet B, Vikkula M. Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients. Genes. 2019; 10(10):833. https://doi.org/10.3390/genes10100833
Chicago/Turabian StyleDemeer, Bénédicte, Nicole Revencu, Raphael Helaers, Cica Gbaguidi, Stéphanie Dakpe, Geneviève François, Bernard Devauchelle, Bénédicte Bayet, and Miikka Vikkula. 2019. "Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients" Genes 10, no. 10: 833. https://doi.org/10.3390/genes10100833
APA StyleDemeer, B., Revencu, N., Helaers, R., Gbaguidi, C., Dakpe, S., François, G., Devauchelle, B., Bayet, B., & Vikkula, M. (2019). Likely Pathogenic Variants in One Third of Non-Syndromic Discontinuous Cleft Lip and Palate Patients. Genes, 10(10), 833. https://doi.org/10.3390/genes10100833