Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology


{kind=link}
{kind=link}
Abstract
1. Introduction
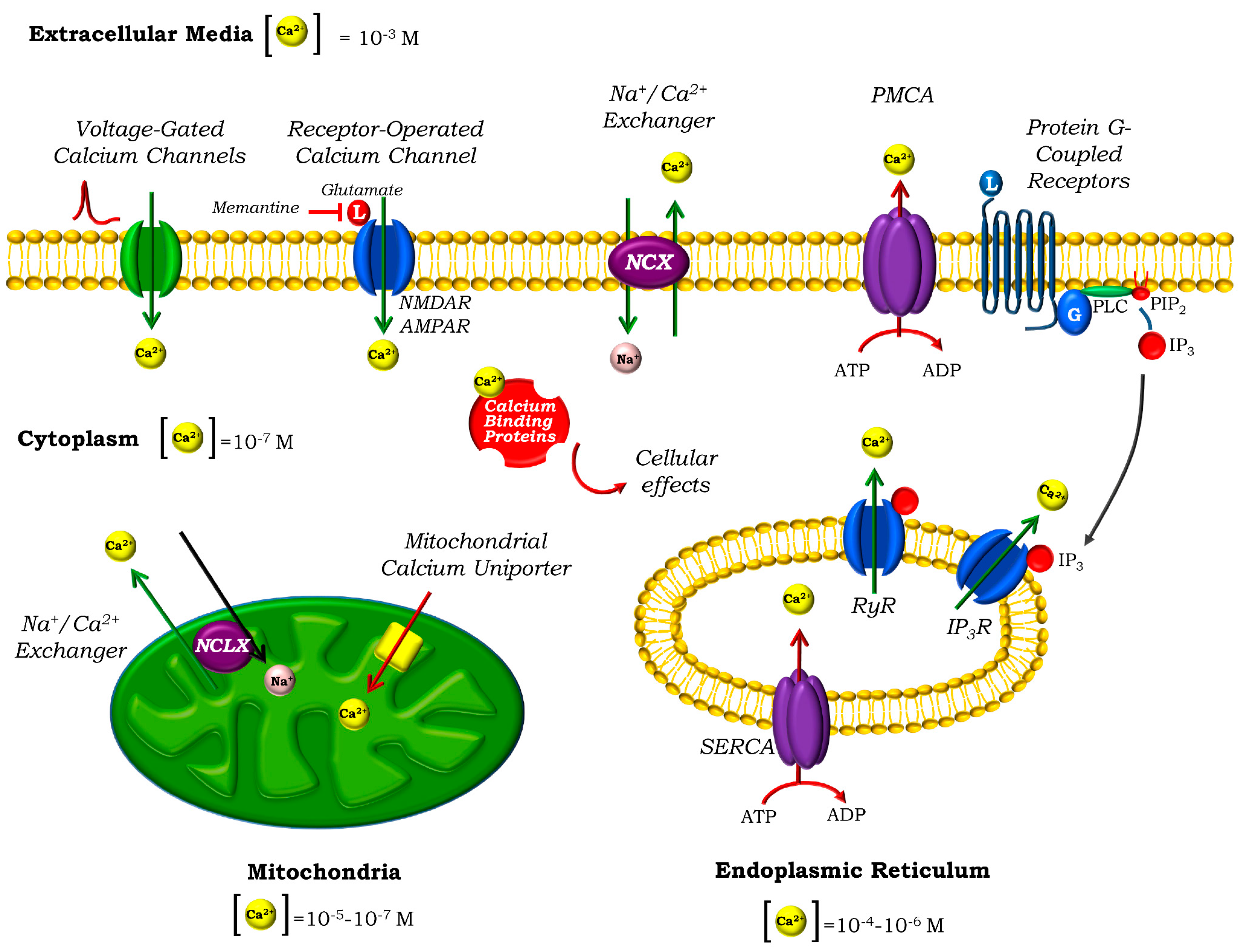
2. Calcium Homeostasis in Neurons
3. Mitochondria and Ca2+ Homeostasis
4. Calcium Homeostasis Impairment in AD and Tauopathies
4.1. Cytosolic Ca2+ Disturbances in AD and Tauopathies
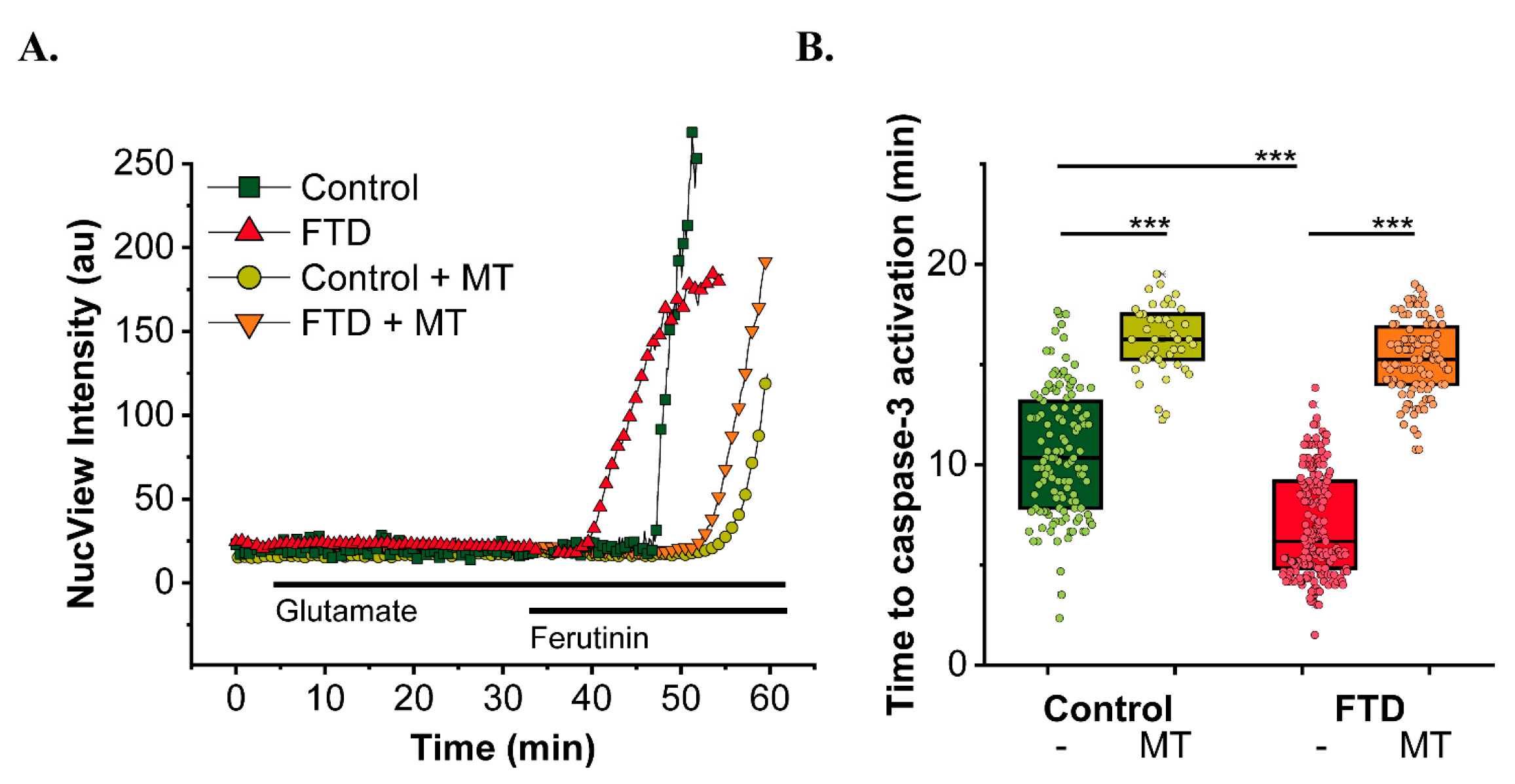
4.2. Mitochondrial Ca2+ Disturbances in AD and Tauopathies
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar] [CrossRef]
- Horigane, S.-I.; Ozawa, Y.; Yamada, H.; Takemoto-Kimura, S. Calcium signalling: A key regulator of neuronal migration. J. Biochem. 2019, 165, 401–409. [Google Scholar] [CrossRef]
- Wojda, U.; Salinska, E.; Kuźnicki, J. Calcium ions in neuronal degeneration. Iubmb Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup; Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182. [Google Scholar] [CrossRef]
- Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J.; Ros, J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018, 2, NS20180061. [Google Scholar] [CrossRef]
- Abeti, R.; Abramov, A.Y. Mitochondrial Ca2+ in neurodegenerative disorders. Pharm. Res. 2015, 99, 377–381. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Abramov, A.Y. Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci. Lett. 2018, 663, 86–90. [Google Scholar] [CrossRef]
- Semyanov, A.V. Spatiotemporal pattern of calcium activity in astrocytic network. Cell Calcium 2019, 78, 15–25. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid- Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Endoplasmic reticulum calcium signaling in nerve cells. Boil. Res. 2004, 37, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Karagas, N.E.; Venkatachalam, K. Roles for the Endoplasmic Reticulum in Regulation of Neuronal Calcium Homeostasis. Cells 2019, 8, 1232. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nat. 2006, 443, 230–233. [Google Scholar] [CrossRef]
- López, J.J.; Jardin, I.; Sánchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Boil. 2010, 3, a004168. [Google Scholar] [CrossRef]
- Roome, C.J.; Power, E.M.; Empson, R. Transient reversal of the sodium/calcium exchanger boosts presynaptic calcium and synaptic transmission at a cerebellar synapse. J. Neurophysiol. 2013, 109, 1669–1680. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ Buffers. Cold Spring Harb. Perspect. Boil. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Boil. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Angelova, P.R. Cellular mechanisms of complex I-associated pathology. Biochem. Soc. Trans. 2019, 47, 1963–1969. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Boil. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Wu, W.; Shen, Q.; Zhang, R.; Qiu, Z.; Wang, Y.; Zheng, J.; Jia, Z. The structure of the MICU 1- MICU 2 complex unveils the regulation of the mitochondrial calcium uniporter. Embo J. 2020, 10. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nature 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Holmstrom, K.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.A.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Pan, X.; Nguyen, T.; Liu, J.; Holmstrom, K.; Finkel, T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun. 2014, 449, 384–385. [Google Scholar] [CrossRef] [PubMed]
