Raloxifene and n-Acetylcysteine Ameliorate TGF-Signalling in Fibroblasts from Patients with Recessive Dominant Epidermolysis Bullosa

 , ,
, ,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Weaker TGF-β1 Expression in Human RDEB Fibroblasts Following the Pharmacological Stimulation of Endoglin Expression
2.2. Raloxifene and n-Acetylcysteine Regulate Fibrosis Associated Biomarkers
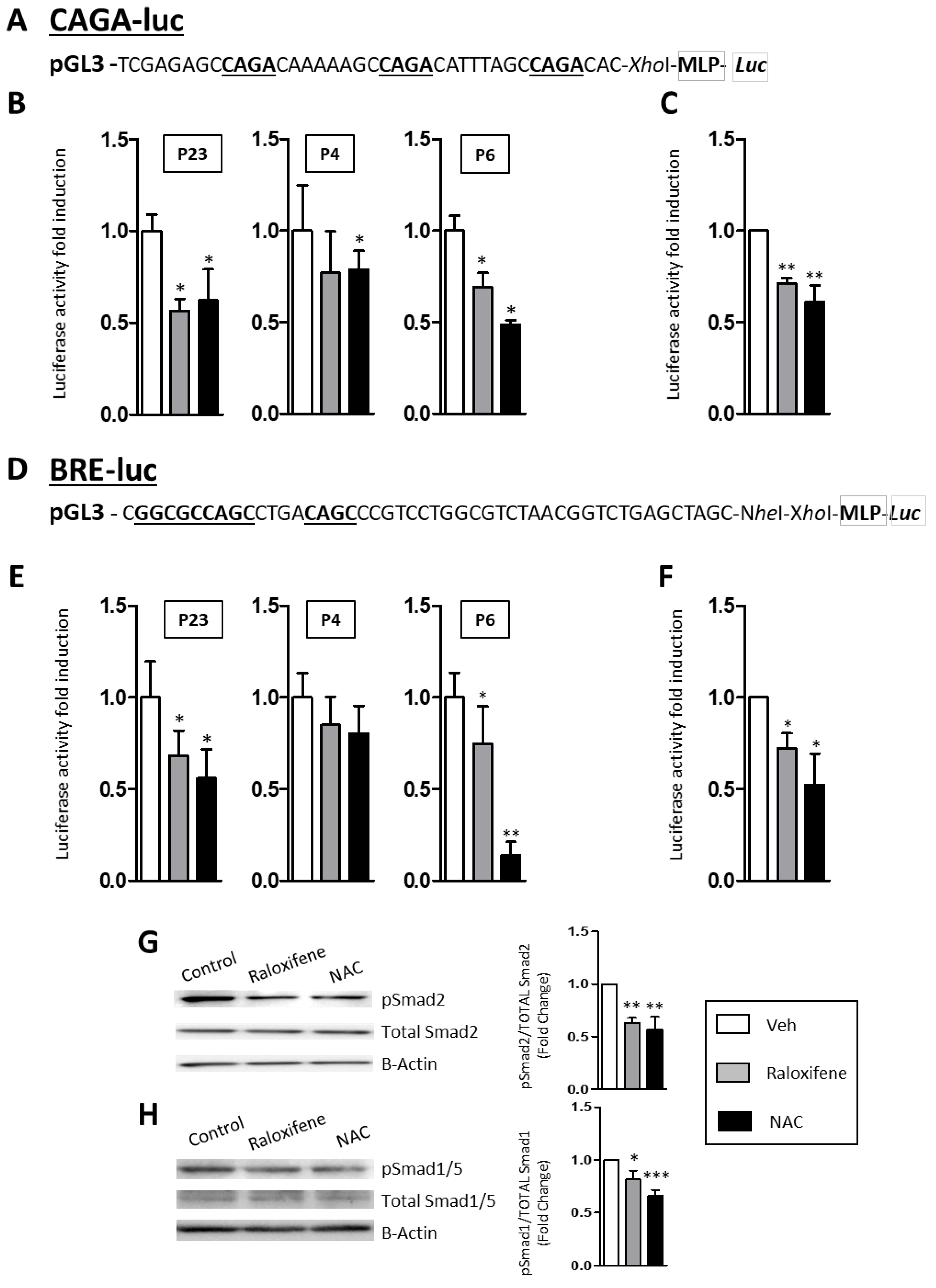
2.3. Raloxifene and NAC Modulate the Canonical TGF-β1/Smad Signaling Pathway
2.4. Raloxifene and NAC Limit the Contractility of Fibroblasts from RDEB Patients in the Collagen Gel Contraction Assay
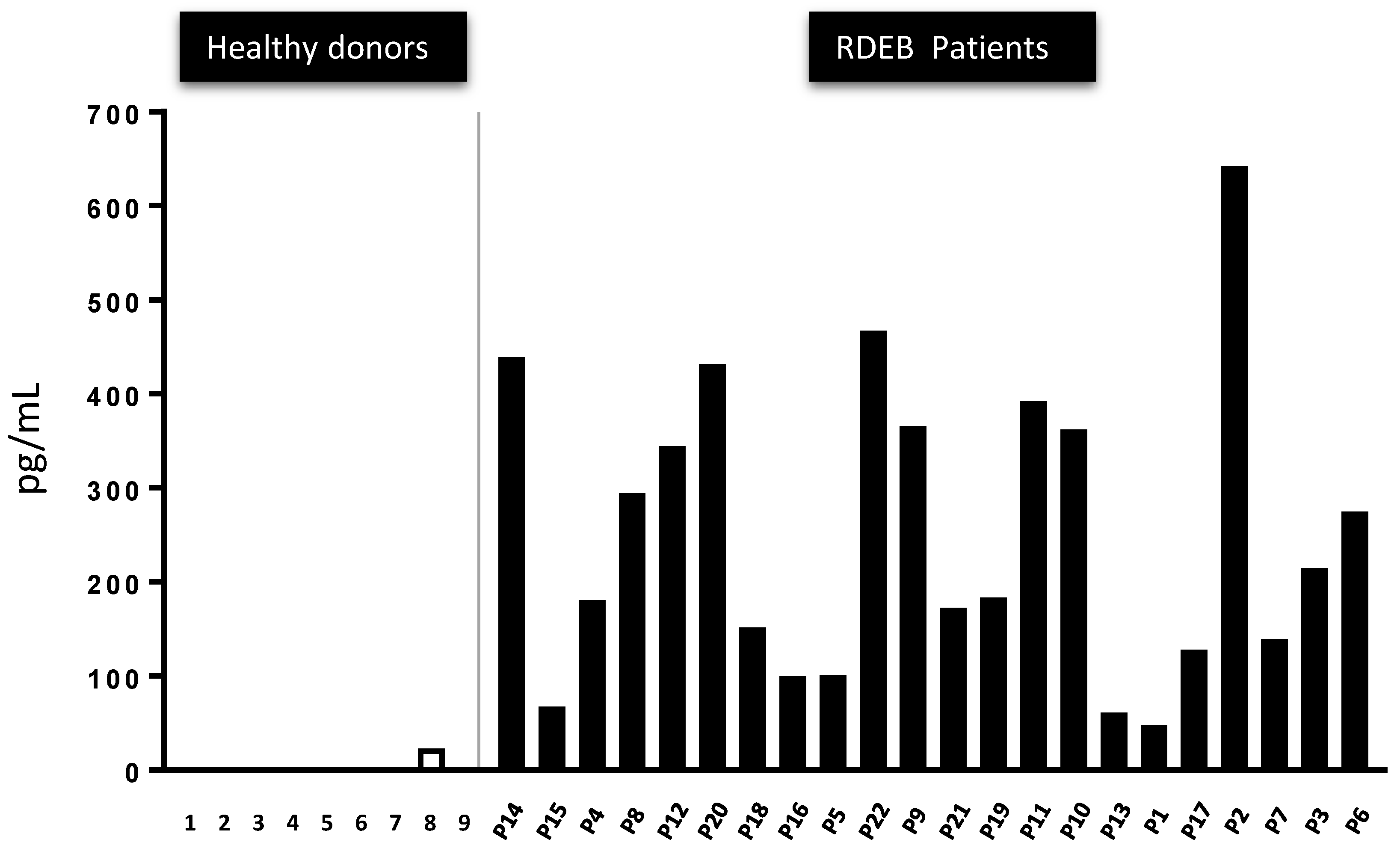
2.5. The Presence of Active TGF-β Peptide in the Serum of 22 RDEB Patients Indicates It May Represent a Disease Biomarker
3. Discussion
4. Methods
4.1. Human Subjects
4.2. Culture of Primary RDEB Patient Fibroblasts
4.3. Primary RDEB Patient Fibroblasts Transfection and Reporter Assay
4.4. Collagen Gel Contraction Assay
4.5. RNA Extraction and Relative Quantification of mRNA
4.6. Western Blotting
4.7. Immunofluorescence Staining
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fine, J.D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef]
- Bauer, J.W.; Koller, J.; Murauer, E.M.; De Rosa, L.; Enzo, E.; Carulli, S.; Bondanza, S.; Recchia, A.; Muss, W.; Diem, A.; et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. J. Investig. Dermatol. 2017, 137, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Mavilio, F.; Pellegrini, G.; Ferrari, S.; Di Nunzio, F.; Di Iorio, E.; Recchia, A.; Maruggi, G.; Ferrari, G.; Provasi, E.; Bonini, C.; et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat. Med. 2006, 12, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Mencia, A.; Chamorro, C.; Bonafont, J.; Duarte, B.; Holguin, A.; Illera, N.; Llames, S.G.; Escamez, M.J.; Hausser, I.; Del Rio, M.; et al. Deletion of a Pathogenic Mutation-Containing Exon of COL7A1 Allows Clonal Gene Editing Correction of RDEB Patient Epidermal Stem Cells. Mol. Ther. Nucleic Acids 2018, 11, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Nystrom, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Loven, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, M.; Ramirez, J.R.; Perez-Gomez, E.; Romero, D.; Velasco, B.; Letarte, M.; Lopez-Novoa, J.M.; Bernabeu, C. Expression of the TGF-beta coreceptor endoglin in epidermal keratinocytes and its dual role in multistage mouse skin carcinogenesis. Oncogene 2003, 22, 5976–5985. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perez-Gomez, E.; Jerkic, M.; Prieto, M.; Del Castillo, G.; Martin-Villar, E.; Letarte, M.; Bernabeu, C.; Perez-Barriocanal, F.; Quintanilla, M.; Lopez-Novoa, J.M. Impaired wound repair in adult endoglin heterozygous mice associated with lower NO bioavailability. J. Investig. Dermatol. 2014, 134, 247–255. [Google Scholar] [CrossRef]
