Jumonji C Demethylases in Cellular Senescence


Abstract
1. Introduction
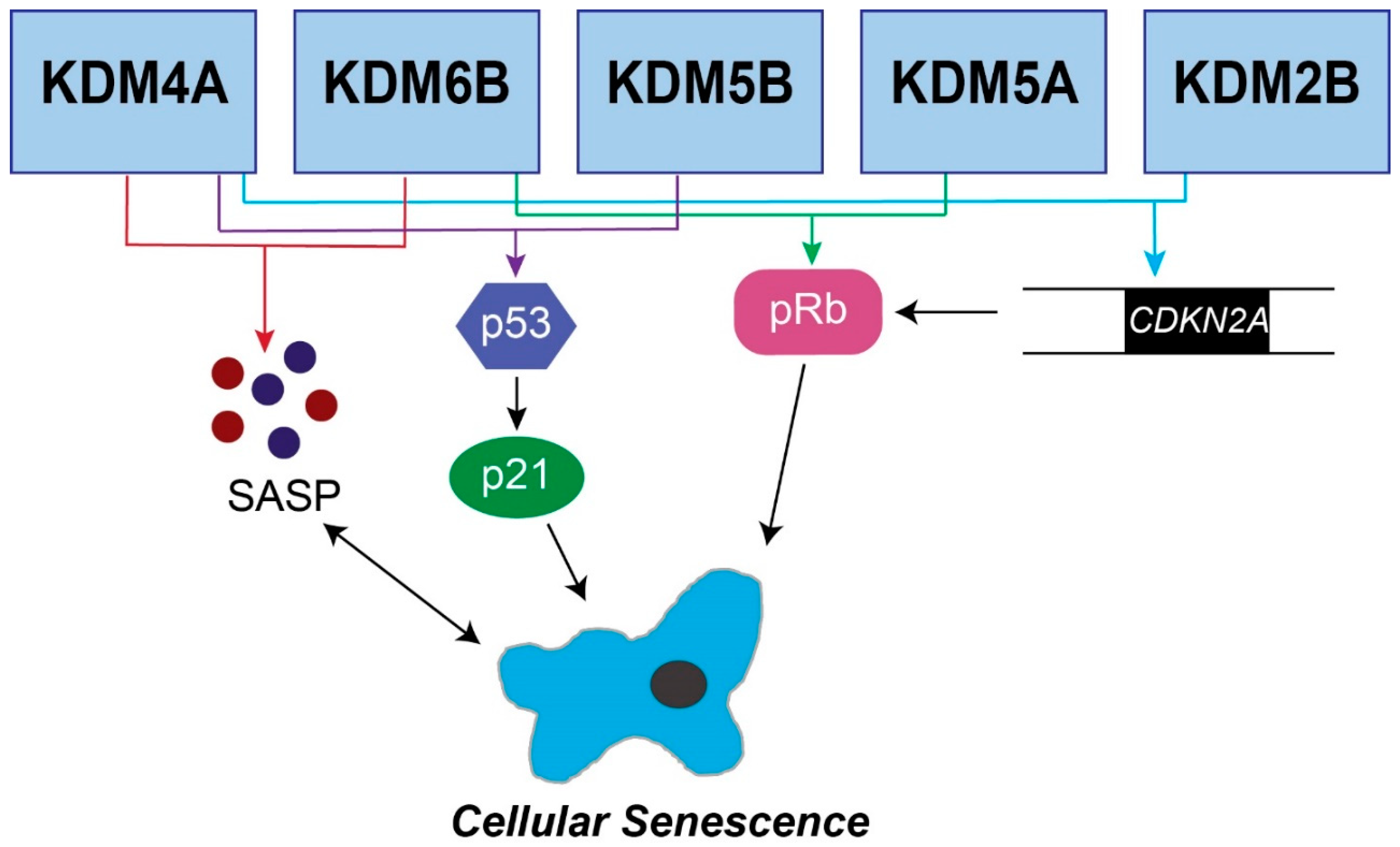
2. Role of Jumonji C Demethylases in Senescence
2.1. Cellular Senescence
2.2. KDM6B
2.3. KDM5B
2.4. KDM5A
2.5. KDM4A
2.6. KDM4B
2.7. KDM2B
3. Jumonji C Histone Demethylases Suppress Senescence during Tumorigenesis
3.1. Jumonji C Demethylases Affect Cell Cycle Regulatory Genes to Suppress Senescence
3.2. JmjC Demethylases Affect the Senescence-Associated Secretory Phenotype
4. Targeting JmjC Demethylases for Cancer Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 a resolution. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Thomas, J.O. Chromatin structure; oligomers of the histones. Science 1974, 184, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Moudrianakis, E.N. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. USA 1995, 92, 11170–11174. [Google Scholar] [CrossRef] [PubMed]
- Kebede, A.F.; Schneider, R.; Daujat, S. Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. FEBS J. 2015, 282, 1658–1674. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Fenley, A.T.; Anandakrishnan, R.; Kidane, Y.H.; Onufriev, A.V. Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenet. Chromatin 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Schneider, J.; Shilatifard, A. Histone demethylation by hydroxylation: Chemistry in action. ACS Chem. Biol. 2006, 1, 75–81. [Google Scholar] [CrossRef]
- Byvoet, P.; Shepherd, G.R.; Hardin, J.M.; Noland, B.J. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972, 148, 558–567. [Google Scholar] [CrossRef]
- Li, T.; Vu, T.H.; Ulaner, G.A.; Yang, Y.; Hu, J.F.; Hoffman, A.R. Activating and silencing histone modifications form independent allelic switch regions in the imprinted Gnas gene. Hum. Mol. Genet. 2004, 13, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Trievel, R.C. Histone modifying enzymes: Structures, mechanisms, and specificities. Biochim. Biophys. Acta 2009, 1789, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Accari, S.L.; Fisher, P.R. Emerging roles of JmjC domain-containing proteins. Int. Rev. Cell Mol. Biol. 2015, 319, 165–220. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Yamazaki, Y.; Katoh-Fukui, Y.; Tsuchiya, R.; Kondo, S.; Motoyama, J.; Higashinakagawa, T. Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes Dev. 1995, 9, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Leissing, T.M.; Chowdhury, R.; Wilkins, S.E.; Lu, X.; Schofield, C.J. Structure-function relationships of human JmjC oxygenases-demethylases versus hydroxylases. Curr. Opin. Struct. Biol. 2016, 41, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Markolovic, S.; Wilkins, S.E.; Schofield, C.J. Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenases. J. Biol. Chem. 2015, 290, 20712–20722. [Google Scholar] [CrossRef] [PubMed]
- Labbe, R.M.; Holowatyj, A.; Yang, Z.Q. Histone lysine demethylase (KDM) subfamily 4: Structures, functions and therapeutic potential. Am. J. Transl. Res. 2013, 6, 1–15. [Google Scholar] [PubMed]
