Modulation of Bacterial sRNAs Activity by Epigenetic Modifications: Inputs from the Eukaryotic miRNAs



Abstract
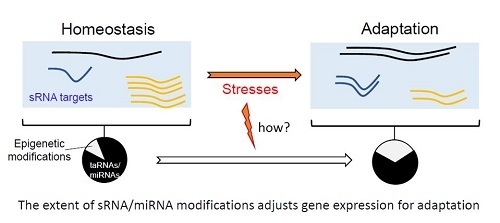
{kind=link}
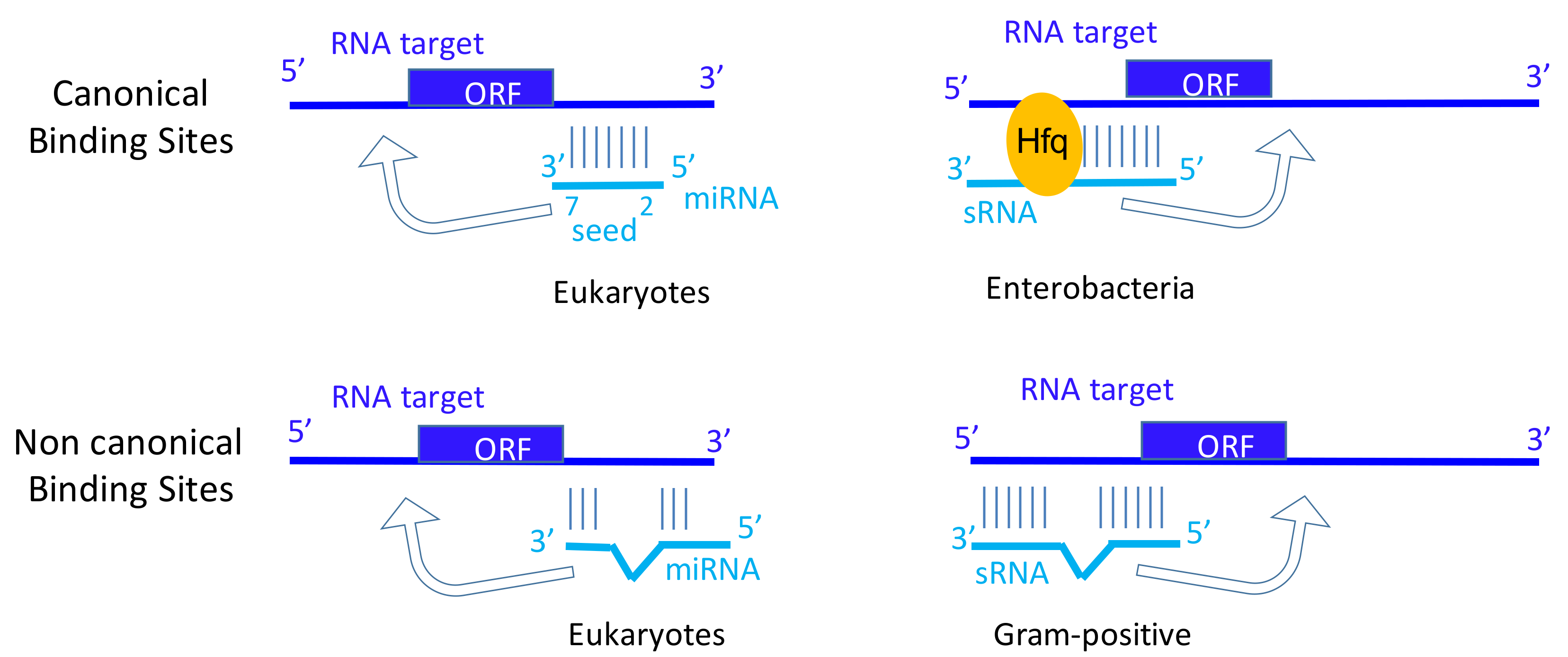{kind=link}
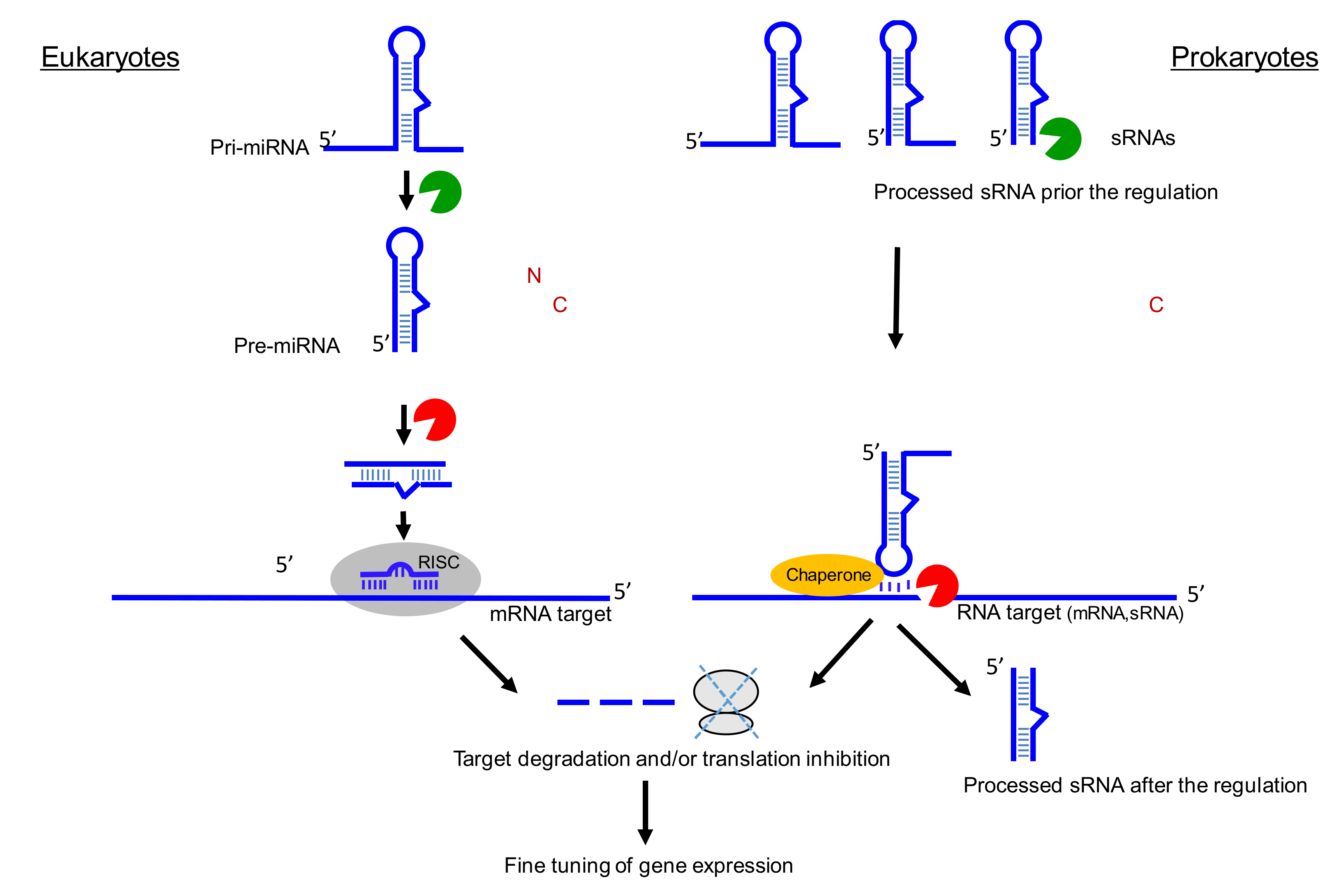{kind=link}
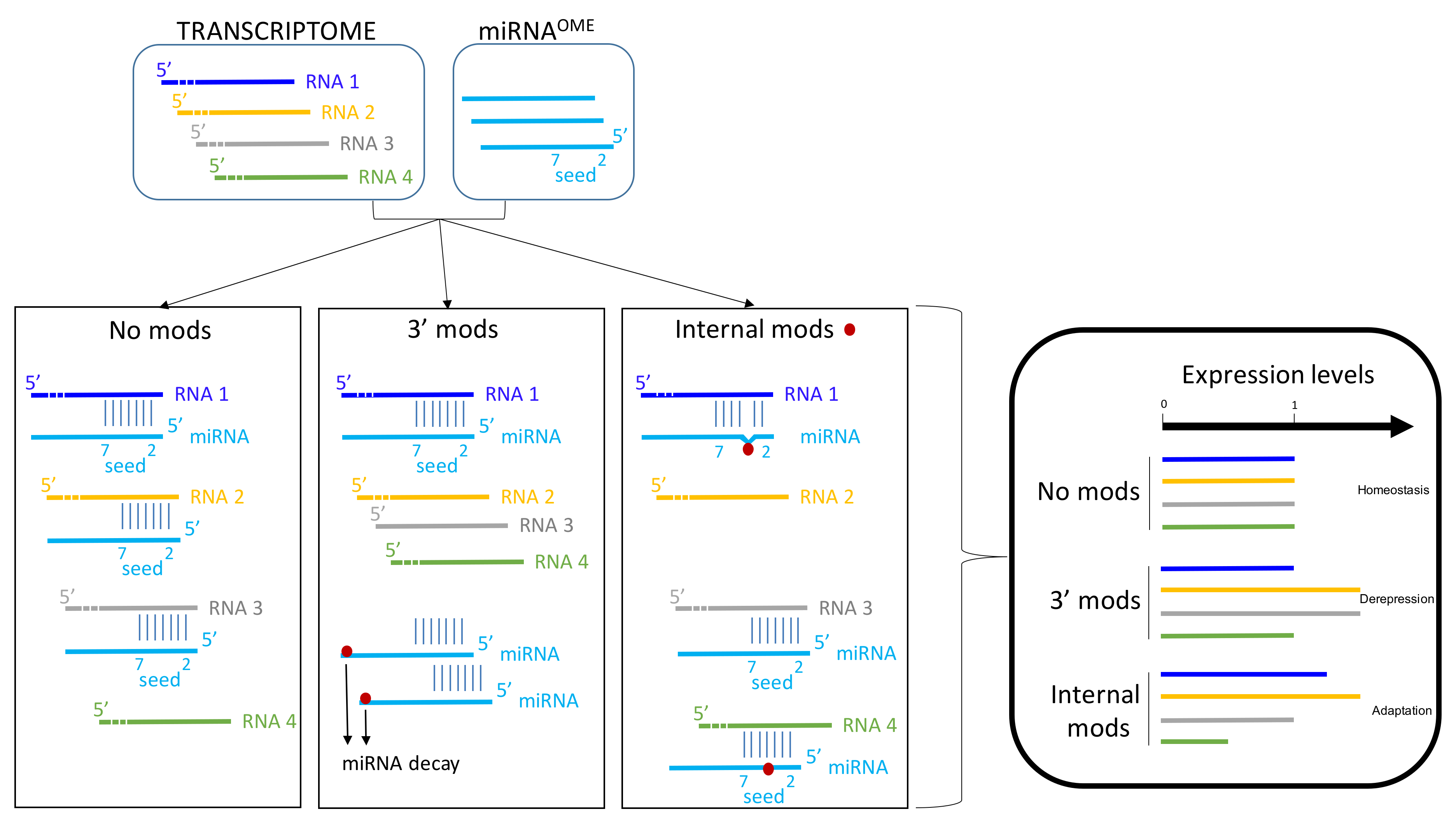{kind=link}
1. Introduction
2. Biogenesis and Functions of Bacterial Trans-Acting RNAs and Eukaryotic micro RNAs
2.1. sRNA Biogenesis
2.2. sRNA Functions
2.3. Micro RNA Genesis
2.4. Micro RNA Functions
3. The Set of RNA Modifications among Bacterial Trans-Acting Small RNAs and Eukaryotic Micro RNAs
3.1. Internal RNA Mods
3.1.1. sRNAs: Pseudouridine, Uracil to Thymidine (C5-Methylation, m5C)
3.1.2. Micro RNAs: A to I Editing, N6-methyladenosine
3.1.3. Adenosines (m6A) and Uridines (m5U) Methylations
3.1.3.1. N6-methyladenosine
3.1.3.2. m6A Expression and Localization
3.1.3.3. m6A Writers
3.1.3.4. m6A Readers
3.1.3.5. m6A Erasers
3.1.3.6. m6A and Micro RNAs
3.1.3.7. Consequences of m6A on Micro RNA Activity
3.1.4. 3′-Terminal Untemplated Nucleotide Addition
3.2. Mods at RNA Ends
3.2.1. sRNAs: 5′-NAD-capped RNA
3.2.2. Micro RNAs: 3′-Uridinylation
3.2.2.1. Micro RNA and Mono-Uridylation
3.2.2.2. Pre-Micro RNA and Oligo-Uridylation
3.2.3. sRNA Stabilization by 3′ Polyadenylation in Prokaryotes
4. Concluding Remarks and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: an answer to the ’genome complexity’ conundrum. Genes Dev. 2007, 21, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Gelsinger, D.R.; DiRuggiero, J. Transcriptional Landscape and Regulatory Roles of Small Noncoding RNAs in the Oxidative Stress Response of the Haloarchaeon Haloferax volcanii. J Bacteriol. 2018, 200, e00779-17. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Storz, G. Regulatory RNAs in bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Georg, J.; Hess, W.R. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev. 2011, 75, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Wagner, E.G.H.; Romby, P. Small RNAs in bacteria and archaea. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2015; pp. 133–208. [Google Scholar]
