Transcriptome Profile Analysis of Mammary Gland Tissue from Two Breeds of Lactating Sheep

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals and RNA Preparation
2.3. cDNA Library Construction and RNA-Seq
2.4. Mapping of Sequencing Reads and Identification of DEGs
2.5. GO and KEGG Pathway Analyses
2.6. Validation of DEGs by Reverse Transcription-Quantitative PCR
3. Results
3.1. RNA-Seq Reads and Mapping to the Reference Genome
3.2. Identification of DEGs
3.3. GO Enrichment and KEGG Pathway Analyses
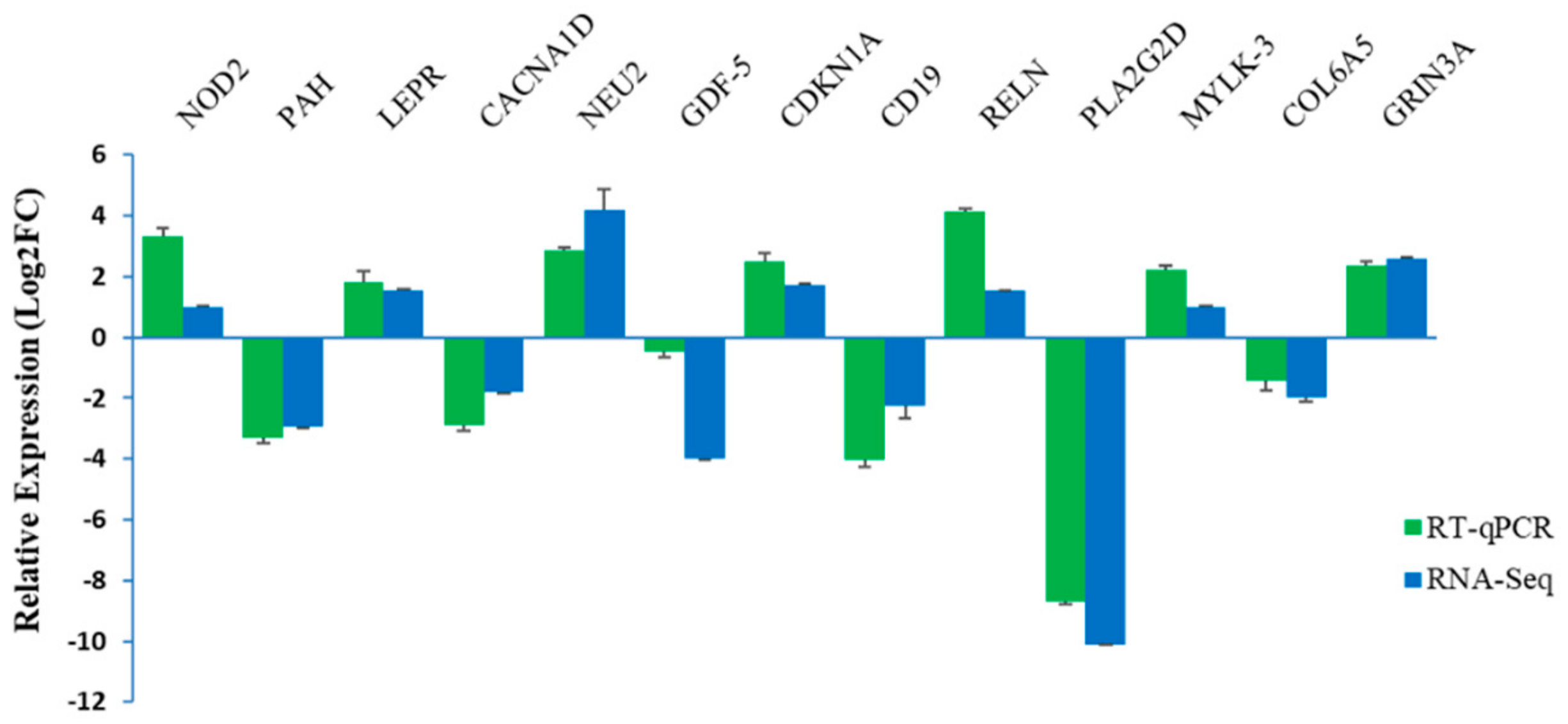
3.4. Validation of Selected DEGs Using RT-qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bittante, G.; Cipolatgotet, C.; Pazzola, M.; Dettori, M.L.; Vacca, G.M.; Cecchinato, A. Genetic analysis of coagulation properties, curd firming modeling, milk yield, composition, and acidity in Sarda dairy sheep. J. Dairy Sci. 2017, 100, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Lemay, D.G.; Ballard, O.A.; Hughes, M.A.; Morrow, A.L.; Horseman, N.D.; Nommsen-rivers, A. RNA sequencing of the human milk fat layer transcriptome reveals distinct gene expression profiles at three stages of lactation. PloS ONE 2013, 8, e67531. [Google Scholar] [CrossRef] [PubMed]
- Lemay, D.G.; Hovey, R.C.; Hartono, S.R.; Hinde, K.; Smilowitz, J.T.; Ventimiglia, F.; Schmidt, K.A.; Lee, J.W.; Islas-trejo, A.; Silva, P.I. Sequencing the transcriptome of milk production: Milk trumps mammary tissue. BMC Genomics 2013, 14, 872. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.G.; Hou, Y.; Yang, S.H.; Xie, Y.; Zhang, S.L.; Zhang, Y.; Zhang, Q.; Lu, X.M.; Liu, G.E.; Sun, D.X. Transcriptional profiling of mammary gland in Holstein cows with extremely different milk protein and fat percentage using RNA sequencing. BMC Genomics 2014, 15, 226. [Google Scholar] [CrossRef]
- Wickramasinghe, S.; Rincon, G.; Islastrejo, A.; Medrano, J.F. Transcriptional profiling of bovine milk using RNA sequencing. BMC Genomics 2012, 13, 45. [Google Scholar] [CrossRef]
- Fan, J.F.; Luo, Y.Z.; Yu, S.J.; Cui, Y.; Xu, G.Q.; Wang, L.B.; Pang, Y.Y.; He, H.H. Transcriptional profiling of two different physiological states of the yak mammary gland using RNA sequencing. PLoS ONE 2018, 13, e0201628. [Google Scholar]