- Guerrero-Esteo, M.; Lastres, P.; Letamendia, A.; Perez-Alvarez, M.J.; Langa, C.; Lopez, L.A.; Fabra, A.; Garcia-Pardo, A.; Vera, S.; Letarte, M.; et al. Endoglin overexpression modulates cellular morphology, migration, and adhesion of mouse fibroblasts. Eur. J. Cell Biol. 1999, 78, 614–623. [Google Scholar] [CrossRef]
- Munoz-Felix, J.M.; Perez-Roque, L.; Nunez-Gomez, E.; Oujo, B.; Arevalo, M.; Ruiz-Remolina, L.; Cuesta, C.; Langa, C.; Perez-Barriocanal, F.; Bernabeu, C.; et al. Overexpression of the short endoglin isoform reduces renal fibrosis and inflammation after unilateral ureteral obstruction. Biochim. Biophys. Acta 2016, 1862, 1801–1814. [Google Scholar] [CrossRef]
- Albinana, V.; Bernabeu-Herrero, M.E.; Zarrabeitia, R.; Bernabeu, C.; Botella, L.M. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb. Haemost. 2010, 103, 525–534. [Google Scholar] [CrossRef]
- Zarrabeitia, R.; Ojeda-Fernandez, L.; Recio, L.; Bernabeu, C.; Parra, J.A.; Albinana, V.; Botella, L.M. Bazedoxifene, a new orphan drug for the treatment of bleeding in hereditary haemorrhagic telangiectasia. Thromb. Haemost. 2016, 115, 1167–1177. [Google Scholar] [CrossRef]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef]
- Zafarullah, M.; Li, W.Q.; Sylvester, J.; Ahmad, M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol. Life Sci. 2003, 60, 6–20. [Google Scholar] [CrossRef]
- Chacon-Solano, E.; Leon, C.; Diaz, F.; Garcia-Garcia, F.; Garcia, M.; Escamez, M.J.; Guerrero-Aspizua, S.; Conti, C.J.; Mencia, A.; Martinez-Santamaria, L.; et al. Fibroblast activation and abnormal extracellular matrix remodelling as common hallmarks in three cancer-prone genodermatoses. Br. J. Dermatol. 2019, 181, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Whillier, S.; Raftos, J.E.; Chapman, B.; Kuchel, P.W. Role of N-acetylcysteine and cystine in glutathione synthesis in human erythrocytes. Redox Rep. 2009, 14, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Zhan, Y.J.; Yang, C.S.; Tzeng, S.F. Oxidative stress-induced attenuation of thrombospondin-1 expression in primary rat astrocytes. J. Cell Biochem. 2011, 112, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; Ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef]
