Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cancer Genomics Analysis via The cBioPortal Website
2.2. Gene Set Enrichment Analysis (GSEA) and Pathway Construction
2.3. Chemosensitivity Profiling in PDAC Cancer Cell Lines and PDAC Patient-Derived Organoids
2.4. Materials
2.5. Cell Culture and MTT Cell Viability Assay
2.6. Real-Time Quantitative Polymerase Chain Reaction (qPCR) and Western Blot Analysis
2.7. Statistical Analysis
3. Results
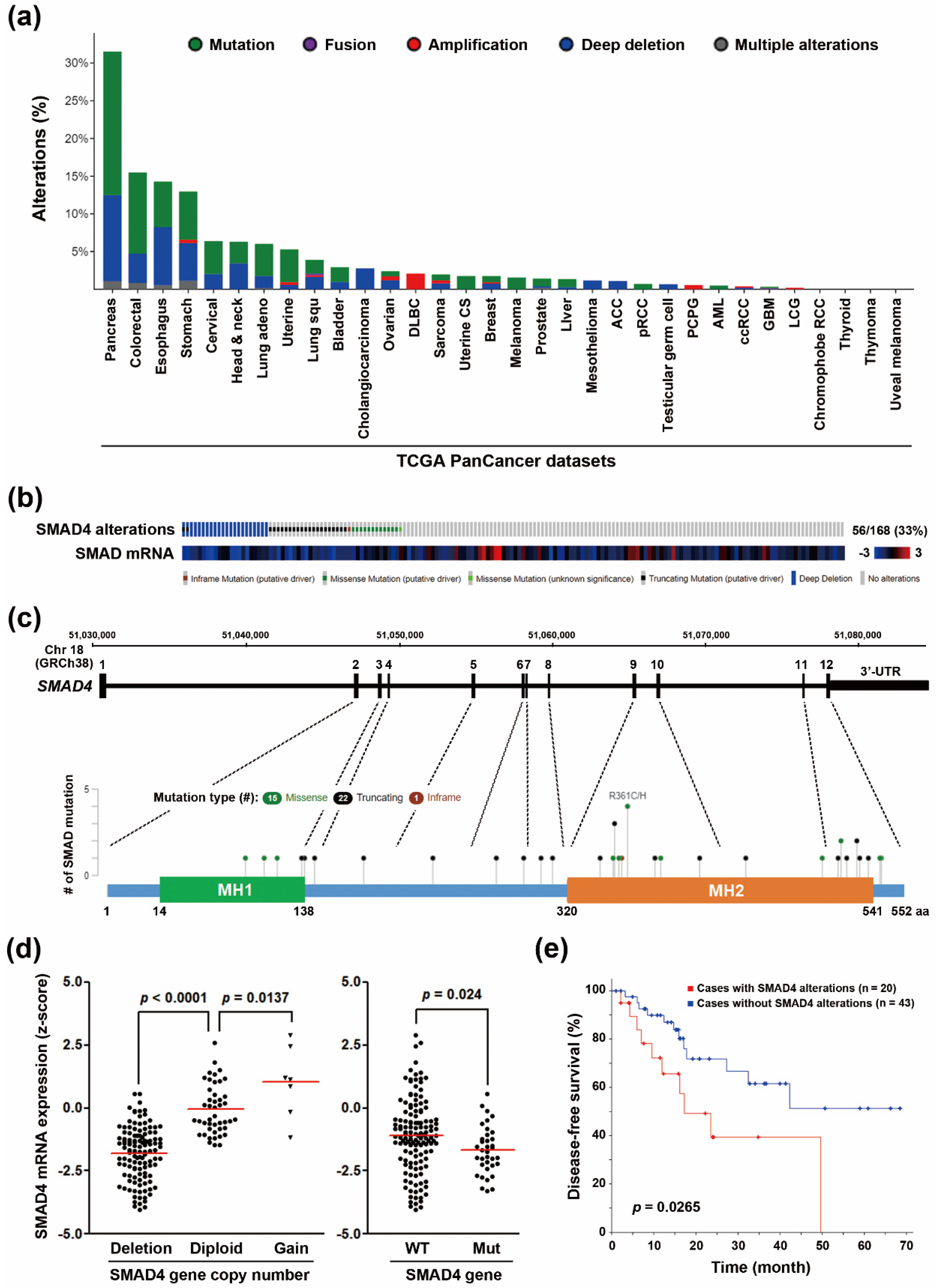
3.1. SMAD4 Gene Deletion Predicts A Poorer Disease-Free Survival in PDAC Patients
3.2. Upregulation Of Cell Cycle-Related Genes in SMAD4-Deleted PDAC
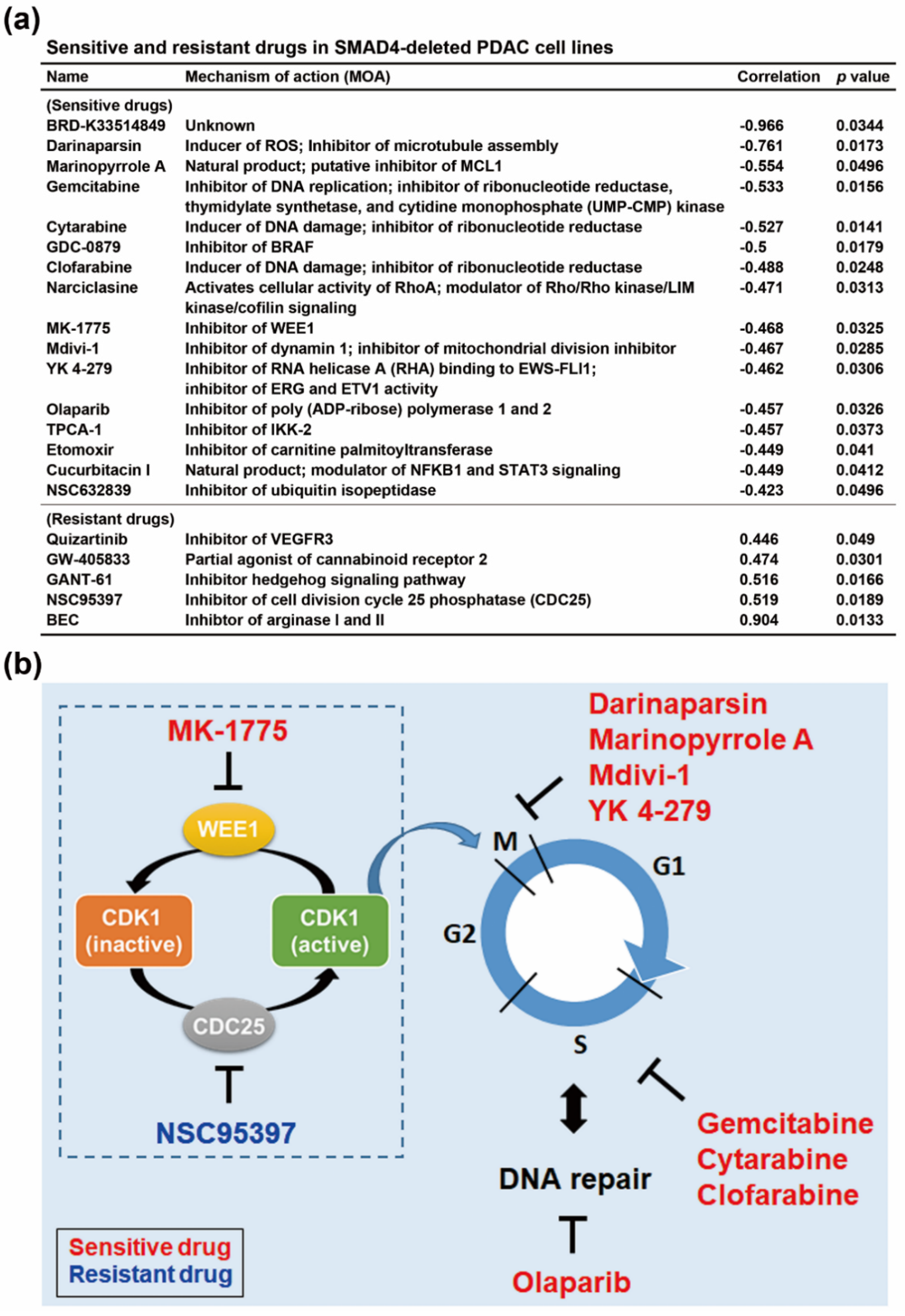
3.3. SMAD4 Gene Deletion is Associated With an Increased Sensitivity To Cell Cycle-Targeting Drugs in PDAC Cell Lines
3.4. Increased Gemcitabine Sensitivity in Both SMAD4-Deleted PDAC Cell Lines and PDAC Patients-Derived Organoids
3.5. The Association Between CDK1 and Genes Correlated with Gemcitabine Sensitivity in SMAD4-Deleted PDAC Patient-Derived Organoids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 12–36. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, V.N.; Luna, A.; Yamade, M.; Loman, L.; Varma, S.; Sunshine, M.; Iorio, F.; Sousa, F.G.; Elloumi, F.; Aladjem, M.I.; et al. CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 2018, 10, 247–264. [Google Scholar] [CrossRef]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Parrilla, A.; Cirillo, L.; Thomas, Y.; Gotta, M.; Pintard, L.; Santamaria, A. Mitotic entry: The interplay between Cdk1, Plk1 and Bora. Cell Cycle 2016, 15, 3177–3182. [Google Scholar] [CrossRef]
- Tsesmetzis, N.; Paulin, C.B.J.; Rudd, S.G.; Herold, N. Nucleobase and Nucleoside Analogues: Resistance and Re-Sensitisation at the Level of Pharmacokinetics, Pharmacodynamics and Metabolism. Cancers 2018, 10, 240. [Google Scholar] [CrossRef]
- Kumar, C.; Rani, N.; Velan Lakshmi, P.T.; Arunachalam, A. A comprehensive look of poly(ADP-ribose) polymerase inhibition strategies and future directions for cancer therapy. Future Med. Chem. 2017, 9, 37–60. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef]
