Rare Genetic Variants in Jewish Patients Suffering from Age-Related Macular Degeneration

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Patients and Clinical Evaluation
2.2. Molecular Studies
- (1)
- Mutation screening for previously reported rare and common variants in our population.
- (2)
- WES and bioinformatics analysis.
- (3)
- Screening an in-house cohort for more cases carrying the identified new variant.
2.3. Mutation Screening and Sanger Sequencing (Phase-1)
2.4. Whole Exome Sequencing and Bioinformatic Analysis (Phase-2)
2.5. Further Screening of an In-House AMD Cohort for a Presumed Pathogenic Variant (Phase-3)
3. Results
3.1. Mutation Screening for Previously Described Mutations in CFI and HMCN1 Genes (Phase-1)
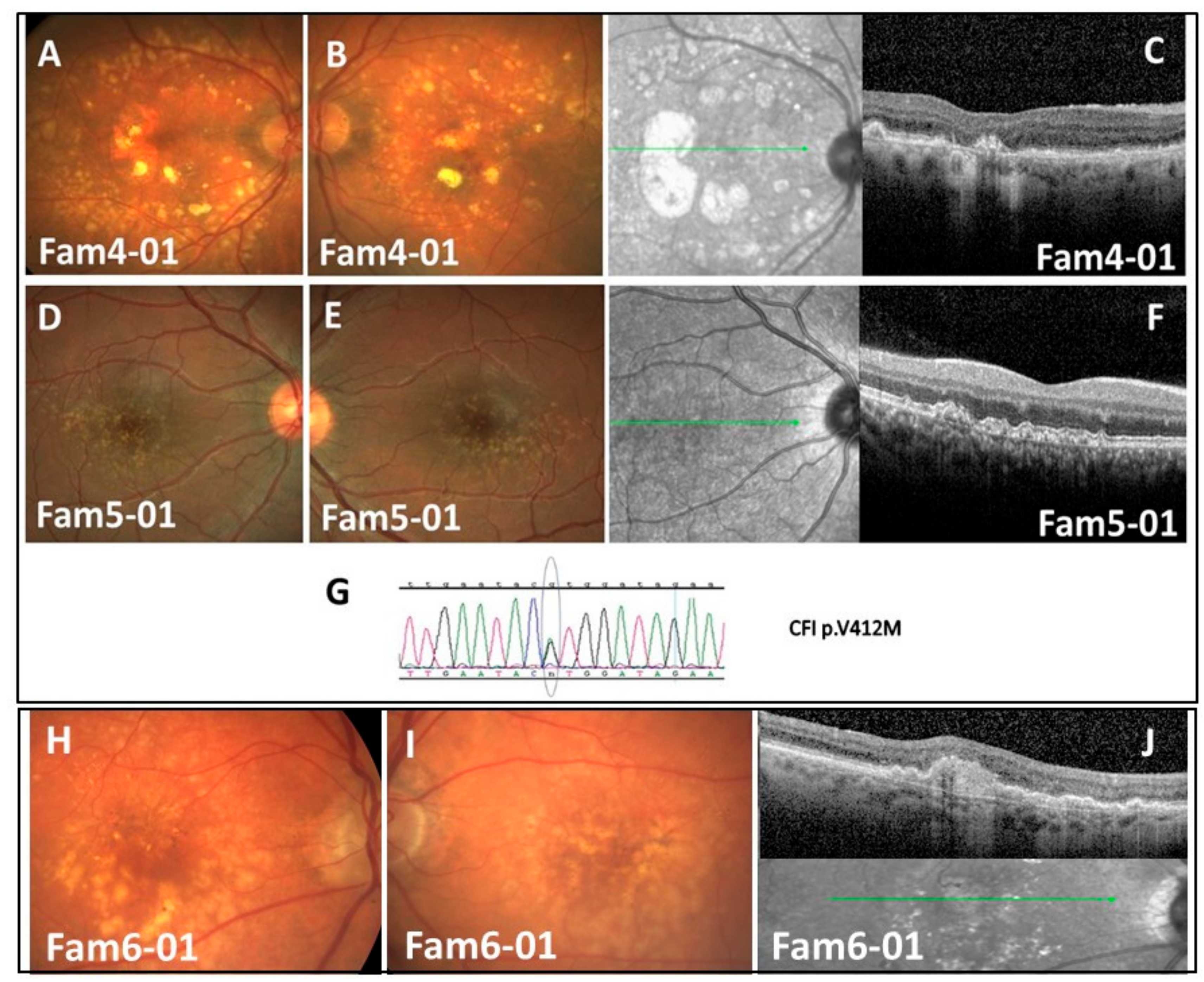
Families 4, 5, and 8 with CFI p.V412M (Figure 1A–G)
3.2. WES Analysis (Phase-2) and Screening More Families for an Identified Mutation (Phase-3)
3.2.1. Family-1 with CFH p.R1210C (Figure 2A–E)
3.2.2. Family-2 with PLEKHA1 P.S177N (Figure 2F–J)
3.2.3. Family-3 with C3 p.R735W; ABCA4 p.G1931E; CFI p.K441R (Phase-2)
3.2.4. Family-6 with C3 p.R735W (phase-3) (Figure 1H–J)
3.2.5. Family 7
3.2.6. Family 8
4. Discussion
4.1. Patients and Gene Selection
4.2. Rare Variants
4.3. Common Variants
4.4. Summary
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Velez-Montoya, R.; Oliver, S.C.N.; Olson, J.L.; Fine, S.L.; Quiroz-Mercado, H.; Mandava, N. Current knowledge and trends in age-related macular degeneration: Genetics, epidemiology, and prevention. Retina 2014, 34, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Joachim, N.; Mitchell, P.; Burlutsky, G.; Kifley, A.; Wang, J.J. The incidence and progression of age-related macular degeneration over 15 years: The Blue Mountains Eye Study. Ophthalmology 2015, 122, 2482–2489. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.A.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Maller, J.; George, S.; Purcell, S.; Fagerness, J.; Altshuler, D.; Daly, M.J.; Seddon, J.M. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat. Genet. 2006, 38, 1055–1059. [Google Scholar] [CrossRef]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 Variant and the Risk of Age-Related Macular Degeneration. New Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Dong, S.; Zhao, C.; Wang, H.; Dai, F.; Yang, J. Cumulative association between age-related macular degeneration and less studied genetic variants in PLEKHA1/ARMS2/HTRA1: A meta and gene-cluster analysis. Mol. Biol. Rep. 2013, 40, 5551–5561. [Google Scholar] [CrossRef]