- Korchynskyi, O.; ten Dijke, P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002, 277, 4883–4891. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Mayorga, G.; Gomez-Lopez, G.; Barbachano, A.; Fernandez-Barral, A.; Pena, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Munoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Ng, Y.Z.; Pourreyron, C.; Salas-Alanis, J.C.; Dayal, J.H.; Cepeda-Valdes, R.; Yan, W.; Wright, S.; Chen, M.; Fine, J.D.; Hogg, F.J.; et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012, 72, 3522–3534. [Google Scholar] [CrossRef]
- Isaka, Y.; Brees, D.K.; Ikegaya, K.; Kaneda, Y.; Imai, E.; Noble, N.A.; Border, W.A. Gene therapy by skeletal muscle expression of decorin prevents fibrotic disease in rat kidney. Nat. Med. 1996, 2, 418–423. [Google Scholar] [CrossRef]
- Hsu, C.K.; Wang, S.P.; Lee, J.Y.; McGrath, J.A. Treatment of hereditary epidermolysis bullosa: Updates and future prospects. Am. J. Clin. Dermatol. 2014, 15, 1–6. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Moffat, J.G.; Vincent, F.; Lee, J.A.; Eder, J.; Prunotto, M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017, 16, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Santibanez, J.F.; Guerrero-Esteo, M.; Langa, C.; Vary, C.P.; Bernabeu, C. Interaction and functional interplay between endoglin and ALK-1, two components of the endothelial transforming growth factor-beta receptor complex. J. Cell Physiol. 2005, 204, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Freedman, A.N.; Yu, B.; Gail, M.H.; Costantino, J.P.; Graubard, B.I.; Vogel, V.G.; Anderson, G.L.; McCaskill-Stevens, W. Benefit/risk assessment for breast cancer chemoprevention with raloxifene or tamoxifen for women age 50 years or older. J. Clin. Oncol. 2011, 29, 2327–2333. [Google Scholar] [CrossRef]
- Vachon, C.M.; Schaid, D.J.; Ingle, J.N.; Wickerham, D.L.; Kubo, M.; Mushiroda, T.; Goetz, M.P.; Carlson, E.E.; Paik, S.; Wolmark, N.; et al. A polygenic risk score for breast cancer in women receiving tamoxifen or raloxifene on NSABP P-1 and P-2. Breast Cancer Res. Treat. 2015, 149, 517–523. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Koch, D.C.; Bisson, W.H.; Jang, H.S.; Kolluri, S.K. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014, 5, e1038. [Google Scholar] [CrossRef]
- O’Donnell, E.F.; Jang, H.S.; Pearce, M.; Kerkvliet, N.I.; Kolluri, S.K. The aryl hydrocarbon receptor is required for induction of p21cip1/waf1 expression and growth inhibition by SU5416 in hepatoma cells. Oncotarget 2017, 8, 25211–25225. [Google Scholar] [CrossRef]
- Ho, T.H.; Nunez-Nateras, R.; Hou, Y.X.; Bryce, A.H.; Northfelt, D.W.; Dueck, A.C.; Wong, B.; Stanton, M.L.; Joseph, R.W.; Castle, E.P. A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2017, 15, 196–202. [Google Scholar] [CrossRef]