- Mason, T.A.; Kolobova, E.; Liu, J.; Roland, J.T.; Chiang, C.; Goldenring, J.R. Darinaparsin is a multivalent chemotherapeutic which induces incomplete stress response with disruption of microtubules and Shh signaling. PLoS ONE 2011, 6, e27699. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Doi, K.; Li, R.; Sung, S.S.; Wu, H.; Liu, Y.; Manieri, W.; Krishnegowda, G.; Awwad, A.; Dewey, A.; Liu, X.; et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J. Biol. Chem. 2012, 287, 10224–10235. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Santana-Santos, L.; Shuda, M.; Sobol, R.W.; Van Houten, B.; Qian, W. A novel strategy for targeted killing of tumor cells: Induction of multipolar acentrosomal mitotic spindles with a quinazolinone derivative mdivi-1. Mol. Oncol. 2015, 9, 488–502. [Google Scholar] [CrossRef]
- Kollareddy, M.; Sherrard, A.; Park, J.H.; Szemes, M.; Gallacher, K.; Melegh, Z.; Oltean, S.; Michaelis, M.; Cinatl, J., Jr.; Kaidi, A.; et al. The small molecule inhibitor YK-4-279 disrupts mitotic progression of neuroblastoma cells, overcomes drug resistance and synergizes with inhibitors of mitosis. Cancer Lett. 2017, 403, 74–85. [Google Scholar] [CrossRef]
- Cui, Y.; Brosnan, J.A.; Blackford, A.L.; Sur, S.; Hruban, R.H.; Kinzler, K.W.; Vogelstein, B.; Maitra, A.; Diaz, L.A., Jr.; Iacobuzio-Donahue, C.A.; et al. Genetically defined subsets of human pancreatic cancer show unique in vitro chemosensitivity. Clin. Cancer Res. 2012, 18, 6519–6530. [Google Scholar] [CrossRef]
- Fullerton, P.T., Jr.; Creighton, C.J.; Matzuk, M.M. Insights Into SMAD4 Loss in Pancreatic Cancer From Inducible Restoration of TGF-β Signaling. Mol. Endocrinol. 2015, 29, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-β in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef]
