Integrating GWAS and Gene Expression Analysis Identifies Candidate Genes for Root Morphology Traits in Maize at the Seedling Stage

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Plant Phenotyping and Data Analysis
2.3. Genotypic Data and Genome-Wide Association Analysis
2.4. Candidate Gene Analysis and Transcriptome Sequencing
3. Results
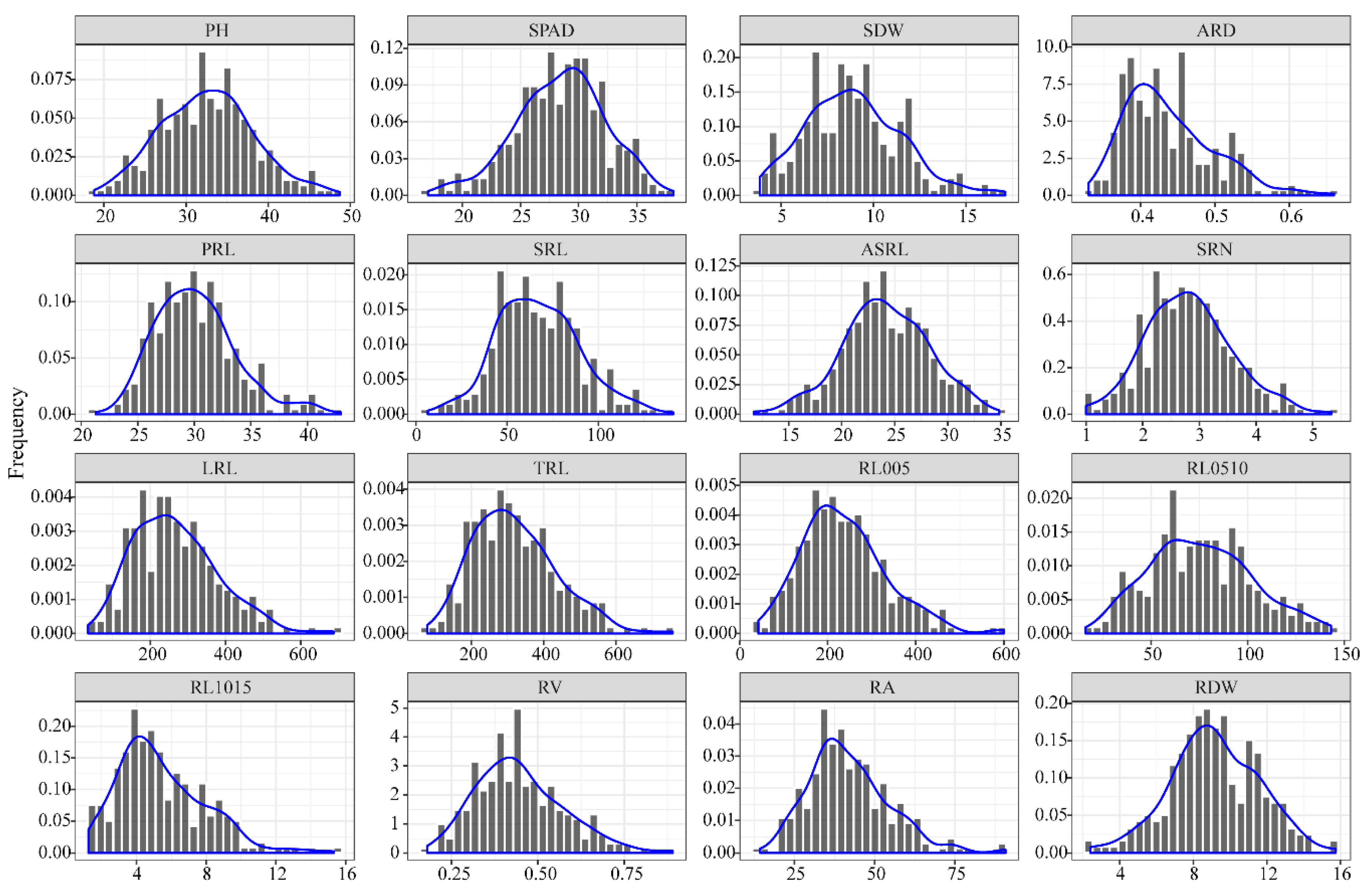
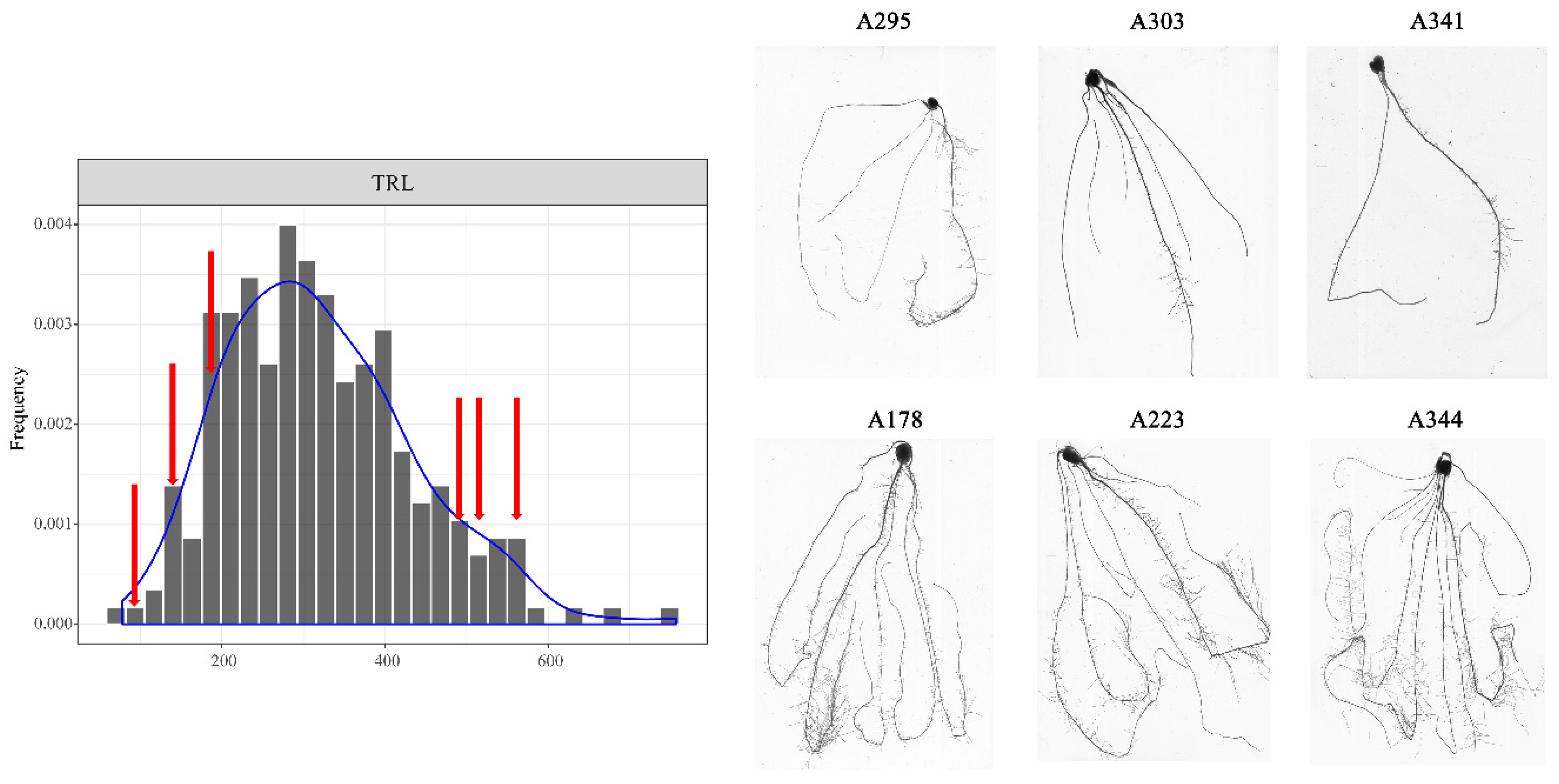
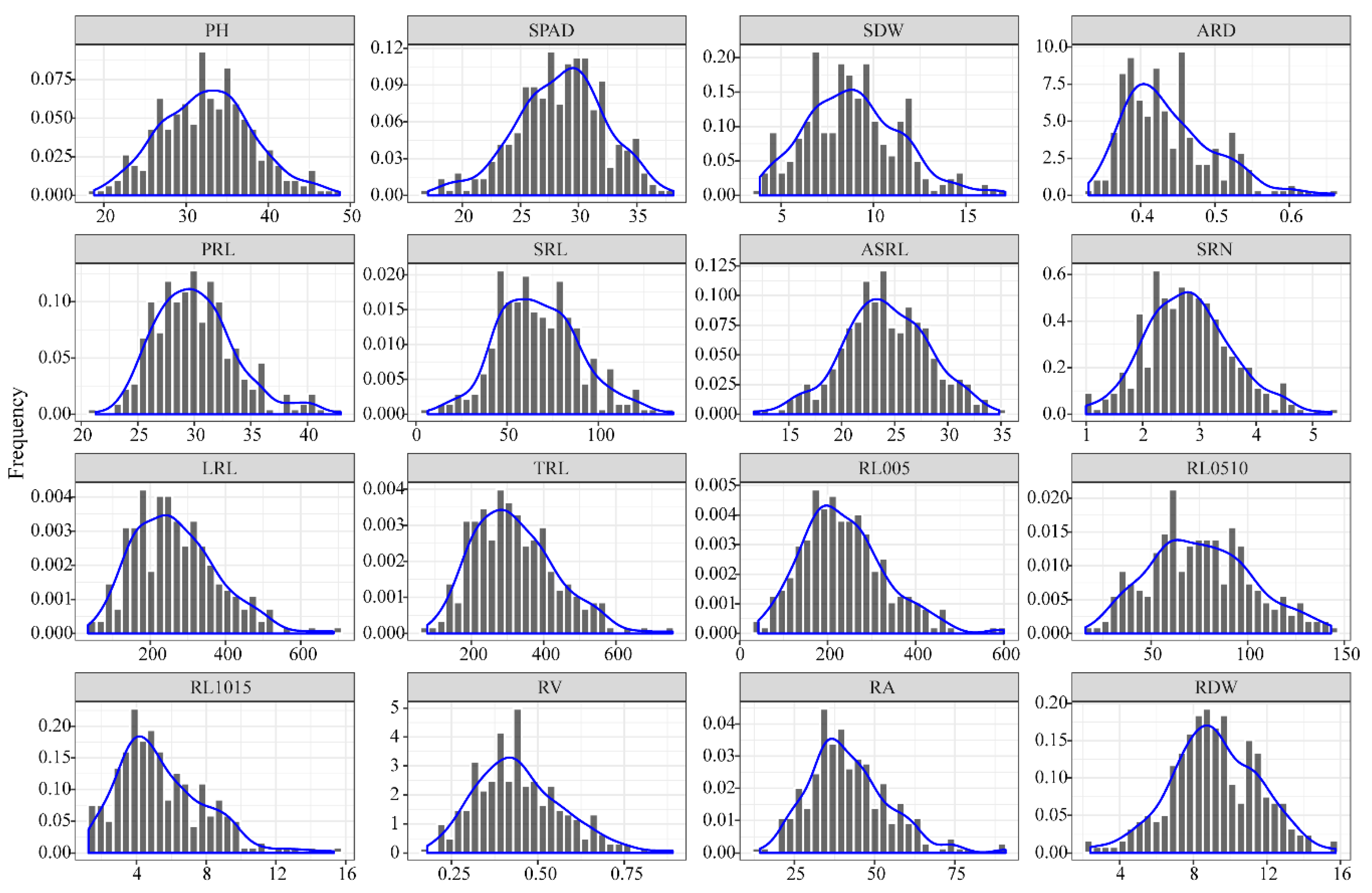
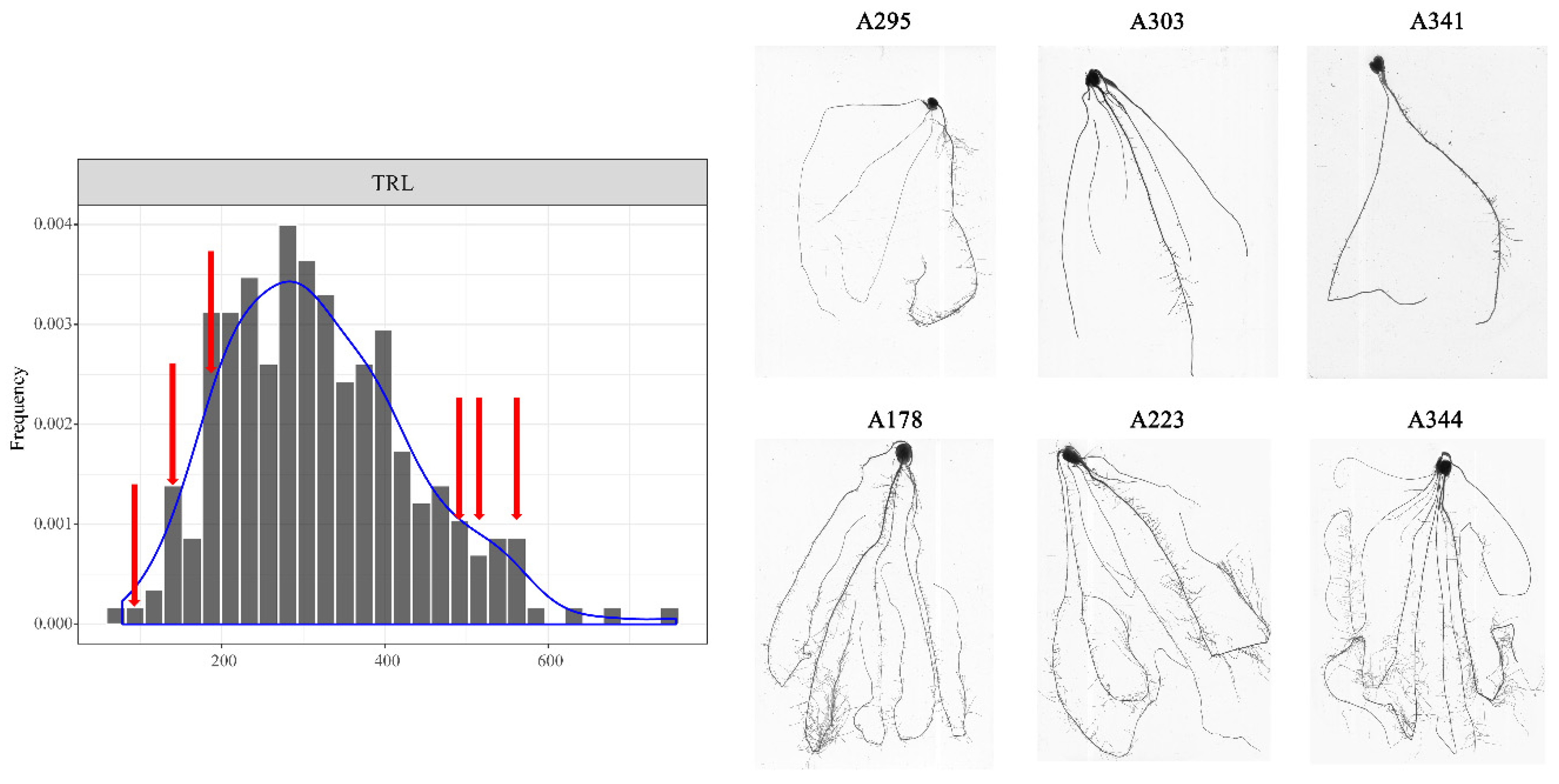
3.1. Phenotypic Analysis of Root Traits at the Seedling Stage
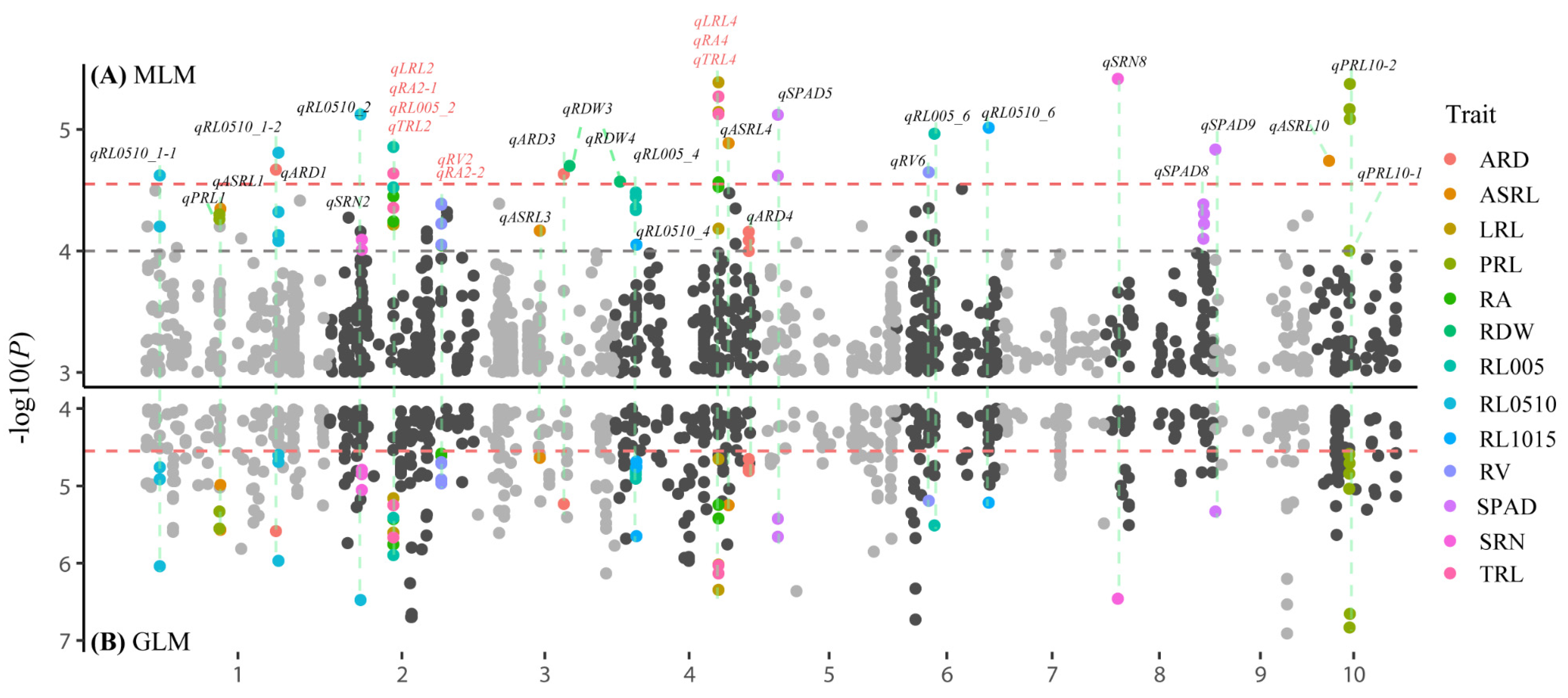
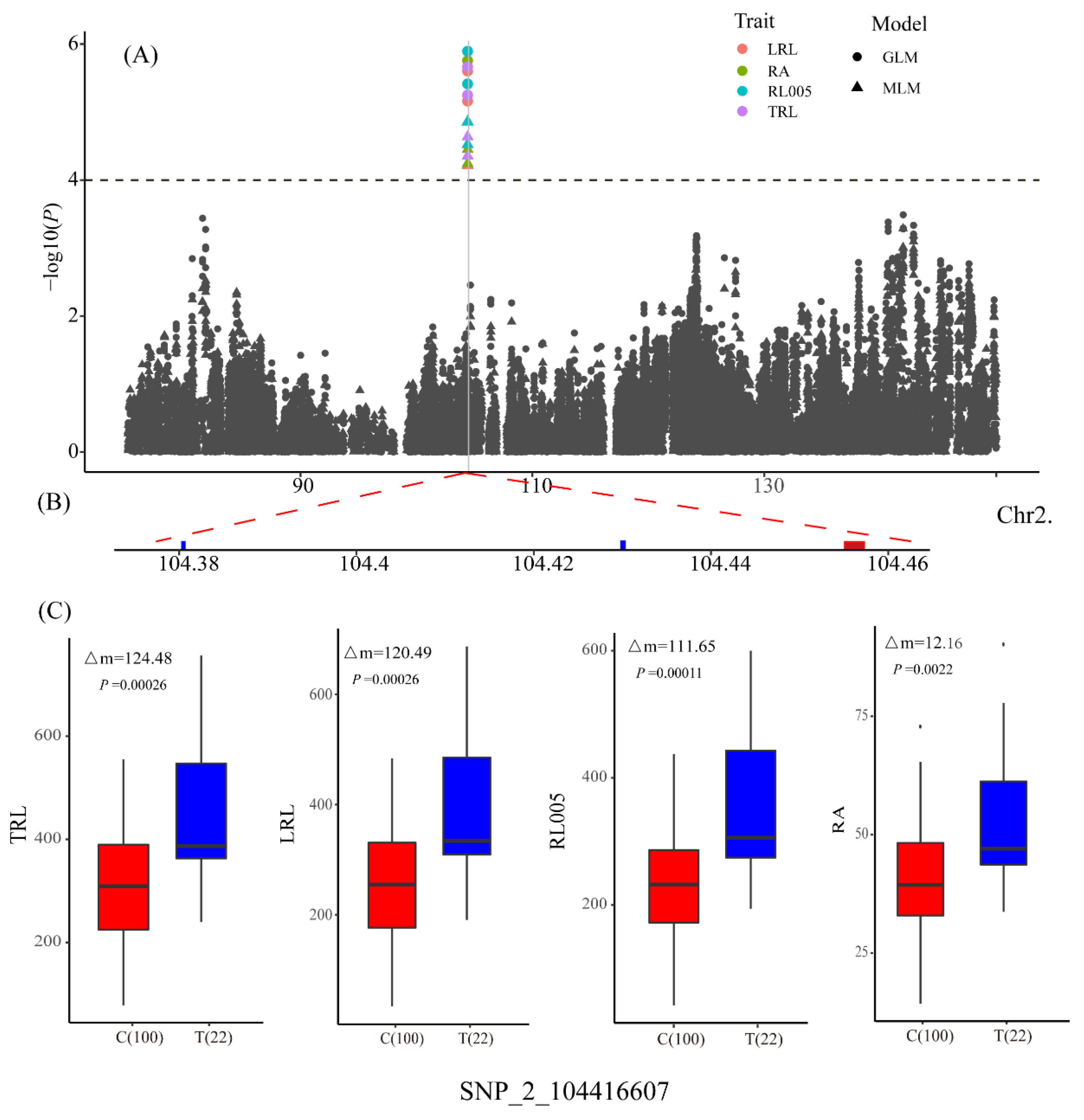
3.2. Genome-Wide Association Studies
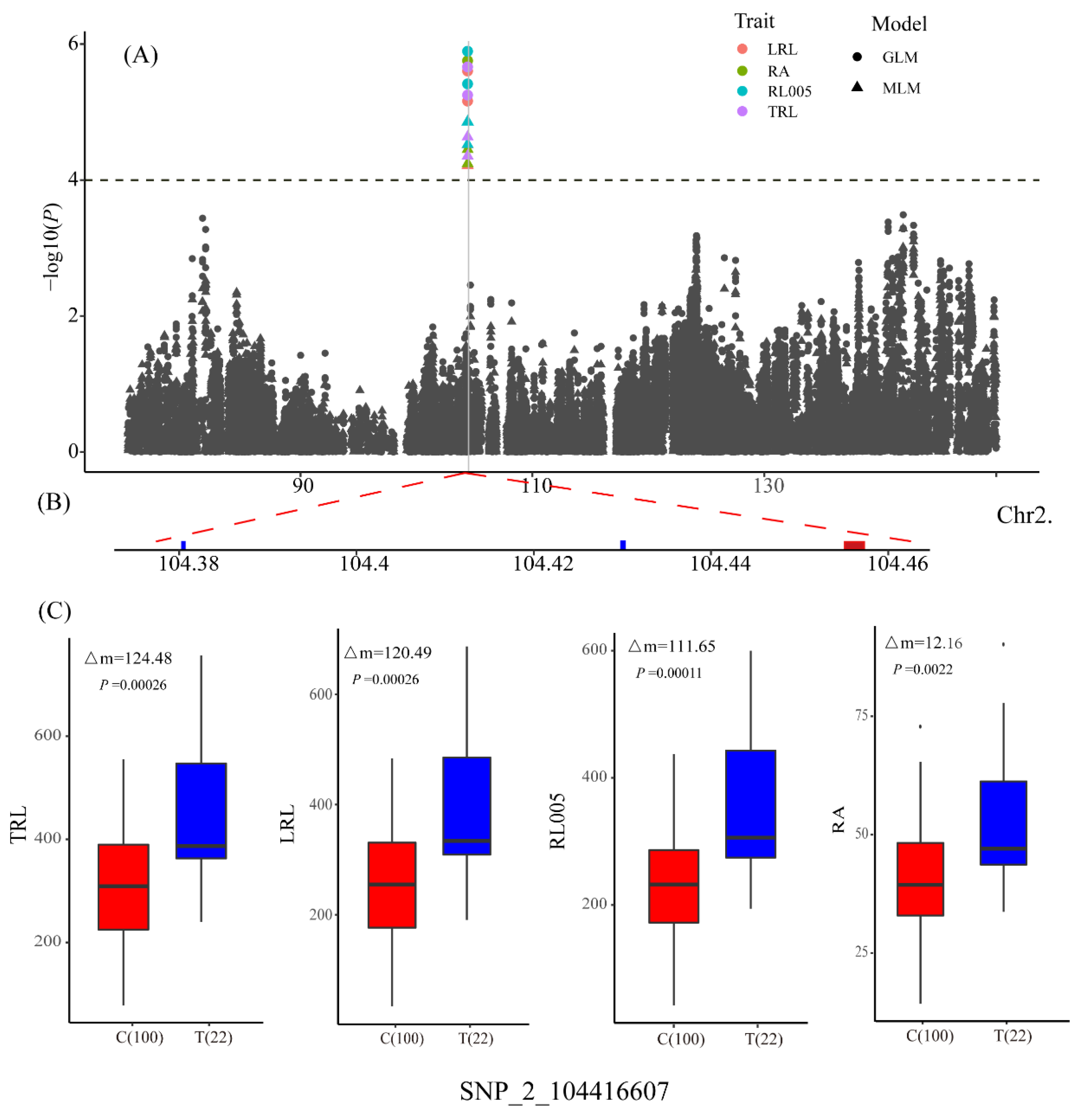
3.3. Determination of Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hickey, L.T.; Amber, N.H.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, T.R. Challenges in breeding for yield increase for drought. Trends Plant Sci. 2011, 16, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.R.; Delhaize, E.; Watt, M.; Richardson, A.E. Plant roots: Understanding structure and function in an ocean of complexity. Ann. Bot. 2016, 118, 555–559. [Google Scholar] [CrossRef]
- Motte, H.; Vanneste, S.; Beeckman, T. Molecular and Environmental Regulation of Root Development. Annu. Rev. Plant Biol. 2019, 70, 465–488. [Google Scholar] [CrossRef] [PubMed]
- Duque, L.O.; Villordon, A. Root Branching and Nutrient Efficiency: Status and Way Forward in Root and Tuber Crops. Front. Plant Sci. 2019, 10, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochholdinger, F.; Yu, P.; Marcon, C. Genetic Control of Root System Development in Maize. Trends Plant Sci. 2018, 23, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Chen, F.J.; Cai, H.G.; Liu, J.C.; Pan, Q.C.; Liu, Z.G.; Gu, R.L.; Mi, G.H.; Zhang, F.S.; Yuan, L.X. A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J. Exp. Bot. 2015, 66, 3175–3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, R.L.; Chen, F.J.; Long, L.Z.; Cai, H.G.; Liu, Z.G.; Yang, J.B.; Wang, L.F.; Li, H.Y.; Li, J.H.; Liu, W.X.; et al. Enhancing phosphorus uptake efficiency through QTL-based selection for root system architecture in maize. J. Genet. Genom. 