- Zhao, X.; Zhang, Y.; Huang, X. Pathogenicity-island-encoded regulatory RNAs regulate bacterial virulence and pathogenesis. Microb. Pathog. 2018, 125, 196–204. [Google Scholar] [CrossRef]
- Felden, B.; Cattoir, V. Bacterial adaptation to antibiotics through regulatory RNAs. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Carrier, M.-C.; Lalaouna, D.; Massé, E. Broadening the definition of bacterial small RNAs: Characteristics and mechanisms of action. Annu. Rev. Microbiol. 2018, 72, 141–161. [Google Scholar] [CrossRef]
- Santiago-Frangos, A.; Woodson, S.A. Hfq chaperone brings speed dating to bacterial sRNA. Wiley interdisciplinary reviews. RNA 2018, 9, e1475. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Exportin 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Juranek, S.; Li, H.; Sheng, G.; Wardle, G.S.; Tuschl, T.; Patel, D.J. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature 2009, 461, 754–761. [Google Scholar] [CrossRef]
- Ameres, S.L.; Zamore, P.D. Diversifying microRNA sequence and function. Nature reviews. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4. [Google Scholar] [CrossRef]
- Seok, H.; Ham, J.; Jang, E.-S.; Chi, S.W. MicroRNA target recognition: Insights from transcriptome-wide non-canonical interactions. Mol. Cells 2016, 39, 375–381. [Google Scholar]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Khorshid, M.; Hausser, J.; Zavolan, M.; van Nimwegen, E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods 2013, 10, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Loeb, G.B.; Khan, A.A.; Canner, D.; Hiatt, J.B.; Shendure, J.; Darnell, R.B.; Leslie, C.S.; Rudensky, A.Y. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol. Cell 2012, 48, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Sung, Y.M.; Park, J.; Kim, S.; Kim, J.; Park, J.; Ha, H.; Bae, J.Y.; Kim, S.; Baek, D. General rules for functional microRNA targeting. Nat. Genet. 2016, 48, 1517–1526. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Huter, P.; Müller, C.; Arenz, S.; Beckert, B.; Wilson, D.N. Structural basis for ribosome rescue in bacteria. Trends Biochem. Sci. 2017, 42, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Felden, B.; Hanawa, K.; Atkins, J.F.; Himeno, H.; Muto, A.; Gesteland, R.F.; McCloskey, J.A.; Crain, P.F. Presence and location of modified nucleotides in Escherichia coli tmRNA: Structural mimicry with tRNA acceptor branches. EMBO J. 1998, 17, 3188–3196. [Google Scholar] [CrossRef] [PubMed]