- Jabbarpoor Bonyadi, M.H.; Yaseri, M.; Nikkhah, H.; Bonyadi, M.; Soheilian, M. Association of risk genotypes of ARMS2/LOC387715 A69S and CFH Y402H with age-related macular degeneration with and without reticular pseudodrusen: A meta-analysis. Acta Ophthalmol. 2018, 96, e105–e110. [Google Scholar] [CrossRef]
- Jabbarpoor Bonyadi, M.H.; Yaseri, M.; Nikkhah, H.; Bonyadi, M.; Nazari, R.; Soheilian, M. Comparison of ARMS2/LOC387715 A69S and CFH Y402H risk effect in wet-type age-related macular degeneration: A meta-analysis. Int. Ophthalmol. 2019, 39, 949–956. [Google Scholar] [CrossRef]
- Priya, R.R.; Chew, E.Y.; Swaroop, A. Genetic studies of age-related macular degeneration: Lessons, challenges, and opportunities for disease management. Ophthalmology 2012, 119, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, C.J.; Klevering, B.J.; Hoyng, C.B.; Zonneveld-Vrieling, M.N.; Nabuurs, S.B.; Blokland, E.; Cremers, F.P.; Hollander, A.I.D. Basal Laminar Drusen Caused by Compound Heterozygous Variants in the CFH Gene. Am. J. Hum. Genet. 2008, 82, 516–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Józsi, M.; Heinen, S.; Hartmann, A.; Ostrowicz, C.W.; Hälbich, S.; Richter, H.; Kunert, A.; Licht, C.; Saunders, R.E.; Perkins, S.J.; et al. Factor H and Atypical Hemolytic Uremic Syndrome: Mutations in the C-Terminus Cause Structural Changes and Defective Recognition Functions. J. Am. Soc. Nephrol. 2005, 17, 170–177. [Google Scholar] [CrossRef]
- Yu, Y.; Triebwasser, M.P.; Wong, E.K.S.; Schramm, E.C.; Thomas, B.; Reynolds, R.; Mardis, E.R.; Atkinson, J.P.; Daly, M.; Raychaudhuri, S.; et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum. Mol. Genet. 2014, 23, 5283–5293. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, J.D.; CookeBailey, J.N.; D’Aoust, L.; Cade, W.; Ayala-Haedo, J.; Fuzzell, D.; Laux, R.; Adams, L.D.; Reinhart-Mercer, L.; Caywood, L.; et al. Rare Complement Factor H Variant Associated with Age-Related Macular Degeneration in the Amish. Investig. Opthalmology Vis. Sci. 2014, 55, 4455–4460. [Google Scholar] [CrossRef]
- Helgason, H.; Sulem, P.; Duvvari, M.R.; Luo, H.; Thorleifsson, G.; Stefansson, H.; Jonsdottir, I.; Masson, G.; Gudbjartsson, D.F.; Walters, G.B.; et al. A rare nonsynonymous sequence variant in C3 is associated with high risk of age-related macular degeneration. Nat. Genet. 2013, 45, 1371–1374. [Google Scholar] [CrossRef]
- Pras, E.; Kristal, D.; Shoshany, N.; Volodarsky, D.; Vulih, I.; Celniker, G.; Isakov, O.; Shomron, N. Rare genetic variants in Tunisian Jewish patients suffering from age-related macular degeneration. J. Med. Genet. 2015, 52, 484–492. [Google Scholar] [CrossRef]