2016, 43, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghani, A.H.; Kumar, B.; Reyes-Matamoros, J.; Gonzalez-Portilla, P.J.; Jansen, C.; San Martin, J.P.; Lee, M.; Lubberstedt, T. Genotypic variation and relationships between seedling and adult plant traits in maize (Zea mays L.) inbred lines grown under contrasting nitrogen levels. Euphytica 2013, 189, 123–133. [Google Scholar] [CrossRef]
- Salvi, S.; Giuliani, S.; Ricciolini, C.; Carraro, N.; Maccaferri, M.; Presterl, T.; Ouzunova, M.; Tuberosa, R. Two major quantitative trait loci controlling the number of seminal roots in maize co-map with the root developmental genes rtcs and rum1. J. Exp. Bot. 2016, 67, 1149–1159. [Google Scholar] [CrossRef]
- Yu, P.; Hochholdinger, F.; Li, C. Plasticity of Lateral Root Branching in Maize. Front. Plant Sci. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Meister, R.; Rajani, M.S.; Ruzicka, D.; Schachtman, D.P. Challenges of modifying root traits in crops for agriculture. Trends Plant. Sci. 2014, 19, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zhou, H.Z.; Wu, Y.; Zhang, H.; Lin, J.; Jiang, X.; He, Q.; Zhu, J.; Li, Y.; Yu, H.; et al. OsSPL3, an SBP-Domain Protein, Regulates Crown Root Development in Rice. Plant Cell 2019, 31, 1257–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Xiang, D.; Zhu, J.; Li, Y.; Mao, C. Molecular Mechanisms of Root Development in Rice. Rice (N Y) 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.P.; Xu, N.; Chen, H.L.; Wang, G.X.; Huang, J.L. OsMADS25 regulates root system development via auxin signalling in rice. Plant J. 2018, 95, 1004–1022. [Google Scholar] [CrossRef] [PubMed]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N.; et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097–1102. [Google Scholar] [CrossRef]