- Alexandrow, M.G.; Moses, H.L. Transforming growth factor β and cell cycle regulation. Cancer Res. 1995, 55, 1452–1457. [Google Scholar] [PubMed]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massague, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.P.; Man, W.Y.; Ma, H.T.; Poon, R.Y. Pharmacological targeting the ATR-CHK1-WEE1 axis involves balancing cell growth stimulation and apoptosis. Oncotarget 2014, 5, 10546–10557. [Google Scholar] [CrossRef]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.B.; Courtin, A.; Boyce, R.J.; Boyle, R.G.; Richards, F.M.; Jodrell, D.I. CHK1 Inhibition Synergizes with Gemcitabine Initially by Destabilizing the DNA Replication Apparatus. Cancer Res. 2015, 75, 3583–3595. [Google Scholar] [CrossRef]
- Wang, H.; Stephens, B.; Von Hoff, D.D.; Han, H. Identification and characterization of a novel anticancer agent with selectivity against deleted in pancreatic cancer locus 4 (DPC4)-deficient pancreatic and colon cancer cells. Pancreas 2009, 38, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, H.; Von Hoff, D.D. Identification of an agent selectively targeting DPC4 (deleted in pancreatic cancer locus 4)-deficient pancreatic cancer cells. Cancer Res. 2006, 66, 9722–9730. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, S.; Zhou, Y.; Hu, X.; Shao, C. MicroRNA 483-3p suppresses the expression of DPC4/Smad4 in pancreatic cancer. FEBS Lett. 2011, 585, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Zhang, S.; Zhou, Y.; Liu, C.; Hu, X.; Shao, C. MicroRNA 421 suppresses DPC4/Smad4 in pancreatic cancer. Biochem. Biophys. Res. Commun. 2011, 406, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, K.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Huang, C.; Zhao, Q.; Qiu, Z. MicroRNA-301a-3p promotes pancreatic cancer progression via negative regulation of SMAD4. Oncotarget 2015, 6, 21046–21063. [Google Scholar] [CrossRef]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg 2013, 258, 336–346. [Google Scholar] [CrossRef]