- Antony, A.N.; Paillard, M.; Moffat, C.; Juskeviciute, E.; Correnti, J.; Bolon, B.; Rubin, E.; Csordás, G.; Seifert, E.L.; Hoek, J.B.; et al. MICU1 regulation of mitochondrial Ca2+ uptake dictates survival and tissue regeneration. Nat. Commun. 2016, 7, 10955. [Google Scholar] [CrossRef] [PubMed]
- Drago, I.; Davis, R.L. Inhibiting the Mitochondrial Calcium Uniporter during Development Impairs Memory in Adult Drosophila. Cell Rep. 2016, 16, 2763–2776. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Boil. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef]
- Elustondo, P.A.; Nichols, M.; Robertson, G.S.; Pavlov, E.V. Mitochondrial Ca2+ uptake pathways. J. Bioenerg. Biomembr. 2016, 49, 113–119. [Google Scholar] [CrossRef]
- De Stefani, D.; Patron, M.; Rizzuto, R. Structure and function of the mitochondrial calcium uniporter complex. Biochim. Et Biophys. Acta (Bba) 2015, 1853, 2006–2011. [Google Scholar] [CrossRef]
- Carafoli, E.; Tiozzo, R.; Lugli, G.; Crovetti, F.; Kratzing, C. The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 1974, 6, 361–371. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2009, 107, 436–441. [Google Scholar] [CrossRef]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Kostić, M.; Ludtmann, M.H.R.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Sekler, I. Functional properties and mode of regulation of the mitochondrial Na+/Ca2+ exchanger, NCLX. Semin. Cell Dev. Boil. 2019, 94, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Deas, E.; Wood, N.W.; Abramov, A.Y. The Role of the Mitochondrial NCX in the Mechanism of Neurodegeneration in Parkinson’s Disease. Neurotransm. Interact. Cogn. Funct. 2012, 961, 241–249. [Google Scholar] [CrossRef]
- Gobbi, P.; Castaldo, P.; Minelli, A.; Salucci, S.; Magi, S.; Corcione, E.; Amoroso, S. Mitochondrial localization of Na+/Ca2+ exchangers NCX1–3 in neurons and astrocytes of adult rat brain in situ. Pharm. Res. 2007, 56, 556–565. [Google Scholar] [CrossRef]
- Sisalli, M.J.; Secondo, A.; Esposito, A.; Valsecchi, V.; Savoia, C.; Di Renzo, G.F.; Annunziato, L.; Scorziello, A. Endoplasmic reticulum refilling and mitochondrial calcium extrusion promoted in neurons by NCX1 and NCX3 in ischemic preconditioning are determinant for neuroprotection. Cell Death Differ. 2014, 21, 1142–1149. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; Del Arco, A.; Saheki, T.; Pardo, B.; Satrústegui, J. Calcium-Regulation of Mitochondrial Respiration Maintains ATP Homeostasis and Requires ARALAR/AGC1-Malate Aspartate Shuttle in Intact Cortical Neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharm. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Potapova, E.; Dremin, V.; Dunaev, A.V. Interaction of Oxidative Stress and Misfolded Proteins in the Mechanism of Neurodegeneration. Life 2020, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Esteras, N.; Abramov, A.Y. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med. Res. Rev. 2020. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease - A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2016, 483, 998–1004. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Alpha-synuclein and beta-amyloid – different targets, same players: Calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem. Biophys. Res. Commun. 2017, 483, 1110–1115. [Google Scholar] [CrossRef]
- Hardy, J. Amyloid, the presenilins and Alzheimer’s disease. Trends Neurosci. 1997, 20, 154–159. [Google Scholar] [CrossRef]
- Hardy, J.; Higgins, G.; Mayford, M.; Barzilai, A.; Keller, F.; Schacher, S.; Kandel, E. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992, 42, 631. [Google Scholar] [CrossRef]
- Rösler, T.W.; Marvian, A.T.; Brendel, M.; Nykänen, N.-P.; Höllerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Prog. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef]
- MacKenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138, 54–70. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s Disease and Brain Aginga. Ann. N. Y. Acad. Sci. 2006, 747, 1–11. [Google Scholar] [CrossRef]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta (BBA) 2004, 1742, 81–87. [Google Scholar] [CrossRef]
- Patel, N.; Ramachandran, S.; Azimov, R.; Kagan, B.L.; Lal, R. Ion Channel Formation by Tau Protein: Implications for Alzheimer’s Disease and Tauopathies. Biochemistry 2015, 54, 7320–7325. [Google Scholar] [CrossRef]
- Esteras, N.; Kundel, F.; Amodeo, G.F.; Pavlov, E.V.; Klenerman, D.; Abramov, A.Y. Insoluble tau aggregates induce neuronal death through modification of membrane ion conductance, activation of voltage-gated calcium channels and NADPH oxidase. FEBS J. 2020. [Google Scholar] [CrossRef]
- Nalbantoglu, J.; Tirado-Santiago, G.; Lahsaïni, A.; Poirier, J.; Gonçalves, O.; Verge, G.; Momoli, F.; Welner, S.A.; Massicotte, G.; Julien, J.-P.; et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature 1997, 387, 500–505. [Google Scholar] [CrossRef]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Mol. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Texidó, L.; Martín-Satué, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.R.; Rego, A.C. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef]
- Rammes, G.; Seeser, F.; Mattusch, K.; Zhu, K.; Haas, L.; Kummer, M.; Heneka, M.; Herms, J.; Parsons, C.G. The NMDA receptor antagonist Radiprodil reverses the synaptotoxic effects of different amyloid-beta (Aβ) species on long-term potentiation (LTP). Neuropharmacology 2018, 140, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wong, T.P.; Aarts, M.M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA Receptor Subunits Have Differential Roles in Mediating Excitotoxic Neuronal Death Both In Vitro and In Vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shen, W.; Su, J.; Cheng, B.; Li, D.; Liu, G.; Zhou, W.; Zhang, Y.-X. Modulating the Balance of Synaptic and Extrasynaptic NMDA Receptors Shows Positive Effects against Amyloid-β-Induced Neurotoxicity. J. Alzheimer’s Dis. 2017, 57, 885–897. [Google Scholar] [CrossRef]