- Gamuyao, R.; Chin, J.H.; Pariasca-Tanaka, J.; Pesaresi, P.; Catausan, S.; Dalid, C.; Slamet-Loedin, I.; Tecson-Mendoza, E.M.; Wissuwa, M.; Heuer, S. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 2012, 488, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Li, H.; Peng, Z.; Yang, X.; Wang, W.; Fu, J.; Wang, J.; Han, Y.; Chai, Y.; Guo, T.; Yang, N.; et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 2013, 45, 43–50. [Google Scholar] [CrossRef]
- Meijon, M.; Satbhai, S.B.; Tsuchimatsu, T.; Busch, W. Genome-wide association study using cellular traits identifies a new regulator of root development in Arabidopsis. Nat. Genet. 2014, 46, 77. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Giehl, R.F.H.; Meyer, R.C.; Altmann, T.; von Wiren, N. Natural variation of BSK3 tunes brassinosteroid signaling to regulate root foraging under low nitrogen. Nat. Commun. 2019, 10, 2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Guo, Z.; Lv, Y.; Cen, X.; Ding, X.; Wu, H.; Li, X.; Huang, J.; Xiong, L. Genetic control of the root system in rice under normal and drought stress conditions by genome-wide association study. PLoS Genet. 2017, 13, e1006889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, H.; Xu, J.; Jiang, C.; Yin, Z.; Xiong, H.; Xie, J.; Wang, X.; Zhu, X.; Li, Y.; et al. Loci and natural alleles underlying robust roots and adaptive domestication of upland ecotype rice in aerobic conditions. PLoS Genet. 2018, 14, e1007521. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.; Gardner, C.; Romay, C.; Ganapathysubramanian, B.; Lubberstedt, T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genom. 2015, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.A.; Pound, M.P.; Bennett, M.J.; Wells, D.M. Uncovering the hidden half of plants using new advances in root phenotyping. Curr. Opin. Biotechnol. 2019, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghani, A.H.; Sanchez, D.L.; Kumar, B.; Lubberstedt, T. Paper Roll Culture and Assessment of Maize Root Parameters. Bio-Protocol 2016, 6, e1926. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, T.T.; Zhou, Y.; Yin, S.Y.; Li, P.C.; Liu, J.; Xu, S.H.; Yang, Z.F.; Xu, C.W. Genome-Wide Association Mapping of Starch Pasting Properties in Maize Using Single-Locus and Multi-Locus Models. Front. Plant Sci. 2018, 9, 1311. [Google Scholar] [CrossRef]
