Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients


 , ,
, ,  , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Skin Biopsy
2.2. DNA Analysis
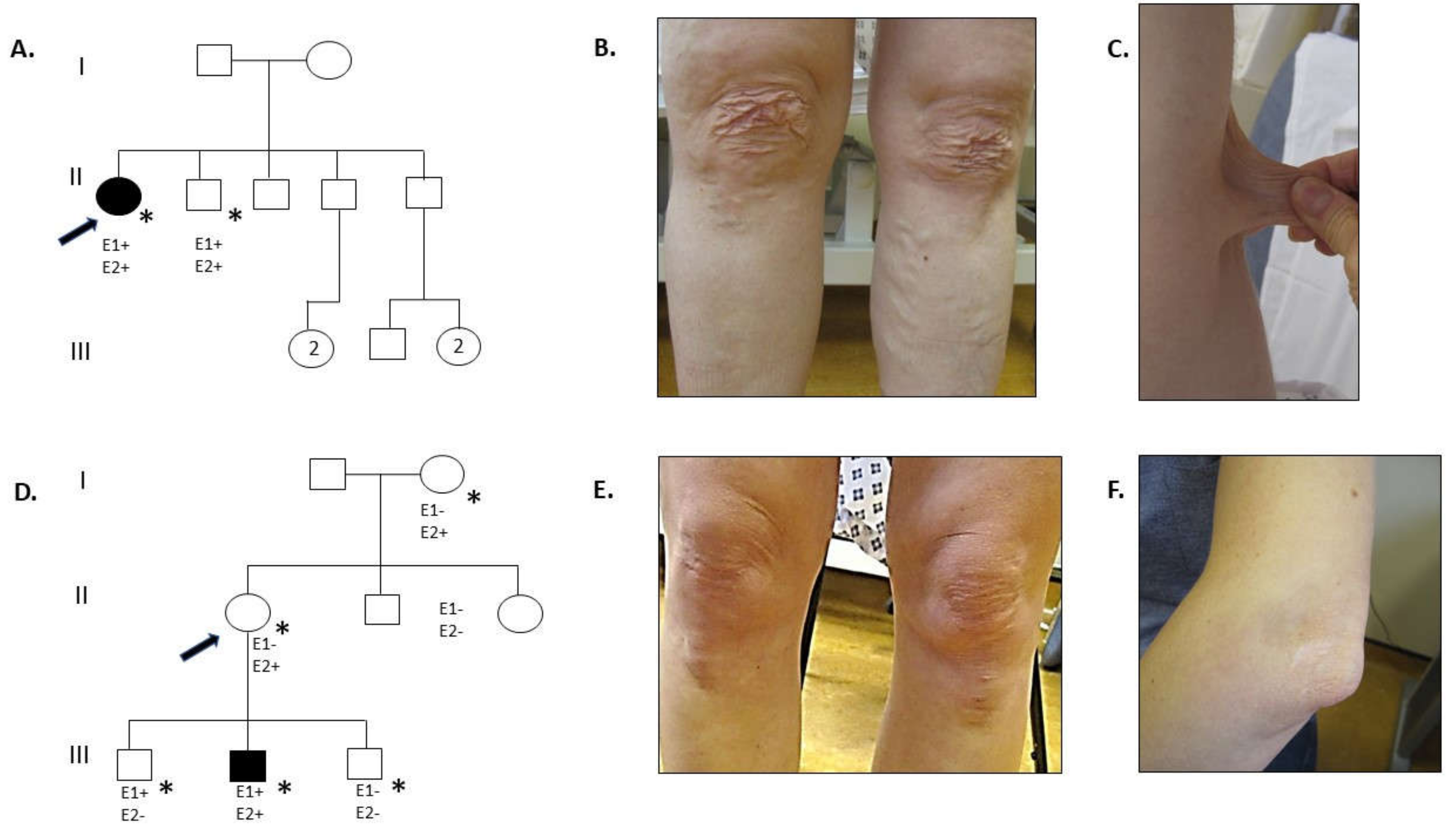
3. Clinical Report
3.1. Family 1
3.2. Family 2
4. Results
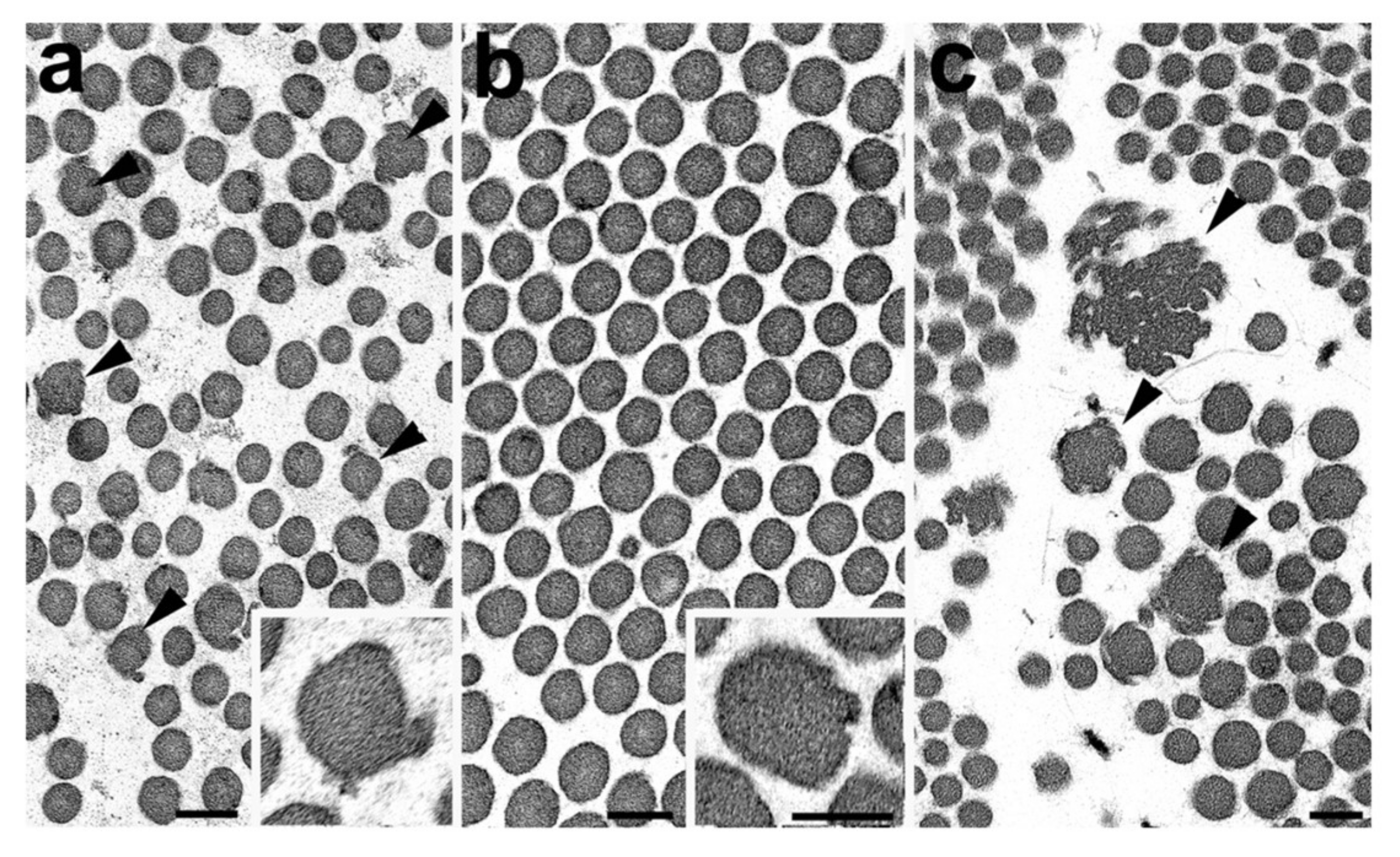
4.1. Skin Biopsy TEM Results
4.2. DNA Analysis
5. Discussion
5.1. Phenotype
5.2. Transmission Electron Microscopy
5.3. COL5A1 Variants
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers–Danlos syndrome, classical type. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-danlos syndromes: Revised nosology, Villefranche, 1997. Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis. 2013, 8, 58. [Google Scholar] [CrossRef]
- Colombi, M.; Dordoni, C.; Cinquina, V.; Venturini, M.; Ritelli, M. A classical Ehlers-Danlos syndrome family with incomplete presentation diagnosed by molecular testing. Eur. J. Med. Genet. 2018, 61, 17–20. [Google Scholar] [CrossRef]
- Dalgleish, R. Ehlers-Danlos Syndrome Variant Database: Collagen, Type V, alpha 1 (COL5A1). Available online: https://eds.gene.le.ac.uk/home.php?select_db=COL5A1 (accessed on 23 August 2019).