- Rotili, D.; Mai, A. Targeting histone demethylases: A new avenue for the fight against cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef]
- Loughery, J.E.; Dunne, P.D.; O’Neill, K.M.; Meehan, R.R.; McDaid, J.R.; Walsh, C.P. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum. Mol. Genet. 2011, 20, 3241–3255. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Pfau, R.; Tzatsos, A.; Kampranis, S.C.; Serebrennikova, O.B.; Bear, S.E.; Tsichlis, P.N. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc. Natl. Acad. Sci. USA 2008, 105, 1907–1912. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Perrigue, P.M.; Silva, M.E.; Warden, C.D.; Feng, N.L.; Reid, M.A.; Mota, D.J.; Joseph, L.P.; Tian, Y.I.; Glackin, C.A.; Gutova, M.; et al. The histone demethylase jumonji coordinates cellular senescence including secretion of neural stem cell-attracting cytokines. Mol. Cancer Res. 2015, 13, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, Y.; Gao, Y.; Geng, P.; Lu, Y.; Liu, X.; Yao, R.; Hou, P.; Liu, D.; Lu, J.; et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 2015, 22, 1630–1640. [Google Scholar] [CrossRef]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 2014, 9, e96545. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Haraguchi, N.; Kano, Y.; Kagawa, Y.; Konno, M.; Nishikawa, S.; Hamabe, A.; Hasegawa, S.; Ogawa, H.; Fukusumi, T.; et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int. J. Oncol. 2013, 42, 1212–1218. [Google Scholar] [CrossRef]
- Chicas, A.; Kapoor, A.; Wang, X.; Aksoy, O.; Evertts, A.G.; Zhang, M.Q.; Garcia, B.A.; Bernstein, E.; Lowe, S.W. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 8971–8976. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zeng, J.; Wang, L.; Fang, M.; Wang, Q.; Zhao, M.; Xu, X.; Liu, Z.; Li, W.; Liu, S.; et al. Histone demethylase retinoblastoma binding protein 2 is overexpressed in hepatocellular carcinoma and negatively regulated by hsa-miR-212. PLoS ONE 2013, 8, e69784. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Ge, Z.; Wang, L.; Li, Q.; Wang, N.; Bjorkholm, M.; Jia, J.; Xu, D. The histone demethylase RBP2 is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [Google Scholar] [CrossRef] [PubMed]
- McBrayer, S.K.; Olenchock, B.A.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Jennings, R.B.; Novak, J.S.; Oser, M.G.; Robbins, A.K.; Modiste, R.; et al. Autochthonous tumors driven by Rb1 loss have an ongoing requirement for the RBP2 histone demethylase. Proc. Natl. Acad. Sci. USA 2018, 115, E3741–E3748. [Google Scholar] [CrossRef] [PubMed]
- Mallette Frédérick, A.; Richard, S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep. 2012, 2, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Sun, J.; Kitagawa, K.; Nakanishi, K.; Kuno, T.; Naiki-Ito, A.; Sawada, Y.; Miyamoto, T.; Okabe, A.; Aburatani, H.; et al. SCFFbxo22-KDM4A targets methylated p53 for degradation and regulates senescence. Nat. Commun. 2016, 7, 10574. [Google Scholar] [CrossRef] [PubMed]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Chen, L.; Pledger, W.J.; Fang, J.; Chen, J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 2014, 33, 734–744. [Google Scholar] [CrossRef]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef]