- Paten, A.M.; Duncan, E.J.; Pain, S.J.; Peterson, S.W.; Kenyon, P.R.; Blair, H.T.; Dearden, P.K. Functional development of the adult ovine mammary gland-insights from gene expression profiling. BMC Genomics 2015, 16, 748. [Google Scholar] [CrossRef]
- Suárez-Vega, A.; Gutiérrez, G.B.; Klopp, C.; Robert, G.C.; Tosser, K.G.; Arranz, J.J. Characterization and comparative analysis of the milk transcriptome in two dairy sheep breeds using RNA sequencing. Sci. Rep. 2015, 5, 18399–18410. [Google Scholar] [CrossRef]
- Suárez-Vega, A.; Gutiérrez, G.B.; Klopp, C.; Tosser-Klopp, G.; Arranz, J.J. Comprehensive RNA-Seq profiling to evaluate lactating sheep mammary gland transcriptome. Sci. Data 2016, 3, 160051. [Google Scholar] [CrossRef]
- Akers, R.M. A 100-year review: Mammary development and lactation. J. Dairy Sci. 2017, 100, 10332–10352. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.Z. Chinese Yang Yang Xue (In Chinese), 1st ed.; China Agricultural Press: Beijing, China, 2013; pp. 90–155. [Google Scholar]
- Li, W.; Guo, J.; Li, F.; Niu, C. Evaluation of crossbreeding of Australian superfine merinos with Gansu Alpine finewool sheep to improve wool characteristics. PLOS ONE 2016, 11, e0166374. [Google Scholar] [CrossRef] [PubMed]
- Paten, A.M.; Pain, S.J.; Peterson, S.W.; Blair, H.T.; Kenyon, P.R.; Dearden, P.K.; Duncan, E.J. Identification of reference genes for RT-qPCR in ovine mammary tissue during late pregnancy and lactation and in response to maternal nutritional programming. Physiol. Genomics 2014, 46, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet Journal. 2011, 17, 3. [Google Scholar] [CrossRef]
- Mayer, K.F.; Waugh, R.; Brown, J.W.; Schulman, A.; Langridge, P.; Platzer, M.; Fincher, G.B.; Muehlbauer, G.J.; Sato, K.; Close, T.J. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar] [PubMed]
- Wang, L.K.; Feng, Z.X.; Wang, X.; Wang, X.W.; Zhang, X.G. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550–571. [Google Scholar] [CrossRef]
- KEGG PATHWAY Database. Available online: https://www.genome.jp/kegg/pathway.html (accessed on 16 June 2018).