- Miyamoto, T.; Stein, L.R.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef]
- Monteiro-Fernandes, D.; Silva, J.; Soares-Cunha, C.; Dalla, C.; Kokras, N.; Arnaud, F.; Billiras, R.; Zhuravleva, V.; Waites, C.; Bretin, S.; et al. Allosteric modulation of AMPA receptors counteracts Tau-related excitotoxic synaptic signaling and memory deficits in stress- and Aβ-evoked hippocampal pathology. Mol. Psychiatry 2020, 1–13. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; Van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.; Siskova, Z.; Mandelkow, E.; Mandelkow, E. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR 2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Bartus, R.; Dean, R.; Beer, B.; Lippa, A. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Hernandez, F.; Ávila, J. The Involvement of Cholinergic Neurons in the Spreading of Tau Pathology. Front. Neurol. 2013, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Kamynina, A.V.; Holmstrom, K.; Koroev, D.O.; Volpina, O.M.; Abramov, A.Y. Acetylcholine and antibodies against the acetylcholine receptor protect neurons and astrocytes against beta-amyloid toxicity. Int. J. Biochem. Cell Boil. 2013, 45, 899–907. [Google Scholar] [CrossRef]
- Ishii, M.; Hiller, A.J.; Pham, L.; McGuire, M.J.; Iadecola, C.; Wang, G. Amyloid-Beta Modulates Low-Threshold Activated Voltage-Gated L-Type Calcium Channels of Arcuate Neuropeptide Y Neurons Leading to Calcium Dysregulation and Hypothalamic Dysfunction. J. Neurosci. 2019, 39, 8816–8825. [Google Scholar] [CrossRef]
- Furukawa, K.; Wang, Y.; Yao, P.J.; Fu, W.; Mattson, M.P.; Itoyama, Y.; Onodera, H.; D’Souza, I.; Poorkaj, P.H.; Bird, T.D.; et al. Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J. Neurochem. 2003, 87, 427–436. [Google Scholar] [CrossRef]
- Lawlor, B.; Segurado, R.; Kennelly, S.; Rikkert, M.O.; Howard, R.J.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef]
- Abdullah, L.; Crawford, F.; Tsolaki, M.; Börjesson-Hanson, A.; Rikkert, M.O.; Pasquier, F.; Wallin, A.; Kennelly, S.; Ait-Ghezala, G.; Paris, D.; et al. The Influence of Baseline Alzheimer’s Disease Severity on Cognitive Decline and CSF Biomarkers in the NILVAD Trial. Front. Neurol. 2020, 11, 149. [Google Scholar] [CrossRef]
- Hwang, D.; Kim, S.; Choi, H.; Oh, I.-H.; Kim, B.S.; Choi, H.R.; Kim, S.Y.; Won, C. Calcium-Channel Blockers and Dementia Risk in Older Adults – National Health Insurance Service – Senior Cohort (2002–2013). Circ. J. 2016, 80, 2336–2342. [Google Scholar] [CrossRef]