- Tzatsos, A.; Pfau, R.; Kampranis, S.C.; Tsichlis, P.N. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc. Natl. Acad. Sci. USA 2009, 106, 2641–2646. [Google Scholar] [CrossRef]
- He, J.; Kallin, E.M.; Tsukada, Y.; Zhang, Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nature Struct. Mol. Biol. 2008, 15, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Tzatsos, A.; Paskaleva, P.; Ferrari, F.; Deshpande, V.; Stoykova, S.; Contino, G.; Wong, K.K.; Lan, F.; Trojer, P.; Park, P.J.; et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J. Clin. Investig. 2013, 123, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Haferkamp, S.; Tran, S.L.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence. Aging 2009, 1, 542–556. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [PubMed]
- Agherbi, H.; Gaussmann-Wenger, A.; Verthuy, C.; Chasson, L.; Serrano, M.; Djabali, M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS ONE 2009, 4, e5622. [Google Scholar] [CrossRef] [PubMed]
- Dey, B.K.; Stalker, L.; Schnerch, A.; Bhatia, M.; Taylor-Papidimitriou, J.; Wynder, C. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol. Cell. Biol. 2008, 28, 5312–53127. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Bayo, J.; Tran, T.A.; Wang, L.; Pena-Llopis, S.; Das, A.K.; Martinez, E.D. Jumonji inhibitors overcome radioresistance in cancer through changes in H3K4 methylation at double-strand breaks. Cell Rep. 2018, 25, 1040–1050.E5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhou, B.; Zhao, X.; Zhu, L.; Xu, J.; Jiang, Z.; Chen, D.; Wei, Q.; Han, M.; Feng, L.; et al. KDM5B demethylates H3K4 to recruit XRCC1 and promote chemoresistance. Int. J. Biol. Sci. 2018, 14, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Yan, Q.; Tothova, Z.; Yamane, K.; Erdjument-Bromage, H.; Tempst, P.; Gilliland, D.G.; Zhang, Y.; Kaelin, W.G., Jr. The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 2007, 128, 889–900. [Google Scholar] [CrossRef]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.K.; Judge, R.A.; Li, L.; Pithawalla, R.; Simanis, J.; Bodelle, P.M.; Marin, V.L.; Henry, R.F.; Petros, A.M.; Sun, C. Targeting lysine specific demethylase 4A (KDM4A) tandem TUDOR domain—A fragment based approach. Bioorg. Med. Chem. Lett. 2018, 28, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Calderas, L.; Gonzalez-Barrios, R.; Patino, C.C.; Alcaraz, N.; Salgado-Albarran, M.; de Leon, D.C.; Hernandez, C.C.; Sanchez-Perez, Y.; Maldonado-Martinez, H.A.; De la Rosa-Velazquez, I.A.; et al. CTCF-KDM4A complex correlates with histone modifications that negatively regulate CHD5 gene expression in cancer cell lines. Oncotarget 2018, 9, 17028–17042. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Munoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppe, J.P.; Campeau, E.; Beausejour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Xiang, L.; Li, S.; Rao, D.; Wang, S.; Yu, K. MiR-10a functions as a tumor suppressor in prostate cancer via targeting KDM4A. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef]
