Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Silico Identification of MIF Proteins
2.2. Classification of Species Taxonomy, Ecology, and Lifestyle
2.3. Statistical Analyses
2.4. Multiple Sequence Alignments and Phylogenetic Reconstructions
2.5. Analysis of Purifying/Diversifying Selection
3. Results
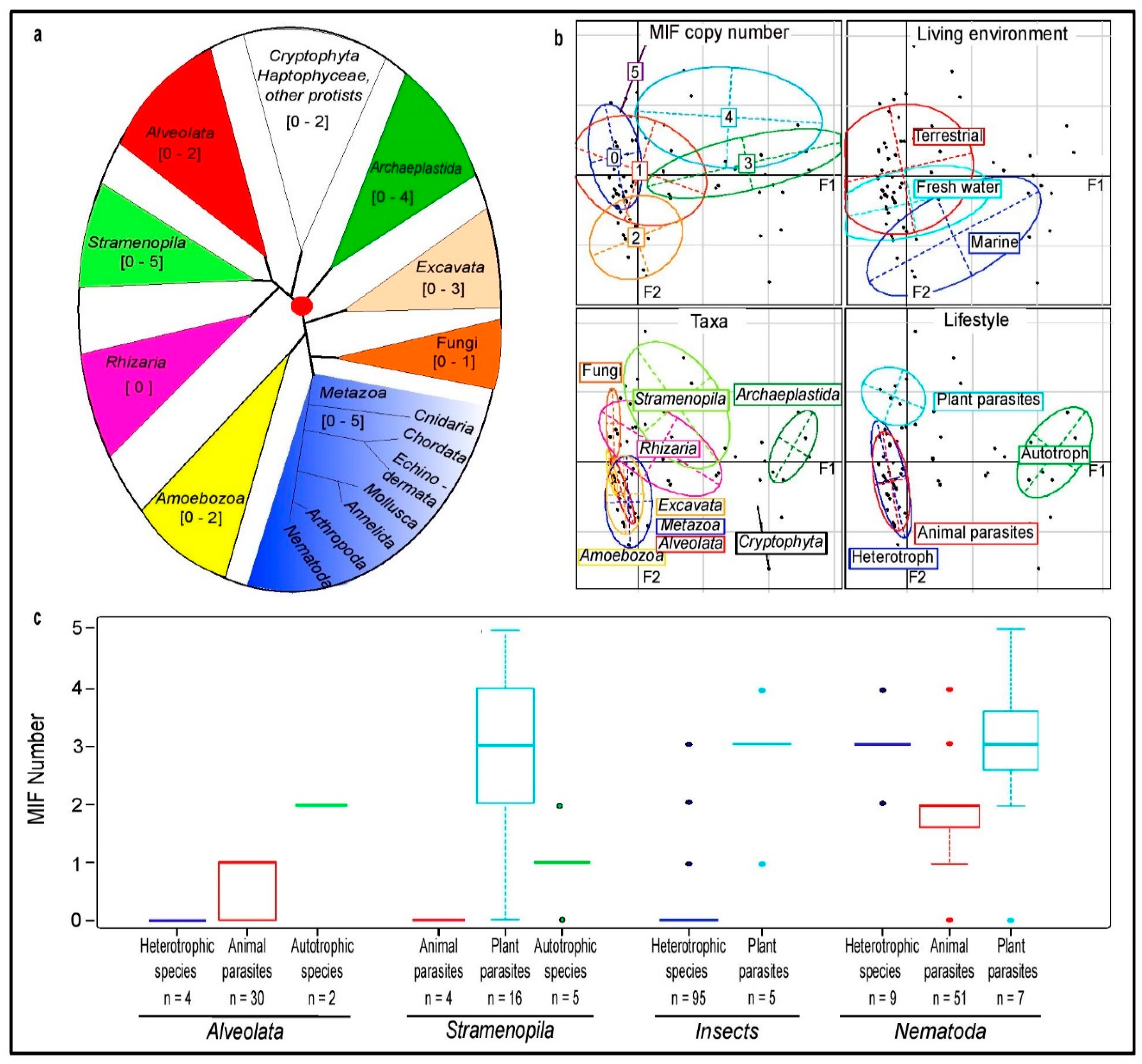
3.1. MIF Presence and Number Across Eukaryotic Kingdoms
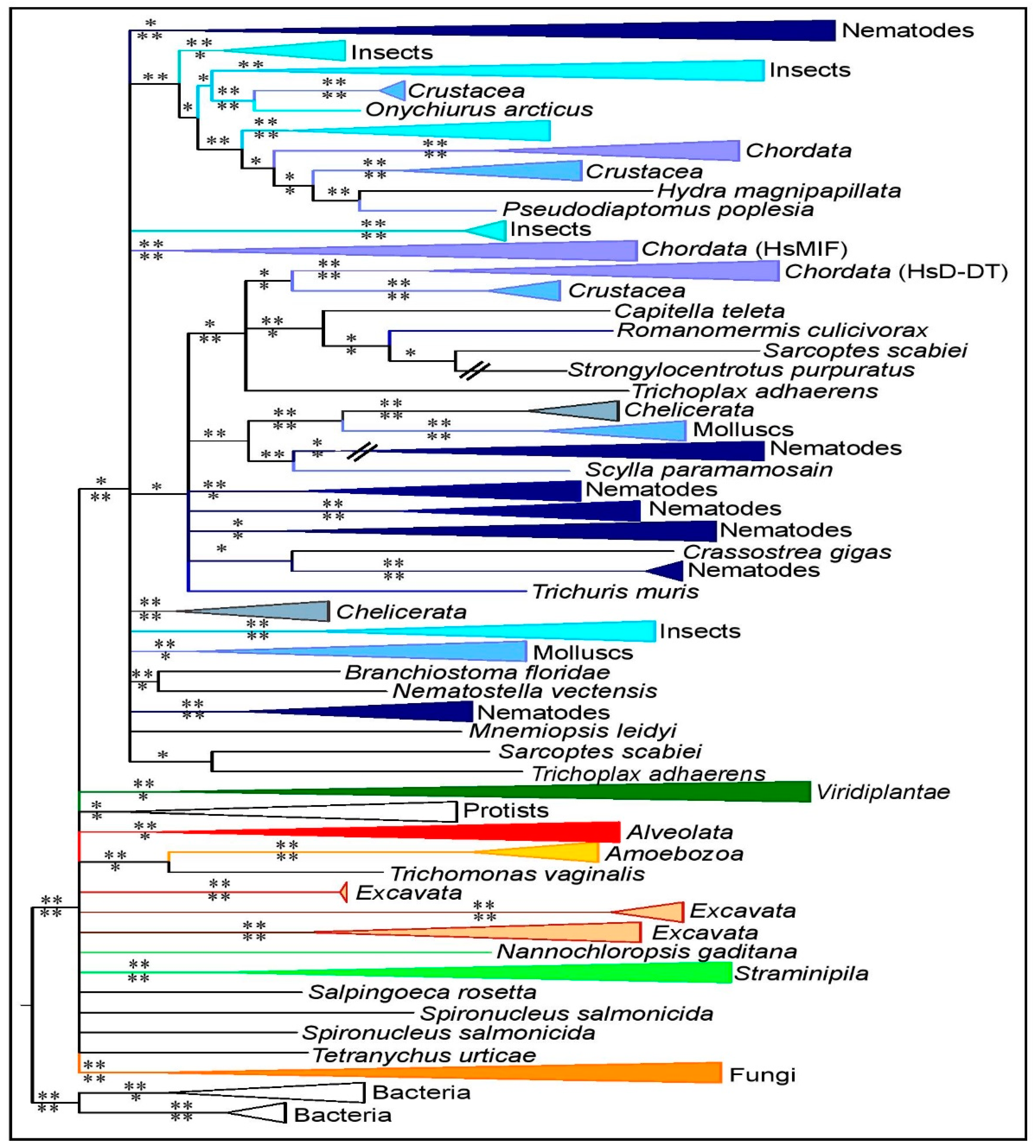
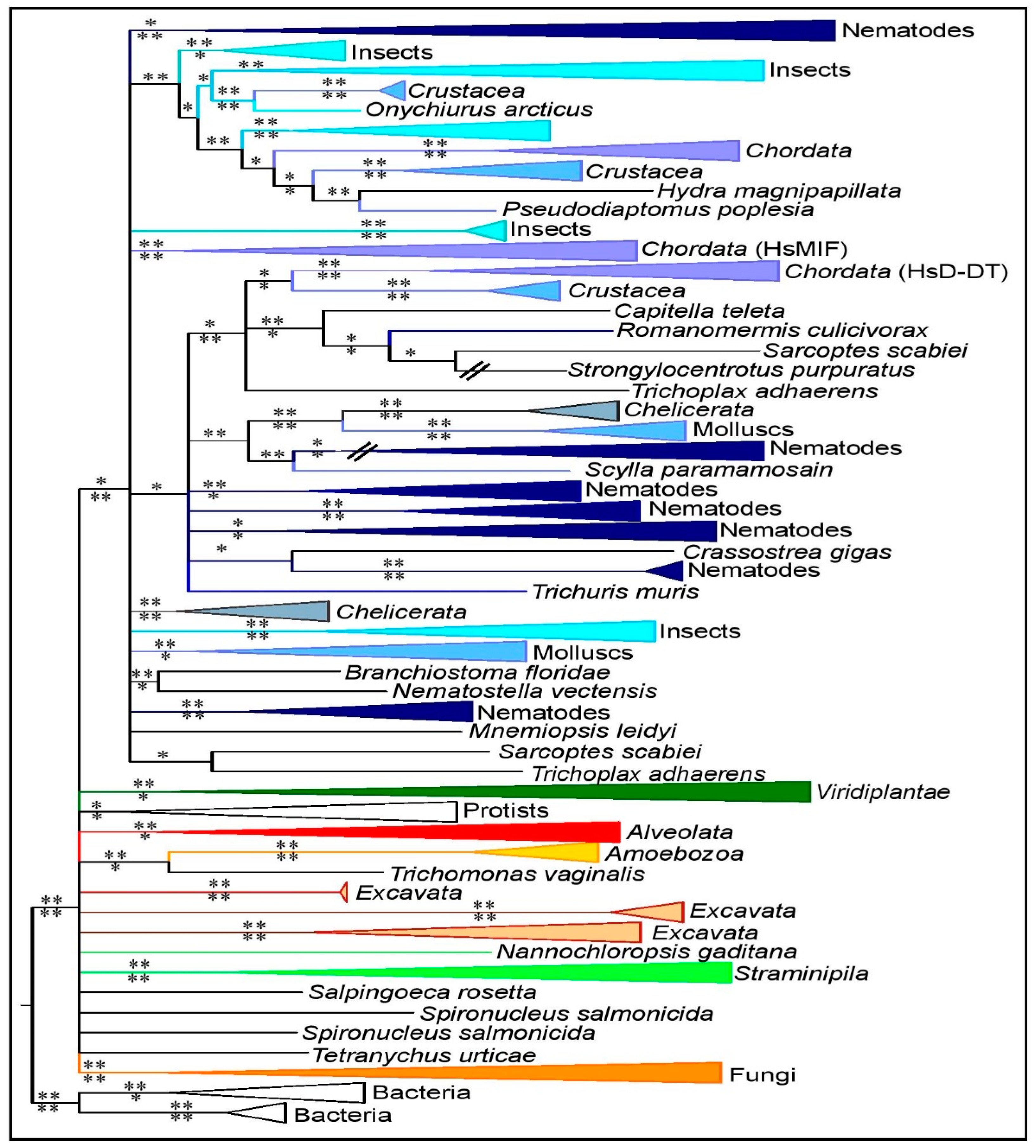
3.2. MIF Phylogenetic Reconstruction across Kingdoms
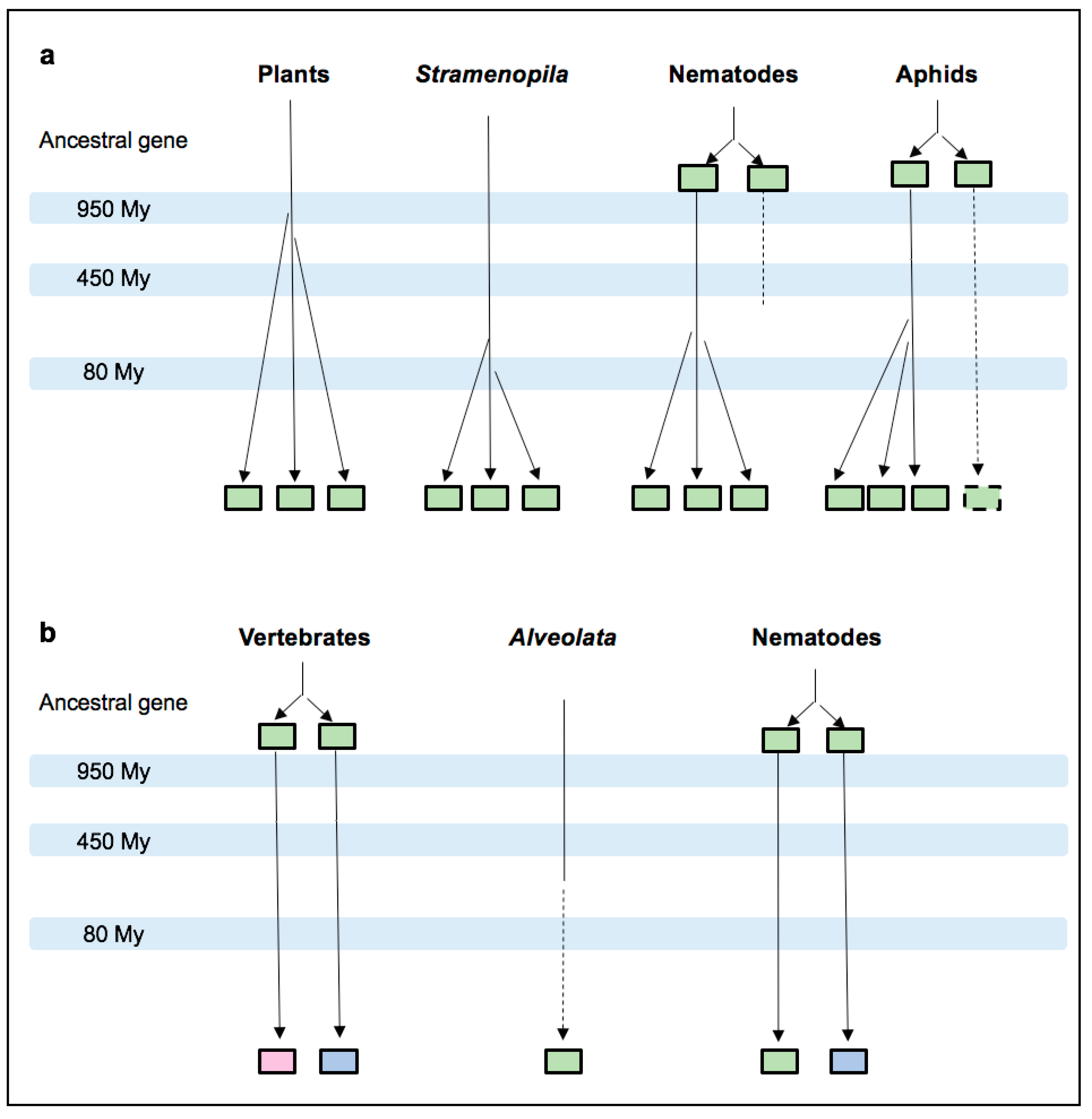
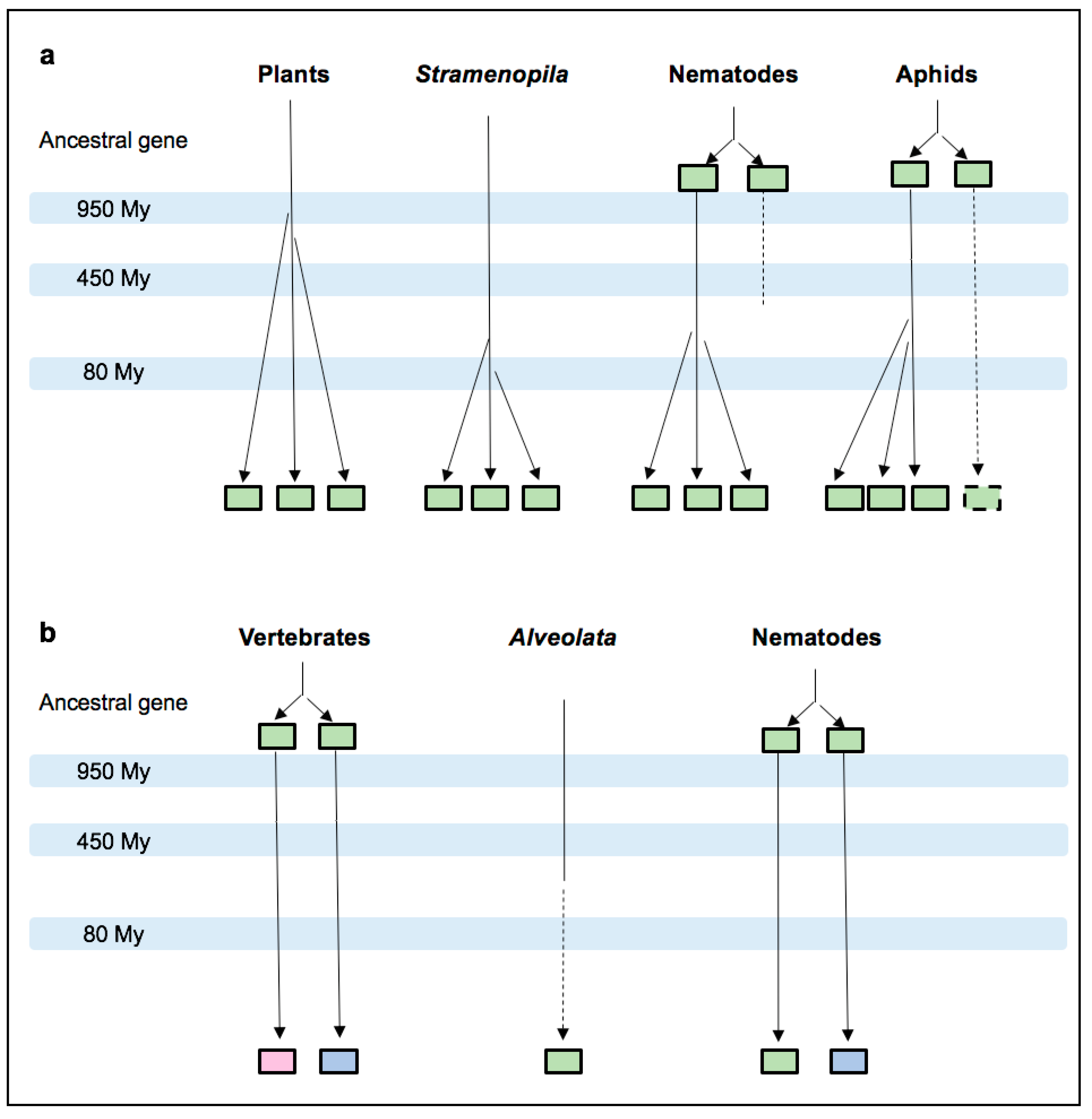
3.3. MIFs in Stramenopila and Alveolata
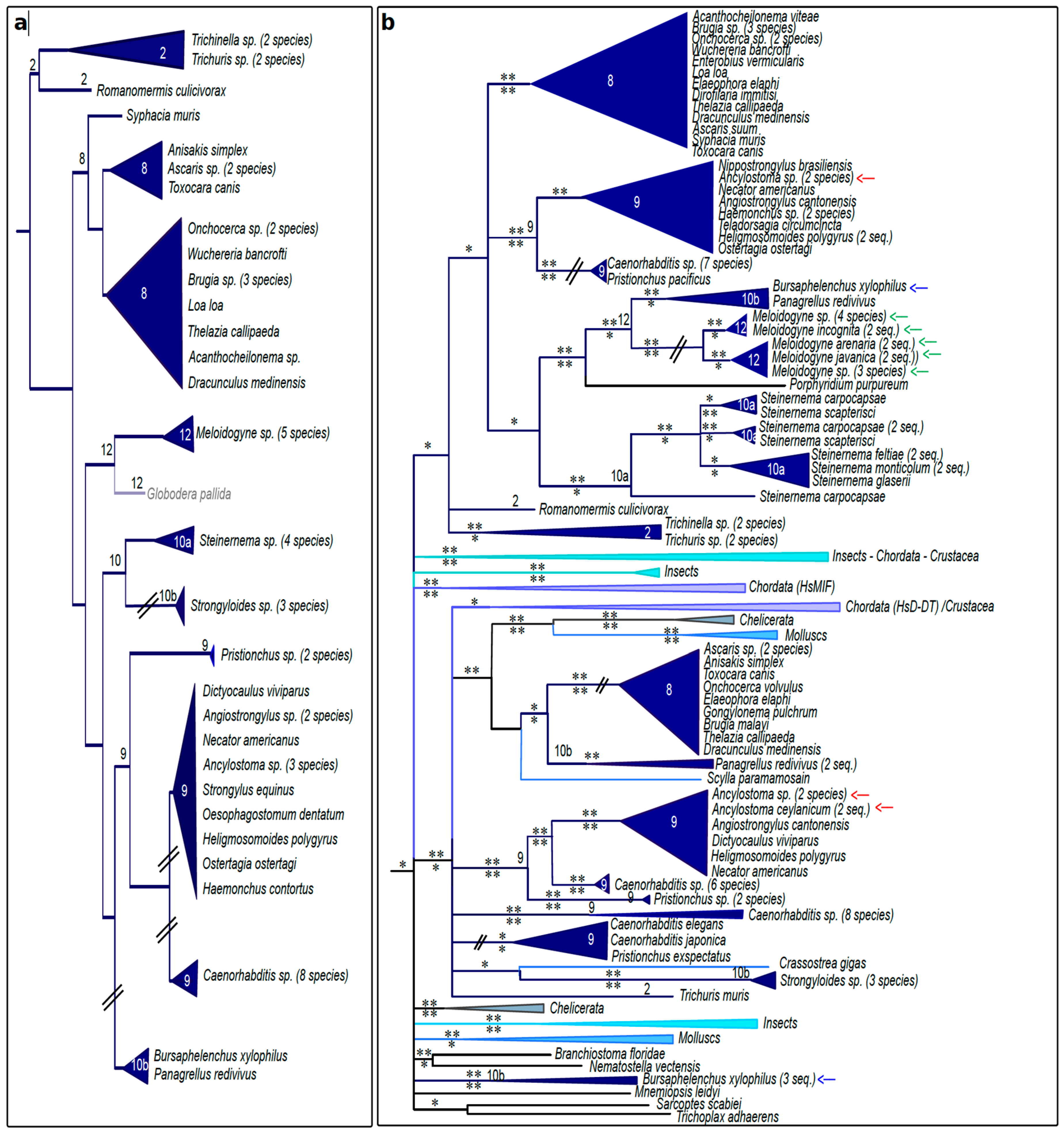
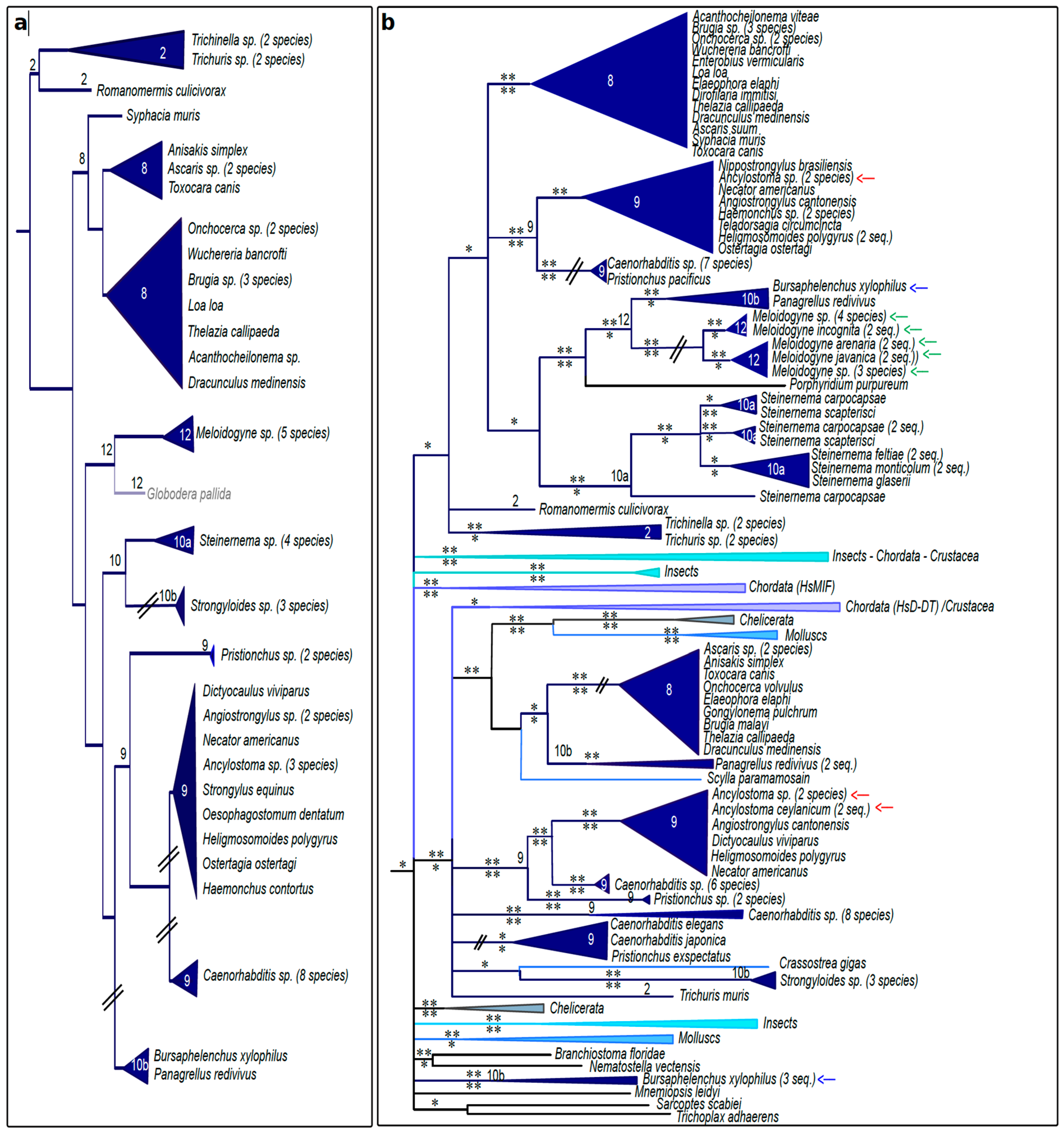
3.4. Nematode MIFs
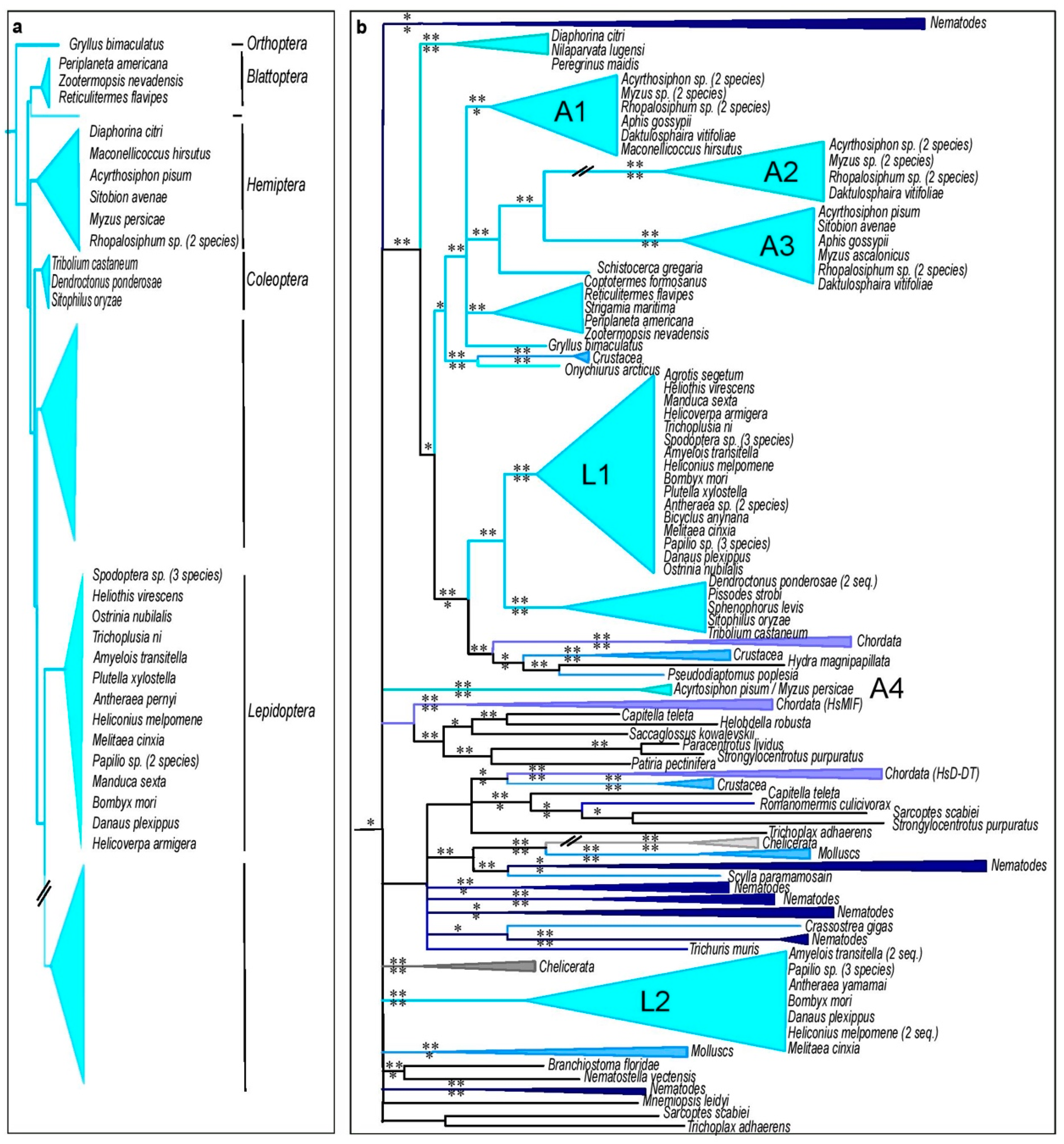
3.5. Insect MIFs
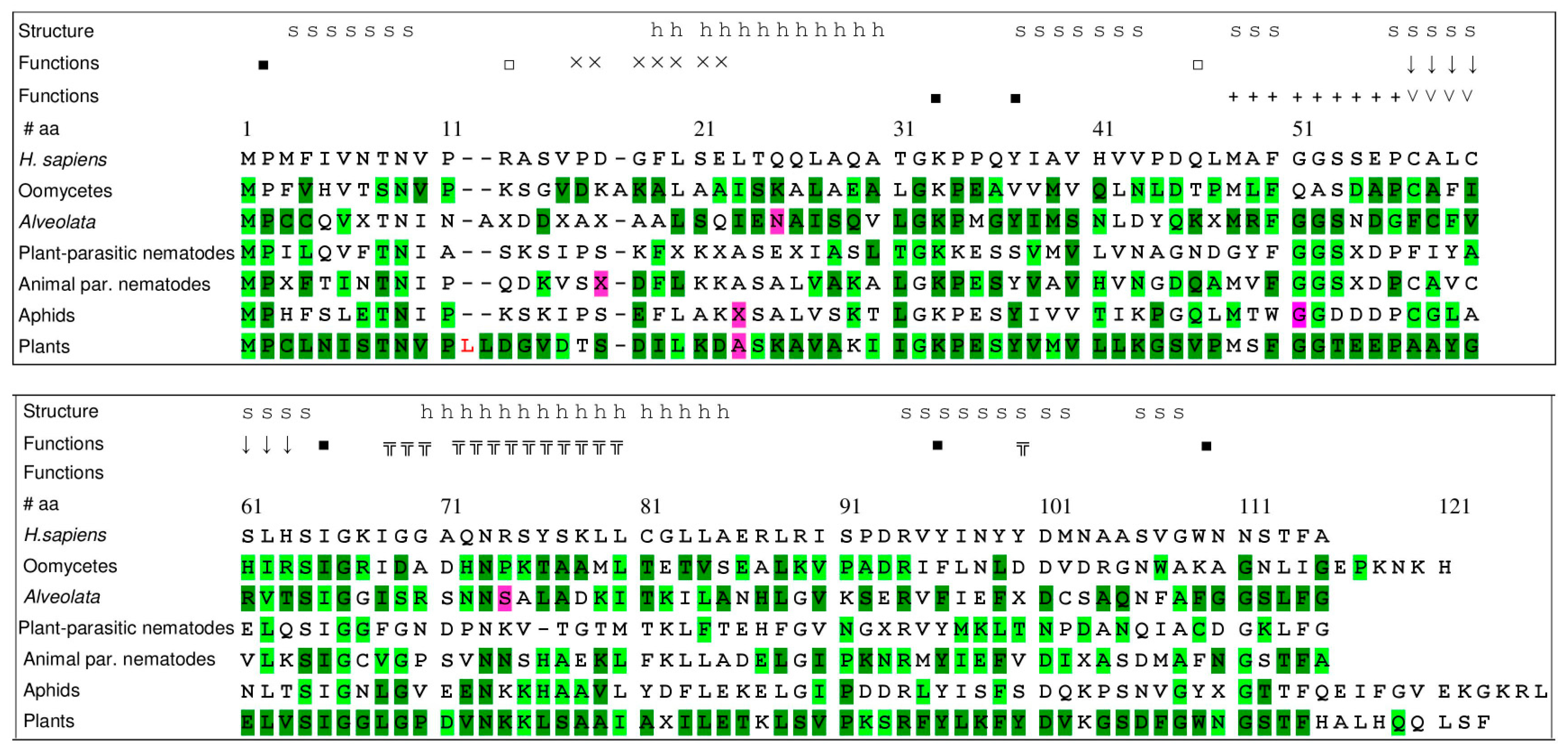
3.6. Conservation of Amino Acids and Motifs within MIF Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bernhagen, J.; Calandra, T.; Mitchell, R.A.; Martin, S.B.; Tracey, K.J.; Voelter, W.; Manogue, K.R.; Cerami, A.; Bucala, R. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature 1993, 365, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Roger, T. Macrophage migration inhibitory factor: A regulator of innate immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399–411. [Google Scholar] [CrossRef]
- Pawig, L.; Klasen, C.; Weber, C.; Bernhagen, J.; Noels, H. Diversity and inter-connections in the CXCR4 chemokine receptor/ligand family: Molecular perspectives. Front. Immunol. 2005, 6, 429. [Google Scholar] [CrossRef]
- Tilstam, P.V.; Qi, D.; Leng, L.; Young, L.; Bucala, R. MIF family cytokines in cardiovascular diseases and prospects for precision-based therapeutics. Expert Opin. Ther. Targets 2017, 21, 671–683. [Google Scholar] [CrossRef]