- Cohn, W.E. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: Isolation, structure, and chemical characteristics. J. Biol. Chem. 1960, 235, 1488–1498. [Google Scholar] [PubMed]
- Edelheit, S.; Schwartz, S.; Mumbach, M.R.; Wurtzel, O.; Sorek, R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013, 9, e1003602. [Google Scholar] [CrossRef]
- Wulff, B.-E.; Nishikura, K. Modulation of MicroRNA expression and function by ADARs. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 91–109. [Google Scholar]
- Bar-Yaacov, D.; Mordret, E.; Towers, R.; Biniashvili, T.; Soyris, C.; Schwartz, S.; Dahan, O.; Pilpel, Y. RNA editing in bacteria recodes multiple proteins and regulates an evolutionarily conserved toxin-antitoxin system. Genome Res. 2017, 27, 1696–1703. [Google Scholar] [CrossRef]
- Ananth, P.; Goldsmith, G.; Yathindra, N. An innate twist between Crick’s wobble and Watson-Crick base pairs. RNA 2013, 19, 1038–1053. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Kong, W.; Yang, H.; He, L.; Zhao, J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Li, L.; Song, Y.; Shi, X.; Liu, J.; Xiong, S.; Chen, W.; Fu, Q.; Huang, Z.; Gu, N.; Zhang, R. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res. 2018, 28, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.; Agranat-Tamir, L.; Light, D.; Ben-Naim Zgayer, O.; Fishman, A.; Lamm, A.T. A-to-I RNA editing promotes developmental stage–specific gene and lncRNA expression. Genome Res. 2017, 27, 462–470. [Google Scholar] [CrossRef]
- Pinto, Y.; Buchumenski, I.; Levanon, E.Y.; Eisenberg, E. Human cancer tissues exhibit reduced A-to-I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2018, 46, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, C.-S.; Varelas, X.; Monti, S. Altered RNA editing in 3′ UTR perturbs microRNA-mediated regulation of oncogenes and tumor-suppressors. Sci. Rep. 2016, 6, 23226. [Google Scholar] [CrossRef] [PubMed]