- Sun, M.; Chen, S.; Adams, S.M.; Florer, J.B.; Liu, H.; Kao, W.W.Y.; Wenstrup, R.J.; Birk, D.E. Collagen V is a dominant regulator of collagen fibrillogenesis: Dysfunctional regulation of structure and function in a corneal-stroma-specific Col5a1-null mouse model. J. Cell Sci. 2011, 124, 4096–4105. [Google Scholar] [CrossRef]
- Colombi, M.; Dordoni, C.; Venturini, M.; Zanca, A.; Calzavara-Pinton, P.; Ritelli, M. Delineation of Ehlers–Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review. Am. J. Med. Genet. Part A 2017, 173, 524–530. [Google Scholar] [CrossRef]
- Malfait, F.; Symoens, S.; Goemans, N.; Gyftodimou, Y.; Holmberg, E.; López-González, V.; Mortier, G.; Nampoothiri, S.; Petersen, M.B.; De Paepe, A. Helical mutations in type i collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome. Orphanet J. Rare Dis. 2013, 8, 78. [Google Scholar] [CrossRef]
- Hausser, I.; Anton-Lamprecht, I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum. Genet. 1994, 93, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Holbrook, K.A.; Steinmann, B.; Gitzelmann, R.; Byres, P.H. Abnormal collagen fibrilstructure in the Gravis Form ( Type I ) of Ehlers-Danlos Syndrome. Lab. Investig. 1979, 40, 201–206. [Google Scholar] [PubMed]
- De Almeida, H.L.; Bicca, E.; Rocha, N.M.; de Castro, L.A.S. Light and Electron Microscopy of Classical Ehlers-Danlos Syndrome. Am. J. Dermatopathol. 2013, 24, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.; Wagner, B.E.; Peres, L.C.; Sobey, G.J.; Parker, M.J.; Dalton, A.; Arundel, P.; Bishop, N.J. Ultrastructural and histological findings on examination of skin in osteogenesis imperfecta: A novel study. Clin. Dysmorphol. 2015, 24, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Weerakkody, R.A.; Vandrovcova, J.; Kanonidou, C.; Mueller, M.; Gampawar, P.; Ibrahim, Y.; Black, H.A. Targeted next-generation sequencing makes new molecular diagnoses and expands genotype-phenotype relationship in Ehlers-Danlos syndrome. Genet. Med. 2016, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Venturini, M.; Ciaccio, C.; Morlino, S.; Chiarelli, N.; Zanca, A.; Calzavara-Pinton, P.; Zoppi, N.; Castori, M.; et al. Spectrum of mucocutaneous, ocular and facial features and delineation of novel presentations in 62 classical Ehlers-Danlos syndrome patients. Clin. Genet. 2017, 92, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Bicca, E.D.B.C.; De Almeida, F.B.; Pinto, G.M.; De Castro, L.A.S.; De Almeida, H.L., Jr. Classical Ehlers-Danlos syndrome: Clinical, Histological and ultrastructural aspects. An. Bras. Dermatol. 2011, 86, 164–167. [Google Scholar] [CrossRef] [PubMed]
- D’Hondt, S.; Van Damme, T.; Malfait, F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: A systematic review. Genet. Med. 2018, 20, 562. [Google Scholar] [CrossRef] [PubMed]
- Monroe, G.R.; Harakalova, M.; van der Crabben, S.N.; Majoor-Krakauer, D.; Bertoli-Avella, A.M.; Moll, F.L.; Oranen, B.I.; Dooijes, D.; Vink, A.; Knoers, N.V.; et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am. J. Med. Genet. Part A 2015, 167, 1196–1203. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405. [Google Scholar] [CrossRef]
- Malfait, F.; Wenstrup, R.; De Paepe, A. Classic Ehlers-Danlos Syndrome. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1244/ (accessed on 10 February 2017).
- Malfait, F.; De Paepe, A. Molecular genetics in classic Ehlers-Danlos syndrome. Am. J. Med. Genet. Semin. Med. Genet. 2005, 139, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N.P.; Nicholls, A.C.; Richards, A.J.; Luccarini, C.; Harrison, J.B.; Yates, J.R.W.; Pope, F.M. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am. J. Hum. Genet. 1998, 63, 390–398. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angwin, C.; Brady, A.F.; Colombi, M.; Ferguson, D.J.P.; Pollitt, R.; Pope, F.M.; Ritelli, M.; Symoens, S.; Ghali, N.; van Dijk, F.S. Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients. Genes 2019, 10, 762. https://doi.org/10.3390/genes10100762
Angwin C, Brady AF, Colombi M, Ferguson DJP, Pollitt R, Pope FM, Ritelli M, Symoens S, Ghali N, van Dijk FS. Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients. Genes. 2019; 10(10):762. https://doi.org/10.3390/genes10100762
Chicago/Turabian StyleAngwin, Chloe, Angela F. Brady, Marina Colombi, David J. P. Ferguson, Rebecca Pollitt, F. Michael Pope, Marco Ritelli, Sofie Symoens, Neeti Ghali, and Fleur S. van Dijk. 2019. "Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients" Genes 10, no. 10: 762. https://doi.org/10.3390/genes10100762
APA StyleAngwin, C., Brady, A. F., Colombi, M., Ferguson, D. J. P., Pollitt, R., Pope, F. M., Ritelli, M., Symoens, S., Ghali, N., & van Dijk, F. S. (2019). Absence of Collagen Flowers on Electron Microscopy and Identification of (Likely) Pathogenic COL5A1 Variants in Two Patients. Genes, 10(10), 762. https://doi.org/10.3390/genes10100762