- Mitchell, R.A.; Liao, H.; Chesney, J.; Fingerle-Rowson, G.; Baugh, J.; David, J.; Bucala, R. Macrophage Migration Inhibitory Factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: Regulatory role in the innate immune response. Proc. Natl. Acad. Sci. USA 2002, 99, 345–350. [Google Scholar] [CrossRef]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354, aad6872. [Google Scholar] [CrossRef] [PubMed]
- Crichlow, G.V.; Fan, C.; Keeler, C.; Hodsdon, M.; Lolis, E.J. Structural interactions dictate the kinetics of macrophage migration inhibitory factor inhibition by different cancer-preventive isothiocyanates. Biochemistry 2012, 51, 7506–7514. [Google Scholar] [CrossRef] [PubMed]
- Subbannayya, T.; Variar, P.; Advani, J.; Nair, B.; Shankar, S.; Gowda, H.; Saussez, S.; Chatterjee, A.; Prasad, T.S.K. An integrated signal transduction network of macrophage migration inhibitory factor. J. Cell Commun. Signal. 2016, 10, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Sparkes, A.; De Baetselier, P.; Roelants, K.; De Trez, C.; Magez, S.; Van Ginderachter, J.A.; Raes, G.; Bucala, R.; Stijlemans, B. The non-mammalian MIF superfamily. Immunobiology 2017, 222, 473–482. [Google Scholar] [CrossRef]
- Augustijn, K.D.; Kleemann, R.; Thompson, J.; Kooistra, T.; Crawford, C.E.; Reece, S.E.; Pain, A.; Siebum, A.H.G.; Janse, C.J.; Waters, A.P. Functional characterization of the Plasmodium falciparum and P. berghei homologues of Macrophage Migration Inhibitory Factor. Infect. Immun. 2007, 75, 1116–1128. [Google Scholar] [CrossRef]
- Garcia, A.B.; Pierce, R.J.; Gourbal, B.; Werkmeister, E.; Colinet, D.; Reichhart, J.-M.; Dissous, C.; Coustau, C. Involvement of the Cytokine MIF in the Snail Host Immune Response to the Parasite Schistosoma mansoni. PLoS Pathog. 2010, 6, e1001115. [Google Scholar]