- Li, P.; Pan, T.; Wang, H.; Wei, J.; Chen, M.; Hu, X.; Zhao, Y.; Yang, X.; Yin, S.; Xu, Y.; et al. Natural variation of ZmHKT1 affects root morphology in maize at the seedling stage. Planta 2019, 249, 879–889. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Yeung, J.M.Y.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2012, 131, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Calling_Variants_in_RNAseq. Available online: https://software.broadinstitute.org/gatk/documentation/article.php?id=3891 (accessed on 22 September 2019).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Kuijken, R.C.; van Eeuwijk, F.A.; Marcelis, L.F.; Bouwmeester, H.J. Root phenotyping: From component trait in the lab to breeding. J. Exp. Bot. 2015, 66, 5389–5401. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.L.; Liu, S.; Ibrahim, R.; Blanco, M.; Lubberstedt, T. Genome-wide association studies of doubled haploid exotic introgression lines for root system architecture traits in maize (Zea mays L.). Plant Sci. 2018, 268, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, H.; Lu, X.; Opitz, N.; Marcon, C.; Paschold, A.; Lithio, A.; Nettleton, D.; Hochholdinger, F. Transcriptomic and anatomical complexity of primary, seminal, and crown roots highlight root type-specific functional diversity in maize (Zea mays L.). J. Exp. Bot. 2015, 67, 1123–1135. [Google Scholar] [CrossRef]
- Burton, A.L.; Brown, K.M.; Lynch, J.P. Phenotypic Diversity of Root Anatomical and Architectural Traits in Zea Species. Crop. Sci. 2013, 53, 1042–1055. [Google Scholar] [CrossRef]
- Bray, A.L.; Topp, C.N. The Quantitative Genetic Control of Root Architecture in Maize. Plant Cell Physiol. 2018, 59, 1919–1930. [Google Scholar] [CrossRef]
- Singh, J.; Gezan, S.A.; Vallejos, C.E. Developmental Pleiotropy Shaped the Roots of the Domesticated Common Bean (Phaseolus vulgaris). Plant Physiol. 