- Feldman, L.; Vinker, S.; Efrati, S.; Beberashvili, I.; Gorelik, O.; Wasser, W.; Shani, M. Amlodipine treatment of hypertension associates with a decreased dementia risk. Clin. Exp. Hypertens. 2016, 38, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Bohlken, J.; Jacob, L.; Kostev, K. The Relationship Between the Use of Antihypertensive Drugs and the Incidence of Dementia in General Practices in Germany. J. Alzheimer’s Dis. 2019, 70, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Parker, I.; LaFerla, F. Enhanced Ryanodine-Mediated Calcium Release in Mutant PS1-Expressing Alzheimer’s Mouse Models. Ann. New York Acad. Sci. 2007, 1097, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Paula-Lima, A.C.; Adasme, T.; Sanmartín, C.; Sebollela, A.; Hetz, C.; Carrasco, M.A.; Ferreira, J.; Hidalgo, C. Amyloid β-Peptide Oligomers Stimulate RyR-Mediated Ca2+ Release Inducing Mitochondrial Fragmentation in Hippocampal Neurons and Prevent RyR-Mediated Dendritic Spine Remodeling Produced by BDNF. Antioxid. Redox Signal. 2011, 14, 1209–1223. [Google Scholar] [CrossRef]
- DeMuro, A.; Parker, I. Cytotoxicity of intracellular aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.-Y.; Foskett, J.K. Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Bojarski, L.; Pomorski, P.; Szybinska, A.; Drab, M.; Skibinska-Kijek, A.; Gruszczynska-Biegala, J.; Kuźnicki, J. Presenilin-dependent expression of STIM proteins and dysregulation of capacitative Ca2+ entry in familial Alzheimer’s disease. Biochim. Biophys. Acta 2009, 1793, 1050–1057. [Google Scholar] [CrossRef]
- Tong, B.C.-K.; Lee, C.S.-K.; Cheng, W.-H.; Lai, K.-O.; Foskett, J.K.; Cheung, K.-H. Familial Alzheimer’s disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+entry. Sci. Signal. 2016, 9, ra89. [Google Scholar] [CrossRef]
- Ye, J.; Yin, Y.; Yin, Y.; Zhang, H.; Wan, H.; Wang, L.; Zuo, Y.; Gao, D.; Li, M.; Li, J.; et al. Tau-induced upregulation of C/EBPβ-TRPC1-SOCE signaling aggravates tauopathies: A vicious cycle in Alzheimer neurodegeneration. Aging Cell 2020, e13209. [Google Scholar] [CrossRef]
- Mata, A.M. Functional interplay between plasma membrane Ca 2+ -ATPase, amyloid β-peptide and tau. Neurosci. Lett. 2018, 663, 55–59. [Google Scholar] [CrossRef]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca2+-ATPase. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1822, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; A Derrico, C.; Hatem, L.; A Colvin, R. Alzheimer’s amyloid-beta peptide inhibits sodium/calcium exchange measured in rat and human brain plasma membrane vesicles. Neuroscience 1997, 80, 675–684. [Google Scholar] [CrossRef]
- Pannaccione, A.; Piccialli, I.; Secondo, A.; Ciccone, R.; Molinaro, P.; Boscia, F.; Annunziato, L. The Na+/Ca2+exchanger in Alzheimer’s disease. Cell Calcium 2020, 87, 102190. [Google Scholar] [CrossRef] [PubMed]
- Atherton, J.; Kurbatskaya, K.; Bondulich, M.; Croft, C.L.; Garwood, C.J.; Chhabra, R.; Wray, S.; Jeromin, A.; Hanger, D.P.; Noble, W. Calpain cleavage and inactivation of the sodium calcium exchanger-3 occur downstream of Aβ in Alzheimer’s disease. Aging Cell 2013, 13, 49–59. [Google Scholar] [CrossRef]
- Kip, S.N.; Strehler, E.E. Rapid downregulation of NCX and PMCA in hippocampal neurons following H2O2 oxidative stress. Ann. New York Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef]