- Pritchard, T.; Rosengren, R.J.; Greish, K.; Taurin, S. Raloxifene nanomicelles reduce the growth of castrate-resistant prostate cancer. J. Drug Target. 2016, 24, 441–449. [Google Scholar] [CrossRef]
- Kyaw, M.; Yoshizumi, M.; Tsuchiya, K.; Izawa, Y.; Kanematsu, Y.; Fujita, Y.; Ali, N.; Ishizawa, K.; Yamauchi, A.; Tamaki, T. Antioxidant effects of stereoisomers of N-acetylcysteine (NAC), L-NAC and D-NAC, on angiotensin II-stimulated MAP kinase activation and vascular smooth muscle cell proliferation. J. Pharmacol. Sci. 2004, 95, 483–486. [Google Scholar] [CrossRef]
- Paravicini, T.M.; Touyz, R.M. Redox signaling in hypertension. Cardiovasc. Res. 2006, 71, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Plzak, J.; Boucek, J.; Bandurova, V.; Kolar, M.; Hradilova, M.; Szabo, P.; Lacina, L.; Chovanec, M.; Smetana, K., Jr. The Head and Neck Squamous Cell Carcinoma Microenvironment as a Potential Target for Cancer Therapy. Cancers 2019, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.D.; Johnson, L.B.; Weiner, M.; Li, K.P.; Suchindran, C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J. Am. Acad. Dermatol. 2009, 60, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Li, M.; Intong-Wheeler, L.R.A.; Tran, K.; Marucci, D.; Murrell, D.F. Epidemiology and Outcome of Squamous Cell Carcinoma in Epidermolysis Bullosa in Australia and New Zealand. Acta Derm. Venereol. 2018, 98, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Eickelberg, O.; Pansky, A.; Mussmann, R.; Bihl, M.; Tamm, M.; Hildebrand, P.; Perruchoud, A.P.; Roth, M. Transforming growth factor-beta1 induces interleukin-6 expression via activating protein-1 consisting of JunD homodimers in primary human lung fibroblasts. J. Biol. Chem. 1999, 274, 12933–12938. [Google Scholar] [CrossRef]
- Seong, G.J.; Hong, S.; Jung, S.A.; Lee, J.J.; Lim, E.; Kim, S.J.; Lee, J.H. TGF-beta-induced interleukin-6 participates in transdifferentiation of human Tenon’s fibroblasts to myofibroblasts. Mol. Vis. 2009, 15, 2123–2128. [Google Scholar]
- Dietz, H.C. TGF-beta in the pathogenesis and prevention of disease: A matter of aneurysmic proportions. J. Clin. Investig. 2010, 120, 403–407. [Google Scholar] [CrossRef]
- Odorisio, T.; Di Salvio, M.; Orecchia, A.; Di Zenzo, G.; Piccinni, E.; Cianfarani, F.; Travaglione, A.; Uva, P.; Bellei, B.; Conti, A.; et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-beta signalling in modifying disease severity. Hum. Mol. Genet. 2014, 23, 3907–3922. [Google Scholar] [CrossRef]
- Eickelberg, O.; Kohler, E.; Reichenberger, F.; Bertschin, S.; Woodtli, T.; Erne, P.; Perruchoud, A.P.; Roth, M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-beta1 and TGF-beta3. Am. J. Physiol. 1999, 276, L814–L824. [Google Scholar] [CrossRef]