2019, 180, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.; Lin, Z.; Wang, J.; Xu, M.; Lai, J.; Yu, J.; Lin, Z. The genetic architecture of nodal root number in maize. Plant. J. 2018, 93, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide association studies in maize: Praise and stargaze. Mol. Plant 2017, 10, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Liu, Y.; Gruber, B.D.; Neumann, K.; Kilian, B.; Graner, A.; von Wiren, N. Genetic Dissection of Root System Architectural Traits in Spring Barley. Front. Plant Sci. 2019, 10, 400. [Google Scholar] [CrossRef]
- Burton, A.L.; Johnson, J.M.; Foerster, J.M.; Hirsch, C.N.; Buell, C.R.; Hanlon, M.T.; Kaeppler, S.M.; Brown, K.M.; Lynch, J.P. QTL mapping and phenotypic variation for root architectural traits in maize (Zea mays L.). Theor. Appl. Genet. 2014, 127, 2293–2311. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, R.J.; Michno, J.M.; Jeffers, J.; Hoekenga, O.; Dilkes, B.; Baxter, I.; Myers, C.L. Integrating Coexpression Networks with GWAS to Prioritize Causal Genes in Maize. Plant Cell 2018, 30, 2922–2942. [Google Scholar] [CrossRef] [Green Version]
- Sekhon, R.S.; Saski, C.; Kumar, R.; Flinn, B.; Luo, F.; Beissinger, T.M.; Ackerman, A.J.; Breitzman, M.W.; Bridges, W.C.; de Leon, N.; et al. Integrated Genome-Scale Analysis Identifies Novel Genes and Networks Underlying Senescence in Maize. Plant Cell 2019. [Google Scholar] [CrossRef]
- Wen, Z.; Tan, R.; Zhang, S.; Collins, P.J.; Yuan, J.; Du, W.; Gu, C.; Ou, S.; Song, Q.; An, Y.C.; et al. Integrating GWAS and gene expression data for functional characterization of resistance to white mould in soya bean. Plant Biotechnol. J. 2018, 16, 1825–1835. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Full Name | Unit | Measuring Method |
|---|---|---|---|
| PH | Plant height | cm | measured from the coleoptilar to yhe tip of the longest leaf |
| SPAD | Leaf chlorophyll concentrations | - | measured using a SPAD-502 PLUS chlorophyll (Minolta, Japan) |
| SDW | Shoot dry weight | 10 mg | measured by electronic balance |
| ARD | Average root diameter | mm | measured by image analysis software (WinRhizo Pro 2009) |
| PRL | Primary root length | cm | measured by a ruler |
| SRL | Seminal roots length | cm | measured by a ruler |