- Brümmer, A.; Yang, Y.; Chan, T.W.; Xiao, X. Structure-mediated modulation of mRNA abundance by A-to-I editing. Nat. Commun. 2017, 8, 1255. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [PubMed]
- Roignant, J.-Y.; Soller, M. m6A in mRNA: An ancient mechanism for fine-tuning gene expression. Trends Genet. 2017, 33, 380–390. [Google Scholar] [CrossRef]
- Huang, J.; Yin, P. Structural insights into N6-methyladenosine (m6A) modification in the transcriptome. Genom. Proteom. Bioinform. 2018, 16, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Bokar, J.A.; Rath-Shambaugh, M.E.; Ludwiczak, R.; Narayan, P.; Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994, 269, 17697–17704. [Google Scholar] [PubMed]
- Edupuganti, R.R.; Geiger, S.; Lindeboom, R.G.H.; Shi, H.; Hsu, P.J.; Lu, Z.; Wang, S.-Y.; Baltissen, M.P.A.; Jansen, P.W.T.C.; Rossa, M.; et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 2018, 71, 973–985.e5. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Chen, K.; Luo, G.-Z.; Weng, X.; Ji, Q.; Zhou, T.; He, C. Widespread occurrence of N6-methyladenosine in bacterial mRNA. Nucleic Acids Res. 2015, 43, 6557–6567. [Google Scholar] [CrossRef] [PubMed]
- Sonnleitner, E.; Haas, D. Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl. Microbiol. Biotechnol. 2011, 91, 63–79. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, H.C.; Scarsdale, J.N.; Rife, J.P. Crystal structure of KsgA, a universally conserved rRNA adenine dimethyltransferase in Escherichia coli. J. Mol. Biol. 2004, 339, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Sergiev, P.V.; Serebryakova, M.V.; Bogdanov, A.A.; Dontsova, O.A. The ybiN gene of Escherichia coli encodes adenine-N6 methyltransferase specific for modification of A1618 of 23 S ribosomal RNA, a methylated residue located close to the ribosomal exit tunnel. J. Mol. Biol. 2008, 375, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Lim, J.; Ha, M.; Kim, V.N. TAIL-seq: Genome-wide determination of poly(A) tail length and 3′ end modifications. Mol. Cell 2014, 53, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, B.; Kim, V.N. Emerging roles of RNA modification: M6A and U-tail. Cell 2014, 158, 980–987. [Google Scholar] [CrossRef]