- Young, L.C.; Hendzel, M.J. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem. Cell Biol. 2013, 91, 369–377. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Nguyen, A.T.; Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 2011, 117, 3869–3880. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, R.; Park, G.; Park, J.W.; Kim, J.E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Harmeyer, K.M.; Facompre, N.D.; Herlyn, M.; Basu, D. JARID1 histone demethylases: Emerging targets in cancer. Trends Cancer 2017, 3, 713–725. [Google Scholar] [CrossRef]
- Plch, J.; Hrabeta, J.; Eckschlager, T. KDM5 demethylases and their role in cancer cell chemoresistance. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Yang, X.; Wang, H.; Shao, Q. The critical role of histone lysine demethylase KDM2B in cancer. Am. J. Transl. Res. 2018, 10, 2222–2233. [Google Scholar]
- Guerra-Calderas, L.; Gonzalez-Barrios, R.; Herrera, L.A.; Cantu de Leon, D.; Soto-Reyes, E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015, 208, 215–224. [Google Scholar] [CrossRef]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Laberge, R.M.; Awad, P.; Campisi, J.; Desprez, P.Y. Epithelial-mesenchymal transition induced by senescent fibroblasts. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2012, 5, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed]
- Lachner, M.; Jenuwein, T. The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 2002, 14, 286–298. [Google Scholar] [CrossRef]
- Silva, J.; Mak, W.; Zvetkova, I.; Appanah, R.; Nesterova, T.B.; Webster, Z.; Peters, A.H.; Jenuwein, T.; Otte, A.P.; Brockdorff, N. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 2003, 4, 481–495. [Google Scholar] [CrossRef]
- Culhane, J.C.; Wang, D.; Yen, P.M.; Cole, P.A. Comparative analysis of small molecules and histone substrate analogues as LSD1 lysine demethylase inhibitors. J. Am. Chem. Soc. 2010, 132, 3164–3176. [Google Scholar] [CrossRef]
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Schmidt, D.M.; McCafferty, D.G.; Shiekhattar, R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem. Biol. 2006, 13, 563–567. [Google Scholar] [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Gunzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef]
- Szewczuk, L.M.; Culhane, J.C.; Yang, M.; Majumdar, A.; Yu, H.; Cole, P.A. Mechanistic analysis of a suicide inactivator of histone demethylase LSD1. Biochemistry 2007, 46, 6892–6902. [Google Scholar] [CrossRef]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef]
- Hillringhaus, L.; Yue, W.W.; Rose, N.R.; Ng, S.S.; Gileadi, C.; Loenarz, C.; Bello, S.H.; Bray, J.E.; Schofield, C.J.; Oppermann, U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 2011, 286, 41616–41625. [Google Scholar] [CrossRef]
- Mackeen, M.M.; Kramer, H.B.; Chang, K.H.; Coleman, M.L.; Hopkinson, R.J.; Schofield, C.J.; Kessler, B.M. Small-molecule-based inhibition of histone demethylation in cells assessed by quantitative mass spectrometry. J. Proteome Res. 2010, 9, 4082–4092. [Google Scholar] [CrossRef]