- Ferreira, A.; Bigio, E.H. Calpain-Mediated Tau Cleavage: A Mechanism Leading to Neurodegeneration Shared by Multiple Tauopathies. Mol. Med. 2011, 17, 676–685. [Google Scholar] [CrossRef]
- Liang, B.; Duan, B.-Y.; Zhou, X.-P.; Gong, J.-X.; Luo, Z.-G. Calpain Activation Promotes BACE1 Expression, Amyloid Precursor Protein Processing, and Amyloid Plaque Formation in a Transgenic Mouse Model of Alzheimer Disease. J. Boil. Chem. 2010, 285, 27737–27744. [Google Scholar] [CrossRef]
- Rao, M.; McBrayer, M.K.; Campbell, J.; Kumar, A.; Hashim, A.; Sershen, H.; Stavrides, P.H.; Ohno, M.; Hutton, M.; Nixon, R.A. Specific Calpain Inhibition by Calpastatin Prevents Tauopathy and Neurodegeneration and Restores Normal Lifespan in Tau P301L Mice. J. Neurosci. 2014, 34, 9222–9234. [Google Scholar] [CrossRef]
- Reinecke, J.B.; Devos, S.L.; McGrath, J.P.; Shepard, A.M.; Goncharoff, D.K.; Tait, D.N.; Fleming, S.R.; Vincent, M.; Steinhilb, M.L. Implicating Calpain in Tau-Mediated Toxicity In Vivo. PLoS ONE 2011, 6, e23865. [Google Scholar] [CrossRef]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef]
- Esteras, N.; Alquézar, C.; De La Encarnación, A.; Villarejo, A.; Bermejo-Pareja, F.; Requero, A.M. Calmodulin levels in blood cells as a potential biomarker of Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Boil. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.A.H.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid -peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, P.F.; Wiehager, B.; Sakai, J.; Frykman, S.; Behbahani, H.; Winblad, B.; Ankarcrona, M. Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J. 2010, 25, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Et Biophys. Acta (Bba) 2018, 1864, 3247–3256. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’Aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 Tau Fragment Targets Neuronal Mitochondria at AD Synapses: Possible Implications for Neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta (BBA) 2008, 1777, 953–964. [Google Scholar] [CrossRef]
- Duchen, M. Mitochondria and Ca2+in cell physiology and pathophysiology. Cell Calcium 2000, 28, 339–348. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. β-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Stout, A.K.; Raphael, H.M.; Kanterewicz, B.I.; Klann, E.; Reynolds, I.J. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat. Neurosci. 1998, 1, 366–373. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Nguyen, H.V.; Winters, C.A.; Brantner, C.A.; Smith, C.L.; Andrews, S.B. Excitotoxic Calcium Overload in a Subpopulation of Mitochondria Triggers Delayed Death in Hippocampal Neurons. J. Neurosci. 2004, 24, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons Against Glutamate Excitotoxicity. Mol. Neurobiol. 2018, 56, 2244–2255. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Valero, R.A.; Rodríguez-Crespo, I.; Villalobos, C.; Nuñez, L. Mitochondrial Ca2+ Overload Underlies Aβ Oligomers Neurotoxicity Providing an Unexpected Mechanism of Neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Calvo-Rodriguez, M.; Caballero, E.; García-Durillo, M.; Nuñez, L.; Villalobos, C. Is it All Said for NSAIDs in Alzheimer’s Disease? Role of Mitochondrial Calcium Uptake. Curr. Alzheimer Res. 2018, 15, 504–510. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.; Luongo, T.S.; Ludtmann, M.H.; Praticò, M.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimer’s Dis. 2016, 55, 1549–1570. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+dysregulation in neurons from transgenic mice expressing mutant presenilin. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Shilling, D.; Mak, D.-O.D.; Kang, D.E.; Foskett, J.K. Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+Leak Channels. J. Boil. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef]
- Parks, J.K.; Smith, T.S.; A Trimmer, P.; Bennett, J.P.; Parker, W.D. Neurotoxic Aβ peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001, 76, 1050–1056. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef]
- Britti, E.; Ros, J.; Esteras, N.; Abramov, A.Y. Tau inhibits mitochondrial calcium efflux and makes neurons vulnerable to calcium-induced cell death. Cell Calcium 2020, 86, 102150. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; A Sosunov, A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Kopach, O.; Esteras, N.; Wray, S.; Rusakov, D.A.; Abramov, A.Y. Maturation and phenotype of pathophysiological neuronal excitability of human cells in tau-related dementia. J. Cell Sci. 2020, 133, jcs241687. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Boil. 2017, 12, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, F.; Esteras, N.; Martin-Requero, A.; Boutoleau-Bretonnière, C.; Vercelletto, M.; Gabelle, A.; Le Ber, I.; Honda, T.; Dinkova-Kostova, A.T.; Hardy, J.; et al. Pathogenic p62/SQSTM1 mutations impair energy metabolism through limitation of mitochondrial substrates. Sci. Rep. 2017, 7, 1666. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.M.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2016, 24, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y. Actions of ionomycin, 4-BrA23187 and a novel electrogenic Ca2+ ionophore on mitochondria in intact cells. Cell Calcium 2003, 33, 101–112. [Google Scholar] [CrossRef]
- Zamaraeva, M.V.; Hagelgans, A.I.; Abramov, A.Y.; Ternovsky, V.I.; Merzlyak, P.G.; Tashmukhamedov, B.A.; Saldkhodzjaev, A.I. Ionophoretic properties of ferutinin. Cell Calcium 1997, 22, 235–241. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteras, N.; Abramov, A.Y. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells 2020, 9, 2135. https://doi.org/10.3390/cells9092135
Esteras N, Abramov AY. Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells. 2020; 9(9):2135. https://doi.org/10.3390/cells9092135
Chicago/Turabian StyleEsteras, Noemi, and Andrey Y. Abramov. 2020. "Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology" Cells 9, no. 9: 2135. https://doi.org/10.3390/cells9092135
APA StyleEsteras, N., & Abramov, A. Y. (2020). Mitochondrial Calcium Deregulation in the Mechanism of Beta-Amyloid and Tau Pathology. Cells, 9(9), 2135. https://doi.org/10.3390/cells9092135