- Goodall, A.J.; Kumar, P.; Tobin, A.K. Identification and expression analyses of cytosolic glutamine synthetase genes in barley (Hordeum vulgare L.). Plant Cell Physiol. 2013, 54, 492–505. [Google Scholar] [CrossRef]
- Canovas, A.; Rincón, G.; Bevilacqua, C.; Islas-Trejo, A.; Brenaut, P.; Hovey, R.C.; Boutinaud, M.; Morgenthaler, C.; VanKlompenberg, M.K.; Martin, P. Comparison of five different RNA sources to examine the lactating bovine mammary gland transcriptome using RNA-Sequencing. Sci. Rep. 2014, 4, 5297. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Tosser-Klopp, G.; Arranz, J.J. Variant discovery in the sheep milk transcriptome using RNA sequencing. BMC Genomics 2017, 18, 170. [Google Scholar] [CrossRef] [PubMed]
- Crisa, A.; Ferre, F.; Chillemi, G.; Moioli, B. RNA-Sequencing for profiling goat milk transcriptome in colostrum and mature milk. BMC Vet. Res. 2016, 12, 264–285. [Google Scholar] [CrossRef] [PubMed]
- Wilk, J.B.; Walter, R.E.; Laramie, J.M.; Gottlieb, D.J.; Connor, G.T.O. Framingham heart study genome-wide association: results for pulmonary function measures. BMC Med. Genet. 2007, 8, S8. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Veile, R.; Allen, M.; Wise, C.A.; Dobbs, M.; Morcuende, J.A.; Szappanos, L.; Herring, J.A.; Bowcock, A.M.; Lovett, M. SNTG1, the gene encoding gamma1-syntrophin: A candidate gene for idiopathic scoliosis. Hum. Genet. 2004, 115, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Gábor, M.; Sophie, E.; Patrik, W.; Johann, S. A genome wide association study for longevity in cattle. Open J. Genet. 2014, 4, 46–55. [Google Scholar]
- Wang, W.D.; Jiang, M.H.; Liu, S.; Zhang, S.K.; Liu, W.; Ma, Y.W.; Zhang, L.F.; Zhang, J.Y.; Cao, X.T. RNF122 suppresses antiviral type I interferon production by targeting RIG-I CARDs to mediate RIG-I degradation. PNAS 2016, 113, 9581–9586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, 605–616. [Google Scholar] [CrossRef]
- Macias, H.; Hinck, L. Mammary gland development. WIREs Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [Green Version]
- Roarty, K.; Serra, R. Wnt5a is required for proper mammary gland development and TGF-β mediated inhibition of ductal growth. Development 2008, 134, 3929–3939. [Google Scholar] [CrossRef]
- Ji, Z.B.; Chao, T.L.; Zhang, C.L.; Liu, Z.H.; Hou, L.; Wang, J.; Wang, A.L.; Wang, Y.; Zhou, J.; Xuan, R. Transcriptome analysis of dairy goat mammary gland tissues from different lactation stages. DNA Cell Biol. 2019, 38, 129–143. [Google Scholar] [CrossRef]
- Jellusova, J.; Nitschke, L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front. Immunol. 2011, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| NOD2 | GAATTACCGGTCCCATTGGC | ACACTTCTTCCAGGCACAGA |
| PLA2G2D | AACCCAGAGATGCCACAGAC | AAGCCAACGTCTTGTCACAG |
| LEPR | TGTTGCTTTGGAGTGAGGA | TCCAGTGTGCACCTGTTTGT |
| NEU2 | GACGAGCAAGAAGGATGAGC | CGGGGATGGCAATGAAGAAG |