| ASRL | Average seminal root length | cm | estimated by SRL and SRN |
| SRN | Seminal roots numbers | number | Count of the seminal roots |
| LRL | Lateral root length | cm | estimated by TRL, PRL and SRL |
| TRL | Total root length | cm | measured by image analysis software |
| RL005 | L ≤ 0.5 (Root length between 0 and 0.5 mm in diameter) | cm | measured by image analysis software |
| RL0510 | 0.5 < L ≤ 1.0 (Root length between 0.5 mm and 1 mm in diameter) | cm | measured by image analysis software |
| RL1015 | 1.0 < L ≤ 1.5 (Root length greater than 1 mm in diameter) | cm | measured by image analysis software |
| RV | Root volume | cm3 | measured by image analysis software |
| RA | Root surface area | cm2 | measured by image analysis software |
| RDW | Root dry weight | 10 mg | measured by electronic balance |
| Trait | Mean | SD | Min | Max | Fold Change | CV a | ANOVA | ||
|---|---|---|---|---|---|---|---|---|---|
| MS (Genotype) | MS (Error) | F (Genotype) | |||||||
| PH | 32.47 | 5.74 | 18.80 | 48.69 | 2.59 | 17.67% | 63.36 | 7.70 | 8.23 ** b |
| SPAD | 28.47 | 3.86 | 17.05 | 38.15 | 2.24 | 13.55% | 28.82 | 9.23 | 3.12 ** |
| SDW | 8.86 | 2.60 | 3.85 | 17.12 | 4.45 | 29.34% | 12.34 | 2.38 | 5.18 ** |
| ARD | 0.44 | 0.06 | 0.33 | 0.66 | 2.00 | 14.16% | 0.01 | 0.00 | 11.79 ** |
| PRL | 30.12 | 3.62 | 21.27 | 42.98 | 2.02 | 12.01% | 25.68 | 4.11 | 6.24 ** |
| SRL | 67.08 | 22.71 | 6.60 | 141.00 | 21.36 | 33.85% | 967.54 | 313.63 | 3.09 ** |
| ASRL | 23.95 | 4.40 | 11.71 | 34.83 | 2.97 | 18.39% | 31.87 | 7.55 | 4.22 ** |
| SRN | 2.81 | 0.76 | 1.00 | 5.33 | 5.33 | 27.19% | 1.09 | 0.60 | 1.83 ** |
| LRL | 265.88 | 111.46 | 34.35 | 687.12 | 20.00 | 41.92% | 23267.84 | 1826.34 | 12.74 ** |
| TRL | 319.86 | 114.79 | 78.31 | 756.81 | 9.66 | 35.89% | 24982.71 | 1914.59 | 13.05 ** |
| RL005 | 236.78 | 96.32 | 42.79 | 599.84 | 14.02 | 40.68% | 17497.93 | 1603.34 | 10.91 ** |
| RL0510 | 75.60 | 26.60 | 16.03 | 143.26 | 8.94 | 35.18% | 1372.08 | 173.74 | 7.90 ** |
| RL1015 | 5.43 | 2.51 | 1.24 | 15.32 | 12.35 | 46.22% | 12.23 | 2.39 | 5.12 ** |
| RV | 0.45 | 0.13 | 0.18 | 0.89 | 4.94 | 28.60% | 296.39 | 30.66 | 9.67 ** |