- Huang, S.; Cao, Y.; Lu, M.; Peng, W.; Lin, J.; Tang, C.; Tang, L. Identification and functional characterization of Oncomelania hupensis macrophage migration inhibitory factor involved in the snail host innate immune response to the parasite Schistosoma japonicum. Int. J. Parasitol. 2017, 47, 485–499. [Google Scholar] [CrossRef]
- Furukawa, R.; Tamaki, K.; Kaneko, H. Two Macrophage Migration Inhibitory Factors regulate starfish larval immune cell chemotaxis. Immunol. Cell Biol. 2017, 94, 315–321. [Google Scholar] [CrossRef]
- Panstruga, R.; Baumgarten, K.; Bernhagen, J. Phylogeny and evolution of plant macrophage migration inhibitory factor/D-dopachrome tautomerase-like proteins. BMC Evol. Biol. 2015, 15, 64. [Google Scholar] [CrossRef]
- Naessens, E.; Dubreuil, G.; Giordanengo, P.; Baron, O.L.; Minet-Kebdani, N.; Keller, H.; Coustau, C. A Secreted MIF Cytokine Enables Aphid Feeding and Represses Plant Immune Responses. Curr. Biol. 2015, 25, 1898–1903. [Google Scholar] [CrossRef] [Green Version]
- Thomma, B.P.H.J.; Eggermont, K.; Penninckx, I.A.M.A.; Mauch-Mani, B.; Vogelsang, R.; Cammue, B.P.A.; Broekaert, W.F. Separate jasmonate-dependent and salicylate-dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc. Natl. Acad. Sci. USA 1998, 95, 15107–15111. [Google Scholar] [CrossRef]
- Dubreuil, G.; Deleury, E.; Crochard, D.; Simon, J.-C.; Coustau, C. Diversification of MIF immune regulators in aphids: Link with agonistic and antagonistic interactions. BMC Genom. 2014, 15, 762. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Cho, Y.; Jones, B.F.; Vermeire, J.J.; Leng, L.; DiFedele, L.; Harrison, L.M.; Xiong, H.; Kwong, Y.