- Ye, Q.; Holowatyj, A.; Wu, J.; Liu, H.; Zhang, L.; Suzuki, T.; Yang, Z.Q. Genetic alterations of KDM4 subfamily and therapeutic effect of novel demethylase inhibitor in breast cancer. Am. J. Cancer Res. 2015, 5, 1519–1530. [Google Scholar]
- Franci, G.; Sarno, F.; Nebbioso, A.; Altucci, L. Identification and characterization of PKF118-310 as a KDM4A inhibitor. Epigenetics 2017, 12, 198–205. [Google Scholar] [CrossRef]
- Schiller, R.; Scozzafava, G.; Tumber, A.; Wickens, J.R.; Bush, J.T.; Rai, G.; Lejeune, C.; Choi, H.; Yeh, T.L.; Chan, M.C.; et al. A cell-permeable ester derivative of the JmjC histone demethylase inhibitor IOX1. ChemMedChem 2014, 9, 566–571. [Google Scholar] [CrossRef]
- Easmon, J.; Heinisch, G.; Purstinger, G.; Langer, T.; Osterreicher, J.K.; Grunicke, H.H.; Hofmann, J. Azinyl and diazinyl hydrazones derived from aryl N-heteroaryl ketones: Synthesis and antiproliferative activity. J. Med. Chem. 1997, 40, 4420–4425. [Google Scholar] [CrossRef]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Pena-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 2035. [Google Scholar] [CrossRef]
- Cascella, B.; Lee, S.G.; Singh, S.; Jez, J.M.; Mirica, L.M. The small molecule JIB-04 disrupts O2 binding in the Fe-dependent histone demethylase KDM4A/JMJD2A. Chem. Commun. 2017, 53, 2174–2177. [Google Scholar] [CrossRef]


{kind=link}
| JmjC Histone Demethylase | Target(s) | Implications in Senescence | Role in Suppressing Senescence to Promote Tumorigenesis |
|---|---|---|---|
| KDM6B | H3K27me3/2 H3K9me2/1 H4K20me1 | Overexpression increases SASP gene expression [27]; promotes SAHF formation through pRb [28]; regulation of p53 [29] | Overexpressed in glioma cells to promote SASP expression [27] |
| KDM5B | H3K4me3/2 | Silences E2F target genes [30,31]; knockdown increases H3K4 methylation at the CDKN2A locus [30] | |
| KDM5A | H3K4me3/2 | Knockdown or depletion induces senescence by increasing p21, p27, p16 [32,33] | Expression is required in pRb-defective cancer [34] |
| KDM4A | H3K36me3/2 H3K9me3/2 | Downregulation activates the p53 pathway and induces PML body accumulation [35] | Cooperates with RAS to promote transformation [35]; affects the SASP [36] |
| KDM4B | H3K36me3/2 H3K9me3/2 | No direct evidence; p53-responsive [37,38]; important for DSB repair [38,39] | |
| KDM2B | H3K36me2/1 H3K4me3 H3K79me3/2 | Demethylates CDKN2A [40] and CDKN2B loci [41] | Cooperates with KRAS to promote pancreatic cancer [42]; immortalizes MEFs [24,40] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leon, K.E.; Aird, K.M. Jumonji C Demethylases in Cellular Senescence. Genes 2019, 10, 33. https://doi.org/10.3390/genes10010033
Leon KE, Aird KM. Jumonji C Demethylases in Cellular Senescence. Genes. 2019; 10(1):33. https://doi.org/10.3390/genes10010033
Chicago/Turabian StyleLeon, Kelly E., and Katherine M. Aird. 2019. "Jumonji C Demethylases in Cellular Senescence" Genes 10, no. 1: 33. https://doi.org/10.3390/genes10010033
APA StyleLeon, K. E., & Aird, K. M. (2019). Jumonji C Demethylases in Cellular Senescence. Genes, 10(1), 33. https://doi.org/10.3390/genes10010033