| RA | 41.97 | 12.72 | 14.34 | 90.75 | 6.33 | 30.31% | 0.03 | 0.00 | 8.23 ** |
| RDW | 9.15 | 2.43 | 2.40 | 15.75 | 6.56 | 26.52% | 11.74 | 1.66 | 7.09 ** |
| Trait | QTL | Lead SNP | Chr. | Pos. a | Allele b | −log10(P) | R2 |
|---|---|---|---|---|---|---|---|
| ARD | qARD1 | SNP_1_213315833 | 1 | 213315833 | G/T | 4.67 | 8.50 |
| qARD3 | SNP_3_147397047 | 3 | 147397047 | A/C | 4.63 | 7.96 | |
| qARD4 | SNP_4_219099674 | 4 | 219099674 | C/T | 4.16 | 12.61 | |
| ASRL | qASRL1 | SNP_1_121996211 | 1 | 121996211 | C/T | 4.35 | 9.49 |
| qASRL3 | SNP_3_107899717 | 3 | 107899717 | C/T | 4.17 | 6.33 | |
| qASRL4 | SNP_4_185942913 | 4 | 185942913 | A/T | 4.89 | 7.01 | |
| qASRL10 | SNP_10_34244943 | 10 | 34244943 | A/G | 4.74 | 7.05 | |
| LRL | qLRL2 | SNP_2_104416607 | 2 | 104416607 | C/T | 4.52 | 14.97 |
| qLRL4 | SNP_4_168917747 | 4 | 168917747 | C/T | 5.39 | 9.27 | |
| PRL | qPRL1 | SNP_1_120308712 | 1 | 120308712 | C/T | 4.30 | 6.34 |
| qPRL10-1 | SNP_10_67350334 | 10 | 67350334 | G/T | 4.00 | 6.54 | |
| qPRL10-2 | SNP_10_68587247 | 10 | 68587247 | A/G | 5.38 | 9.29 | |
| RA | qRA2-1 | SNP_2_104416607 | 2 | 104416607 | C/T | 4.45 | 14.65 |
| qRA2-2 | SNP_2_184016997 | 2 | 184016997 | C/T | 4.05 | 6.99 | |
| qRA4 | SNP_4_168917747 | 4 | 168917747 | C/T | 4.57 | 7.58 | |
| RDW | qRDW3 | SNP_3_156838240 | 3 | 156838240 | C/T | 4.70 | 8.29 |
| qRDW4 | SNP_4_7135819 | 4 | 7135819 | A/C | 4.57 | 8.39 | |
| RL005 | qRL005_2 | SNP_2_104416607 | 2 | 104416607 | C/T | 4.86 | 15.87 |
| qRL005_4 | SNP_4_33081784 | 4 | 33081784 | C/T | 4.48 | 12.29 | |
| qRL005_6 | SNP_6_63998692 | 6 | 63998692 | A/G | 4.97 | 10.66 | |
| RL0510 | qRL0510_1-1 | SNP_1_22179639 | 1 | 22179639 | A/G | 4.62 | 9.52 |
| qRL0510_1-2 | SNP_1_217582572 | 1 | 217582572 | G/T | 4.81 | 10.10 | |
| qRL0510_2 | SNP_2_50742849 | 2 | 50742849 | A/G | 5.12 | 12.96 | |
| RL1015 | qRL0510_4 | SNP_4_34568124 | 4 | 34568124 | G/T | 4.05 | 11.43 |
| qRL0510_6 | SNP_6_153696612 | 6 | 153696612 | A/G | 5.01 | 13.90 | |
| RV | qRV2 | SNP_2_184016997 | 2 | 184016997 | C/T | 4.39 | 7.65 |