-K.A.; Chen, Y.; Bucala, R.; et al. Structural and functional characterization of a secreted hookworm Macrophage Migration Inhibitory Factor (MIF) that interacts with the human MIF receptor CD74. J. Biol. Chem. 2007, 282, 23447–23456. [Google Scholar] [CrossRef]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 40, D23–D28. [Google Scholar] [CrossRef]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, D26–D31. [Google Scholar] [CrossRef] [PubMed]
- Stajich, J.E.; Harris, T.; Brunk, B.P.; Brestelli, J.; Fischer, S.; Harb, O.S.; Kissinger, J.C.; Li, W.; Nayak, V.; Pinney, D.F.; et al. FungiDB: An integrated functional genomics database for fungi. Nucleic Acids Res. 2012, 40, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Attrill, H.; Falls, K.; Goodman, J.L.; Millburn, G.H.; Antonazzo, G.; Rey, A.J.; Marygold, S.J.; Consortium, F. FlyBase: Establishing a gene group resource for Drosophila melanogaster. Nucleic Acids Res. 2016, 44, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Tayal, A.; Diesh, C.M.; Unni, D.R.; Emery, M.L.; Nguyen, H.N.; Hagen, D.E. Hymenoptera genome database: Integrating genome annotations in HymenopteraMine. Nucleic Acids Res. 2016, 44, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.-P.; Zasadzinski, A.; Rispe, C.; Legeai, F.; Tagu, D. AphidBase: A database for aphid genomic resources. Bioinformatics 2007, 23, 783–784. [Google Scholar] [CrossRef]
- Howe, K.L.; Bolt, B.J.; Cain, S.; Chan, J.; Chen, W.J.; Davis, P.; Done, J.; Down, T.; Gao, S.; Grove, C.; et al. WormBase 2016: Expanding to enable helminth genomic research. Nucleic Acids Res. 2016, 44, 774–780. [Google Scholar] [CrossRef]
- Howe, K.L.; Bolt, B.J.; Shafie, M.; Kersey, P.; Berriman, M. WormBase ParaSite—A comprehensive resource for helminth genomics. Mol. Biochem. Parasitol. 2017, 215, 2–10. [Google Scholar] [CrossRef]