| CDKN1A | GAGAGCGATGGAACTTCGAC | AGTGGTCCTCCTGAGACGTG |
| COL6A5 | GAGACCATCGCAGGGGATAA | ACCATGTCAGAGCCATCACA |
| RELN | ACTCTGGGCCAAACTGCTAT | TTGTCTCACTGTGGATCCCC |
| MYLK-3 | GCTGGCCAGAAGATACAAGC | CGGGAACGAGACAAACTCAT |
| GR1N3A | GCAAATATGGAGCCTGGAAA | CTGGCTTCGTGCAGTATTGA |
| PAH | CGCTGTCCAGGAGTATACGA | TTGTGGCAGCAAAGTTCCTC |
| CACNA1D | TTCCCAGCTCAACAAATGCC | TGCCCGTTTTCAGACACAAG |
| GDF-5 | GGGCTGGGATGACTGGATTA | GGCTGAGTCGATGAAGAGGA |
| CD19 | AGATGCAGCTGAAGGTCACT | CAGGGAAGTCAGGCAGAAGA |
| β-actin | AGCCTTCCTTCCTGGGCATGGA | GGACAGCACCGTGTTGGCGTAGA |
| GAPDH | ATCTCGCTCCTGGAAGATG | TCGGAGTGAACGGATTCG |
| Sample | Useful Reads | Map Event | Map Reads | Map Reads (%) | Multiple Reads | Multiple Reads (%) | Unique Reads | Unique Reads (%) |
|---|---|---|---|---|---|---|---|---|
| GAM-1 | 141244574 | 119373530 | 110324120 | 78.11 | 3934715 | 3.57 | 106389405 | 96.43 |
| GAM-2 | 139563102 | 118260385 | 107968116 | 77.36 | 4219681 | 3.91 | 103748435 | 96.09 |
| GAM-3 | 139407058 | 117075040 | 106817466 | 77.62 | 4707008 | 4.41 | 102110458 | 95.59 |
| STH-1 | 140828556 | 117044888 | 108043045 | 76.72 | 3664975 | 3.39 | 104378070 | 96.61 |
| STH-2 | 136465766 | 113483944 | 103333734 | 75.72 | 4688735 | 4.54 | 98644999 | 95.46 |
| STH-3 | 136990336 | 111826264 | 103170887 | 75.31 | 3926392 | 3.81 | 99244495 | 96.19 |
| KEGG Pathway 1 | Upregulated Genes | Down-Regulated Genes | Corrected p-Value |
|---|---|---|---|
| Hematopoietic cell lineage | CD22, CD19, CD13 | 0.012 | |
| Oxytocin signaling pathway | MYLK-3, CDKN1A | RGS2, CACNA1D | 0.018 |
| Neuroactive ligand-receptor interaction | GIPR, LEPR, GRIN3A | HTR4, LPAR4 | 0.031 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | PAH | 0.034 | |
| cAMP signaling pathway | GIPR, GRIN3A | CACNA1D, HTR4 | 0.048 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Z.; Zhou, H.; Hickford, J.G.H.; Gong, H.; Wang, J.; Hu, J.; Liu, X.; Li, S.; Zhao, M.; Luo, Y. Transcriptome Profile Analysis of Mammary Gland Tissue from Two Breeds of Lactating Sheep. Genes 2019, 10, 781. https://doi.org/10.3390/genes10100781
Hao Z, Zhou H, Hickford JGH, Gong H, Wang J, Hu J, Liu X, Li S, Zhao M, Luo Y. Transcriptome Profile Analysis of Mammary Gland Tissue from Two Breeds of Lactating Sheep. Genes. 2019; 10(10):781. https://doi.org/10.3390/genes10100781
Chicago/Turabian StyleHao, Zhiyun, Huitong Zhou, Jon G.H. Hickford, Hua Gong, Jiqing Wang, Jiang Hu, Xiu Liu, Shaobin Li, Mengli Zhao, and Yuzhu Luo. 2019. "Transcriptome Profile Analysis of Mammary Gland Tissue from Two Breeds of Lactating Sheep" Genes 10, no. 10: 781. https://doi.org/10.3390/genes10100781
APA StyleHao, Z., Zhou, H., Hickford, J. G. H., Gong, H., Wang, J., Hu, J., Liu, X., Li, S., Zhao, M., & Luo, Y. (2019). Transcriptome Profile Analysis of Mammary Gland Tissue from Two Breeds of Lactating Sheep. Genes, 10(10), 781. https://doi.org/10.3390/genes10100781