| qRV6 | SNP_6_54506432 | 6 | 54506432 | A/G | 4.65 | 9.37 | |
| SPAD | qSPAD5 | SNP_5_25046214 | 5 | 25046214 | C/T | 5.12 | 8.17 |
| qSPAD8 | SNP_8_160371425 | 8 | 160371425 | A/G | 4.38 | 6.36 | |
| qSPAD9 | SNP_9_4832814 | 9 | 4832814 | A/T | 4.84 | 8.36 | |
| SRN | qSRN2 | SNP_2_52559869 | 2 | 52559869 | C/T | 4.01 | 5.65 |
| qSRN8 | SNP_8_19589120 | 8 | 19589120 | C/T | 5.42 | 9.39 | |
| TRL | qTRL2 | SNP_2_104416607 | 2 | 104416607 | C/T | 4.64 | 15.22 |
| qTRL4 | SNP_4_168917747 | 4 | 168917747 | C/T | 5.13 | 8.60 |
| QTL | Candidate Genes | log2(FC) a | P | SNP | Amino Acid Polymorphism | Annotation |
|---|---|---|---|---|---|---|
| qARD3 | GRMZM2G138338 | −1.38 | 0.032 | 39 | 8 | Leucine-rich receptor-like protein kinase family protein |
| qASRL4 | GRMZM2G174797 | −1.44 | 0.00017 | 21 | 6 | ELMO/CED-12 family protein |
| GRMZM2G476902 | 3.74 | 0.00079 | 4 | 1 | Armadillo/beta-catenin repeat family protein | |
| qLRL2/ qRA2-1/ qRL005_2/ qTRL2 | GRMZM2G397965 | −2.78 | 0.0082 | 8 | 3 | Vignain precursor/ (SAG12) senescence-associated gene 12 |
| qRDW3 | GRMZM2G031528 | −2.48 | 0.00017 | 12 | 1 | Heavy metal transport/detoxification superfamily protein |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Wei, J.; Li, P.; Wang, Y.; Ge, Z.; Qian, J.; Fan, Y.; Ni, J.; Xu, Y.; Yang, Z.; et al. Integrating GWAS and Gene Expression Analysis Identifies Candidate Genes for Root Morphology Traits in Maize at the Seedling Stage. Genes 2019, 10, 773. https://doi.org/10.3390/genes10100773
Wang H, Wei J, Li P, Wang Y, Ge Z, Qian J, Fan Y, Ni J, Xu Y, Yang Z, et al. Integrating GWAS and Gene Expression Analysis Identifies Candidate Genes for Root Morphology Traits in Maize at the Seedling Stage. Genes. 2019; 10(10):773. https://doi.org/10.3390/genes10100773
Chicago/Turabian StyleWang, Houmiao, Jie Wei, Pengcheng Li, Yunyun Wang, Zhenzhen Ge, Jiayi Qian, Yingying Fan, Jinran Ni, Yang Xu, Zefeng Yang, and et al. 2019. "Integrating GWAS and Gene Expression Analysis Identifies Candidate Genes for Root Morphology Traits in Maize at the Seedling Stage" Genes 10, no. 10: 773. https://doi.org/10.3390/genes10100773