- Logan-Klumpler, F.J.; De Silva, N.; Boehme, U.; Rogers, M.B.; Velarde, G.; McQuillan, J.A.; Carver, T.; Aslett, M.; Olsen, C.; Subramanian, S.; et al. GeneDB—An annotation database for pathogens. Nucleic Acids Res. 2012, 40, D98–D108. [Google Scholar] [CrossRef]
- Price, D.C.; Chan, C.X.; Yoon, H.S.; Yang, E.C.; Qiu, H.; Weber, A.P.M.; Schwacke, R.; Gross, J.; Blouin, N.A.; Lane, C.; et al. Cyanophora paradoxa Genome Elucidates Origin of Photosynthesis in Algae and Plants. Science 2012, 335, 843–847. [Google Scholar] [CrossRef]
- Heiges, M. CryptoDB: A Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 2006, 34, D419–D422. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Rahlf, T. Data Visualisation with R: 100 Examples; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; ISBN 978-3-319-49750-1. [Google Scholar]
- Le Roux, B.L.; Rouanet, H. Geometric Data Analysis: From Correspondence Analysis to Structured Data Analysis; Kluwer Academic Publishers: London, UK, 2004. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.-B. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22. [Google Scholar] [CrossRef]
- Madsen, H. Introduction to General and Generalized Linear Models; CRC Press: Boca Raton, FL, USA, 2010; ISBN 978142009155. [Google Scholar]
- Zeileis, A.; Kleiber, C.; Jackman, S. Regression Models for Count Data in R. J. Stat. Softw. 2008, 27, 1–25. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Clamp, M.; Cuff, J.; Searle, S.M.; Barton, G.J. The Jalview Java alignment editor. Bioinformatics 2004, 20, 426–427. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Yilmaz, P.; Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O.; Dufayard, J.-F. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Doron-Faigenboim, A.; Erez, E.; Martz, E.; Bacharach, E.; Pupko, T. Selecton 2007: Advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res. 2007, 35, W506–W511. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Pond, S.L.K. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Mitchell, R.A.; Endres, S.; Bucala, R. D-dopachrome tautomerase (D-DT or MIF-2): Doubling the MIF cytokine family. Cytokine 2012, 59, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burki, F. The Eukaryotic Tree of Life from a Global Phylogenomic Perspective. Cold Spring Harb. Perspect. Biol. 2014, 6, a016147. [Google Scholar] [CrossRef] [PubMed]
- Matari, N.H.; Blair, J.E. A multilocus timescale for oomycete evolution estimated under three distinct molecular clock models. BMC Evol. Biol. 2014, 14, 101. [Google Scholar] [CrossRef] [PubMed]
- Elsen, S.V.D.; Holovachov, O.; Karssen, G.; Van Megen, H.; Helder, J.; Bongers, T.; Bakker, J.; Holterman, M.; Mooyman, P. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 2009, 11, 927–950. [Google Scholar] [CrossRef]
- Giorgi, C. Structural and evolutionary analysis of the ribosomal genes of the parasitic nematode Meloidogyne artiellia suggests its ancient origin. Mol. Biochem. Parasitol. 2002, 124, 91–94. [Google Scholar] [CrossRef]
- Kim, H.; Lee, S.; Jang, Y. Macroevolutionary Patterns in the Aphidini Aphids (Hemiptera: Aphididae): Diversification, Host Association, and Biogeographic Origins. PLoS ONE 2011, 6, e24749. [Google Scholar] [CrossRef]
- Wasiel, A.A.; Rozeboom, H.J.; Hauke, D.; Baas, B.-J.; Zandvoort, E.; Quax, W.J.; Thunnissen, A.-M.W.H.; Poelarends, G.J. Structural and Functional Characterization of a Macrophage Migration Inhibitory Factor Homologue from the Marine Cyanobacterium Prochlorococcus marinus. Biochemistry 2010, 49, 7572–7581. [Google Scholar] [CrossRef]