- Database of Single Nucleotide Polymorphisms (dbSNP). National Center for Biotechnology Information, National Library of Medicine; Database of Single Nucleotide Polymorphisms: Bethesda, MD, USA, 2000. [Google Scholar]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). Available online: http://evs.gs.washington.edu/ (accessed on 10 April 2019).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nat. 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Barricarte, R.; Pianetti, G.; Gautard, R.; Misselwitz, J.; Strain, L.; Fremeaux-Bacchi, V.; Skerka, C.; Zipfel, P.F.; Goodship, T.; Noris, M.; et al. The complement factor H R1210C mutation is associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2008, 19, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Reynolds, R.; Yu, Y.; Rosner, B. Three New Genetic Loci (R1210C in CFH, Variants in COL8A1 and RAD51B) Are Independently Related to Progression to Advanced Macular Degeneration. PLoS ONE 2014, 9, e87047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, L.-Y.; Wang, Y.-F.; Wu, C.-R.; Lei, C.-L.; Wang, M.-X.; Ma, L. Associations of the G1961E and D2177N variants in ABCA4 and the risk of age-related macular degeneration. Gene 2015, 567, 51–57. [Google Scholar] [CrossRef]
- Duvvari, M.R.; Paun, C.C.; Buitendijk, G.H.S.; Saksens, N.T.M.; Volokhina, E.B.; Ristau, T.; Schoenmaker-Koller, F.E.; Van De Ven, J.P.H.; Groenewoud, J.M.M.; Heuvel, L.P.; et al. Analysis of Rare Variants in the C3 Gene in Patients with Age-Related Macular Degeneration. PLoS ONE 2014, 9, e94165. [Google Scholar] [CrossRef]
- Caycı, F.S.; Cakar, N.; Hancer, V.S.; Uncu, N.; Acar, B.; Gur, G. Eculizumab therapy in a child with hemolytic uremic syndrome and CFI mutation. Pediatr. Nephrol. 2012, 27, 2327–2331. [Google Scholar] [CrossRef]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- International HapMap 3 Consortium; Altshuler, D.M.; Gibbs, R.A.; Peltonen, L.; Dermitzakis, E.; Schaffner, S.F.; Yu, F.; Bonnen, P.E.; de Bakker, P.I.; Deloukas, P.; et al. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52–58. [Google Scholar] [CrossRef]
- Yu, Y.; Wagner, E.K.; Souied, E.H.; Seitsonen, S.; Immonen, I.J.; Häppölä, P.; Raychaudhuri, S.; Daly, M.J.; Seddon, J.M. Protective coding variants in CFH and PELI3 and a variant near CTRB1 are associated with age-related macular degeneration. Hum. Mol. Genet. 2016, 25, 5276–5285. [Google Scholar] [CrossRef] [PubMed]
- Duvvari, M.R.; van de Ven, J.P.; Geerlings, M.J.; Saksens, N.T.; Bakker, B.; Henkes, A.; Neveling, K.; del Rosario, M.; Westra, D.; van den Heuvel, L.P.; et al. Whole Exome Sequencing in Patients with the Cuticular Drusen Subtype of Age-Related Macular Degeneration. PLoS ONE 2016, 11, e0152047. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Lambris, J.D. Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 2001, 180, 35–48. [Google Scholar] [CrossRef]