- Winter, J.M.; Tang, L.H.; Klimstra, D.S.; Liu, W.; Linkov, I.; Brennan, M.F.; D’Angelica, M.I.; DeMatteo, R.P.; Fong, Y.; Jarnagin, W.R.; et al. Failure patterns in resected pancreas adenocarcinoma: Lack of predicted benefit to SMAD4 expression. Ann. Surg. 2013, 258, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Haas, M.; Remold, A.; Kruger, S.; Holdenrieder, S.; Kirchner, T.; Heinemann, V.; Boeck, S. The Impact of SMAD4 Loss on Outcome in Patients with Advanced Pancreatic Cancer Treated with Systemic Chemotherapy. Int. J. Mol. Sci. 2017, 18, 1094. [Google Scholar] [CrossRef]
- Guenot, D.; Guerin, E.; Aguillon-Romain, S.; Pencreach, E.; Schneider, A.; Neuville, A.; Chenard, M.P.; Duluc, I.; Du Manoir, S.; Brigand, C.; et al. Primary tumour genetic alterations and intra-tumoral heterogeneity are maintained in xenografts of human colon cancers showing chromosome instability. J. Pathol. 2006, 208, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Duregon, E.; Schneider, J.; DeMarzo, A.M.; Hooper, J.E. Rapid research autopsy is a stealthy but growing contributor to cancer research. Cancer 2019, 125, 2915–2919. [Google Scholar] [CrossRef]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.M.; Jabbour, S.K.; Lin, S.H.; Deek, M.P.; Hsu, C.C.; Fishman, E.K.; Kim, S.; Cameron, J.L.; Chekmareva, M.; Laheru, D.A.; et al. Smad4 Loss Correlates With Higher Rates of Local and Distant Failure in Pancreatic Adenocarcinoma Patients Receiving Adjuvant Chemoradiation. Pancreas 2018, 47, 208–212. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-Y.; Liu, T.-P.; Chou, C.-J.; Chen, H.-Y.; Lee, K.-H.; Yang, P.-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766. https://doi.org/10.3390/genes10100766
Hsieh Y-Y, Liu T-P, Chou C-J, Chen H-Y, Lee K-H, Yang P-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes. 2019; 10(10):766. https://doi.org/10.3390/genes10100766
Chicago/Turabian StyleHsieh, Yao-Yu, Tsang-Pai Liu, Chia-Jung Chou, Hsin-Yi Chen, Kuen-Haur Lee, and Pei-Ming Yang. 2019. "Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma" Genes 10, no. 10: 766. https://doi.org/10.3390/genes10100766
APA StyleHsieh, Y.-Y., Liu, T.-P., Chou, C.-J., Chen, H.-Y., Lee, K.-H., & Yang, P.-M. (2019). Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes, 10(10), 766. https://doi.org/10.3390/genes10100766