- Rajasekaran, D.; Gröning, S.; Schmitz, C.; Zierow, S.; Drucker, N.; Bakou, M.; Kohl, K.; Mertens, A.; Lue, H.; Weber, C.; et al. Macrophage Migration Inhibitory Factor-CXCR4 Receptor Interactions. J. Biol. Chem. 2016, 291, 15881–15895. [Google Scholar] [CrossRef] [Green Version]
- Thiele, M.; Bernhagen, J. Link between Macrophage Migration Inhibitory Factor and Cellular Redox Regulation. Antioxid. Redox Signal. 2005, 7, 1234–1248. [Google Scholar] [CrossRef]
- Weber, C.; Kraemer, S.; Drechsler, M.; Lue, H.; Koenen, R.R.; Kapurniotu, A.; Zernecke, A.; Bernhagen, J. Structural determinants of MIF functions in CXCR2-mediated inflammatory and atherogenic leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2008, 105, 16278–16283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, M.; Kontos, C.; Brandhofer, M.; Hille, K.; Gröning, S.; Sinitski, D.; Bourilhon, P.; Rosenberg, E.; Krammer, C.; Thavayogarajah, T.; et al. Identification of an Arg-Leu-Arg tripeptide that contributes to the binding interface between the cytokine MIF and the chemokine receptor CXCR4. Sci. Rep. 2018, 8, 5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantouris, G.; Ho, J.; Shah, D.; Syed, M.A.; Leng, L.; Bhandari, V.; Bucala, R.; Batista, V.S.; Loria, J.P.; Lolis, E.J. Nanosecond Dynamics Regulate the MIF-Induced Activity of CD74. Angew. Chem. Int. Ed. 2018, 57, 7116–7119. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Zierow, S.; Leng, L.; Das, R.; Du, X.; Schulte, W.; Fan, J.; Lue, H.; Chen, Y.; Xiong, H.; et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. USA 2011, 108, E577–E585. [Google Scholar] [CrossRef]