- Mercado-Gomez, O.; Landgrave-Gomez, J.; Arriaga-Avila, V.; Nebreda-Corona, A.; Guevara-Guzman, R. Role of TGF-beta signaling pathway on Tenascin C protein upregulation in a pilocarpine seizure model. Epilepsy Res. 2014, 108, 1694–1704. [Google Scholar] [CrossRef]
- Ummarino, D. Systemic sclerosis: Tenascin C perpetuates tissue fibrosis. Nat. Rev. Rheumatol. 2016, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Tong, S.; Zhao, X.; Ding, W.; Gou, Y.; Xu, K.; Sun, C.; Xia, G. Periostin Mediates TGF-beta-Induced Epithelial Mesenchymal Transition in Prostate Cancer Cells. Cell Physiol. Biochem. 2015, 36, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.S.; Yuan, S.; Innes, A.L.; Kerr, S.; Woodruff, P.G.; Hou, L.; Muller, S.J.; Fahy, J.V. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 14170–14175. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.K.; Bozyk, P.D.; Bentley, J.K.; Popova, A.P.; Birch, C.M.; Wilke, C.A.; Fry, C.D.; White, E.S.; Sisson, T.H.; Tayob, N.; et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L1046–L1056. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.W.; Zhao, Z.; Ge, S.F.; Zhou, J.; Yuan, F.; Yuan, Y.Z. Circulating levels of periostin may help identify patients with more aggressive colorectal cancer. Int. J. Oncol. 2009, 34, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Zhang, L.; Jia, L.; Ji, W.; Wang, Z.; Ren, L.; Niu, R.; Zhou, Y. Identification of Serum Periostin as a Potential Diagnostic and Prognostic Marker for Colorectal Cancer. Clin. Lab. 2018, 64, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Ono, J.; Masuoka, M.; Ohta, S.; Izuhara, K.; Ikezawa, Z.; Aihara, M.; Takahashi, K. Serum periostin levels are correlated with progressive skin sclerosis in patients with systemic sclerosis. Br. J. Dermatol. 2013, 168, 717–725. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Periostin in Skin Tissue Skin-Related Diseases. Allergol. Int. 2014, 63, 161–170. [Google Scholar] [CrossRef]
- Yang, L.; Serada, S.; Fujimoto, M.; Terao, M.; Kotobuki, Y.; Kitaba, S.; Matsui, S.; Kudo, A.; Naka, T.; Murota, H.; et al. Periostin facilitates skin sclerosis via PI3K/Akt dependent mechanism in a mouse model of scleroderma. PLoS ONE 2012, 7, e41994. [Google Scholar] [CrossRef]
- Fernandez, L.A.; Garrido-Martin, E.M.; Sanz-Rodriguez, F.; Pericacho, M.; Rodriguez-Barbero, A.; Eleno, N.; Lopez-Novoa, J.M.; Duwell, A.; Vega, M.A.; Bernabeu, C.; et al. Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum. Mol. Genet. 2007, 16, 1515–1533. [Google Scholar] [CrossRef] [PubMed]