- Cahová, H.; Winz, M.-L.; Höfer, K.; Nübel, G.; Jäschke, A. NAD captureSeq indicates NAD as a bacterial cap for a subset of regulatory RNAs. Nature 2015, 519, 374–377. [Google Scholar] [CrossRef]
- Chen, Y.G.; Kowtoniuk, W.E.; Agarwal, I.; Shen, Y.; Liu, D.R. LC/MS analysis of cellular RNA reveals NAD-linked RNA. Nat. Chem. Biol. 2009, 5, 879–881. [Google Scholar] [CrossRef]
- Jäschke, A.; Höfer, K.; Nübel, G.; Frindert, J. Cap-like structures in bacterial RNA and epitranscriptomic modification. Curr. Opin. Microbiol. 2016, 30, 44–49. [Google Scholar] [CrossRef]
- De Almeida, C.; Scheer, H.; Zuber, H.; Gagliardi, D. RNA uridylation: A key posttranscriptional modification shaping the coding and noncoding transcriptome. Wiley interdisciplinary reviews. RNA 2018, 9, e1440. [Google Scholar] [CrossRef]
- Menezes, M.R.; Balzeau, J.; Hagan, J.P. 3′ RNA uridylation in epitranscriptomics, gene regulation, and disease. Front. Mol. Biosci. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, X. Regulation of small RNA stability: Methylation and beyond. Cell Res. 2012, 22, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M.; D’Ambrogio, A.; Nottrott, S.; Richter, J.D. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 2011, 473, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Sakaguchi, Y.; Miyauchi, K.; Suzuki, T.; Kashiwabara, S.-I.; Baba, T.; Suzuki, T. Selective stabilization of mammalian microRNAs by 3′ adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009, 23, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Blahna, M.T.; Kozlowski, E.; Matsuura, K.Y.; Ferrari, J.D.; Morris, S.A.; Powers, J.T.; Daley, G.Q.; Quinton, L.J.; Mizgerd, J.P. Zcchc11 uridylates mature miRNAs to enhance neonatal IGF-1 expression, growth, and survival. PLoS Genet. 2012, 8, e1003105. [Google Scholar] [CrossRef]
- Gutiérrez-Vázquez, C.; Enright, A.J.; Rodríguez-Galán, A.; Pérez-García, A.; Collier, P.; Jones, M.R.; Benes, V.; Mizgerd, J.P.; Mittelbrunn, M.; Ramiro, A.R.; et al. 3′ Uridylation controls mature microRNA turnover during CD4 T-cell activation. RNA 2017, 23, 882–891. [Google Scholar] [CrossRef]
- Newman, M.A.; Thomson, J.M.; Hammond, S.M. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 2008, 14, 1539–1549. [Google Scholar] [CrossRef]