- Huang, W.-S.; Duan, L.-P.; Huang, B.; Wang, K.-J.; Zhang, C.-L.; Jia, Q.-Q.; Nie, P.; Wang, T. Macrophage migration inhibitory factor (MIF) family in arthropods: Cloning and expression analysis of two MIF and one D-dopachrome tautomerase (DDT) homologues in mud crabs, Scylla paramamosain. Fish Shellfish Immunol. 2016, 50, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, M.-G.; Toubiana, M.; Mangano, V.; Parrinello, N.; Cammarata, M.; Roch, P. MIF from mussel: Coding sequence, phylogeny, polymorphism, 3D model and regulation of expression. Dev. Comp. Immunol. 2012, 36, 688–696. [Google Scholar] [CrossRef]
- Oh, M.; Kasthuri, S.R.; Wan, Q.; Bathige, S.; Whang, I.; Lim, B.-S.; Jung, H.-B.; Oh, M.-J.; Jung, S.-J.; Kim, S.Y.; et al. Characterization of MIF family proteins: MIF and DDT from rock bream, Oplegnathus fasciatus. Fish Shellfish Immunol. 2013, 35, 458–468. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelet, C.; Danchin, E.G.J.; Jaouannet, M.; Bernhagen, J.; Panstruga, R.; Kogel, K.-H.; Keller, H.; Coustau, C. Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily. Genes 2019, 10, 740. https://doi.org/10.3390/genes10100740
Michelet C, Danchin EGJ, Jaouannet M, Bernhagen J, Panstruga R, Kogel K-H, Keller H, Coustau C. Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily. Genes. 2019; 10(10):740. https://doi.org/10.3390/genes10100740
Chicago/Turabian StyleMichelet, Claire, Etienne G. J. Danchin, Maelle Jaouannet, Jürgen Bernhagen, Ralph Panstruga, Karl-Heinz Kogel, Harald Keller, and Christine Coustau. 2019. "Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily" Genes 10, no. 10: 740. https://doi.org/10.3390/genes10100740
APA StyleMichelet, C., Danchin, E. G. J., Jaouannet, M., Bernhagen, J., Panstruga, R., Kogel, K.-H., Keller, H., & Coustau, C. (2019). Cross-Kingdom Analysis of Diversity, Evolutionary History, and Site Selection within the Eukaryotic Macrophage Migration Inhibitory Factor Superfamily. Genes, 10(10), 740. https://doi.org/10.3390/genes10100740