- Smetana, K., Jr.; Rosel, D.; BrAbek, J. Raloxifene and Bazedoxifene Could Be Promising Candidates for Preventing the COVID-19 Related Cytokine Storm, ARDS and Mortality. In Vivo 2020, 34, 3027–3028. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, V.; Etich, J.; Pitzler, L.; Frie, C.; Koch, M.; Fischer, M.; Rappl, G.; Abken, H.; Tomasek, J.J.; Brachvogel, B. Identification of a myofibroblast-specific expression signature in skin wounds. Matrix Biol. 2018, 65, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Green, H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Meana, A.; Iglesias, J.; Del Rio, M.; Larcher, F.; Madrigal, B.; Fresno, M.F.; Martin, C.; San Roman, F.; Tevar, F. Large surface of cultured human epithelium obtained on a dermal matrix based on live fibroblast-containing fibrin gels. Burns 1998, 24, 621–630. [Google Scholar] [CrossRef]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Langa, C.; Bernabeu, C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J. Biol. Chem. 2002, 277, 43799–43808. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Martin, E.M.; Blanco, F.J.; Fernandez, L.A.; Langa, C.; Vary, C.P.; Lee, U.E.; Friedman, S.L.; Botella, L.M.; Bernabeu, C. Characterization of the human Activin-A receptor type II-like kinase 1 (ACVRL1) promoter and its regulation by Sp1. BMC Mol. Biol. 2010, 11, 51. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguado, T.; García, M.; García, A.; Ferrer-Mayorga, G.; Martínez-Santamaría, L.; del Río, M.; Botella, L.-M.; Sánchez-Puelles, J.-M. Raloxifene and n-Acetylcysteine Ameliorate TGF-Signalling in Fibroblasts from Patients with Recessive Dominant Epidermolysis Bullosa. Cells 2020, 9, 2108. https://doi.org/10.3390/cells9092108
Aguado T, García M, García A, Ferrer-Mayorga G, Martínez-Santamaría L, del Río M, Botella L-M, Sánchez-Puelles J-M. Raloxifene and n-Acetylcysteine Ameliorate TGF-Signalling in Fibroblasts from Patients with Recessive Dominant Epidermolysis Bullosa. Cells. 2020; 9(9):2108. https://doi.org/10.3390/cells9092108
Chicago/Turabian StyleAguado, Tania, Marta García, Adela García, Gemma Ferrer-Mayorga, Lucía Martínez-Santamaría, Marcela del Río, Luisa-María Botella, and José-María Sánchez-Puelles. 2020. "Raloxifene and n-Acetylcysteine Ameliorate TGF-Signalling in Fibroblasts from Patients with Recessive Dominant Epidermolysis Bullosa" Cells 9, no. 9: 2108. https://doi.org/10.3390/cells9092108
APA StyleAguado, T., García, M., García, A., Ferrer-Mayorga, G., Martínez-Santamaría, L., del Río, M., Botella, L.-M., & Sánchez-Puelles, J.-M. (2020). Raloxifene and n-Acetylcysteine Ameliorate TGF-Signalling in Fibroblasts from Patients with Recessive Dominant Epidermolysis Bullosa. Cells, 9(9), 2108. https://doi.org/10.3390/cells9092108