- Loughlin, F.E.; Gebert, L.F.R.; Towbin, H.; Brunschweiger, A.; Hall, J.; Allain, F.H.-T. Structural basis of pre-let-7 miRNA recognition by the zinc knuckles of pluripotency factor Lin28. Nat. Struct. Mol. Biol. 2011, 19, 84–89. [Google Scholar] [CrossRef]
- Hagan, J.P.; Piskounova, E.; Gregory, R.I. Lin28 recruits the TUTase Zcchc11 to inhibit let-7 maturation in mouse embryonic stem cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Triboulet, R.; Thornton, J.E.; Gregory, R.I. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28–let-7 pathway. Nature 2013, 497, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N. Polyadenylation of mRNA in prokaryotes. Annu. Rev. Biochem. 1997, 66, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cohen, S.N. RNA degradation in Escherichia coli regulated by 3′ adenylation and 5′ phosphorylation. Nature 1995, 374, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Matz, L.M.; Cameron, T.A.; De Lay, N.R. Poly(A) polymerase is required for RyhB sRNA stability and function in Escherichia coli. RNA 2018, 24, 1496–1511. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Jeker, L.T.; Carver-Moore, K.; Oh, A.; Liu, H.J.; Cameron, R.; Richards, H.; Li, Z.; Adler, D.; Yoshinaga, Y.; et al. A resource for the conditional ablation of microRNAs in the mouse. Cell Rep. 2012, 1, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Saavedra, E.; Horvitz, H.R. Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol. 2010, 20, 367–373. [Google Scholar] [CrossRef]
- Miska, E.A.; Alvarez-Saavedra, E.; Abbott, A.L.; Lau, N.C.; Hellman, A.B.; McGonagle, S.M.; Bartel, D.P.; Ambros, V.R.; Horvitz, H.R. Most Caenorhabditis elegans microRNAs are individually not essential for development or viability. PLoS Genet. 2007, 3, 2395–2403. [Google Scholar] [CrossRef]
- Reichel, M.; Li, Y.; Li, J.; Millar, A.A. Inhibiting plant microRNA activity: Molecular SPONGEs, target MIMICs and STTMs all display variable efficacies against target microRNAs. Plant Biotechnol. J. 2015, 13, 915–926. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Xin, X.; Kim, T.S.; Cabeza, E.A.; Ren, J.; Nielsen, R.; Wrana, J.L.; Zhang, Z. Evidence for positive selection on a number of microRNA regulatory interactions during recent human evolution. PLoS Genet. 2012, 8, e1002578. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-J.; Li, J.-H.; Liu, S.; Wu, J.; Zhou, H.; Qu, L.-H.; Yang, J.-H. RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016, 44, D259–D265. [Google Scholar] [CrossRef] [PubMed]