- Wang, G. Chromosome 10q26 locus and age-related macular degeneration: A progress update. Exp. Eye Res. 2014, 119, 1–7. [Google Scholar] [CrossRef]
- Conley, Y.P.; Jakobsdottir, J.; Mah, T.; Weeks, D.E.; Klein, R.; Kuller, L.; Ferrell, R.E.; Gorin, M.B. CFH, ELOVL4, PLEKHA1 and LOC387715 genes and susceptibility to age-related maculopathy: AREDS and CHS cohorts and meta-analyses. Hum. Molec. Genet. 2006, 15, 3206–3218. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Family | Ancestry | Analysis Method | Gene | c. Variant | p. Variant | Effect | Predicted Severity | Prevalence | Proband | Relative (Affectation Status +/−) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WES | Mutation Screening | ||||||||||
| 1 | Jewish Ashkenazi | + | CFH | c.3268C>T | p.Arg1210Cys | Nonsyn. SNV | High | 0.000173 | Het | WT Hom (−) | |
| 2 | Jewish Syrian (Oriental) | + | PLEKHA1 | c.530G>A | p.Ser177Asn | Nonsyn. SNV | High | 0.001735 | Het | Het (+) | |
| 3 | Jewish Ashkenazi | + | ABCA4 | c.5882G>A | p.Gly1961Glu | Nonsyn. SNV | High | 0.0023 | Het | Het (+) ** | |
| CFI | c.1346A>G | p.Lys441Arg | Nonsyn. SNV | High | 0.0009 | Het | WT Hom (+) ** | ||||
| C3* | c.2203C>T | p.Arg735Trp | Nonsyn. SNV | High | 0.0005 | Het | WT Hom (+) ** | ||||
| 6 | + | - | |||||||||
| 4 | Jewish Tunisian (North African) | + | CFI | c.1234G>A | p.Val412Met | Nonsyn. SNV | High | 0.000107 | Het | Het (+) | |
| 5 | + | ||||||||||
| 8 | + | - | |||||||||
| Family | Subject | CFH p.Y402H | ARMS2 p.A69S |
|---|---|---|---|
| 1 | Proband | Het | Het |
| Relative | WT Hom | Het | |
| 2 | Proband | WT Hom | WT Hom |
| Relative | WT Hom | WT Hom | |
| 3 | Proband | WT Hom | WT Hom |
| Relative | WT Hom | WT Hom | |
| 4 | Proband | WT Hom | Het |
| Relative | WT Hom | WT Hom | |
| 5 | N/A | ||
| 6 | |||
| 7 | |||
| 8 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoshany, N.; Weiner, C.; Safir, M.; Einan-Lifshitz, A.; Pokroy, R.; Kol, A.; Modai, S.; Shomron, N.; Pras, E. Rare Genetic Variants in Jewish Patients Suffering from Age-Related Macular Degeneration. Genes 2019, 10, 825. https://doi.org/10.3390/genes10100825
Shoshany N, Weiner C, Safir M, Einan-Lifshitz A, Pokroy R, Kol A, Modai S, Shomron N, Pras E. Rare Genetic Variants in Jewish Patients Suffering from Age-Related Macular Degeneration. Genes. 2019; 10(10):825. https://doi.org/10.3390/genes10100825
Chicago/Turabian StyleShoshany, Nadav, Chen Weiner, Margarita Safir, Adi Einan-Lifshitz, Russell Pokroy, Ayala Kol, Shira Modai, Noam Shomron, and Eran Pras. 2019. "Rare Genetic Variants in Jewish Patients Suffering from Age-Related Macular Degeneration" Genes 10, no. 10: 825. https://doi.org/10.3390/genes10100825