- Kleaveland, B.; Shi, C.Y.; Stefano, J.; Bartel, D.P. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 2018, 174, 350–362.e17. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J.C. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Höfer, K.; Jäschke, A. Epitranscriptomics: RNA modifications in bacteria and archaea. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Bråte, J.; Neumann, R.S.; Fromm, B.; Haraldsen, A.A.B.; Tarver, J.E.; Suga, H.; Donoghue, P.C.J.; Peterson, K.J.; Ruiz-Trillo, I.; Grini, P.E.; et al. Unicellular origin of the animal microRNA machinery. Curr. Biol. 2018. [Google Scholar] [CrossRef]
- Kang, S.-M.; Choi, J.-W.; Lee, Y.; Hong, S.-H.; Lee, H.-J. Identification of microRNA-size, small RNAs in Escherichia coli. Curr. Microbiol. 2013, 67, 609–613. [Google Scholar] [CrossRef]
- Nejman-Faleńczyk, B.; Bloch, S.; Licznerska, K.; Dydecka, A.; Felczykowska, A.; Topka, G.; Węgrzyn, A.; Węgrzyn, G. A small, microRNA-size, ribonucleic acid regulating gene expression and development of Shiga toxin-converting bacteriophage Φ24Β. Sci. Rep. 2015, 5, 10080. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Felden, B.; Gilot, D. Modulation of Bacterial sRNAs Activity by Epigenetic Modifications: Inputs from the Eukaryotic miRNAs. Genes 2019, 10, 22. https://doi.org/10.3390/genes10010022
Felden B, Gilot D. Modulation of Bacterial sRNAs Activity by Epigenetic Modifications: Inputs from the Eukaryotic miRNAs. Genes. 2019; 10(1):22. https://doi.org/10.3390/genes10010022
Chicago/Turabian StyleFelden, Brice, and David Gilot. 2019. "Modulation of Bacterial sRNAs Activity by Epigenetic Modifications: Inputs from the Eukaryotic miRNAs" Genes 10, no. 1: 22. https://doi.org/10.3390/genes10010022
APA StyleFelden, B., & Gilot, D. (2019). Modulation of Bacterial sRNAs Activity by Epigenetic Modifications: Inputs from the Eukaryotic miRNAs. Genes, 10(1), 22. https://doi.org/10.3390/genes10010022