
PI3Kδ Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Geo Dataset
2.2. Human Subjects
2.3. Keratinocyte Cultures and Treatments
2.4. IMQ-Induced Psoriasiform-Like Model
2.5. Immunohistochemistry
2.6. RNA Isolation and Real-Time Polymerase Chain Reaction (PCR)
2.7. Immunoblotting and Densitometry
2.8. Cytotoxicity Test
2.9. Proliferation Assay
2.10. Scratch Assay
2.11. Apoptosis Analysis
2.12. Statistical Analysis
3. Results
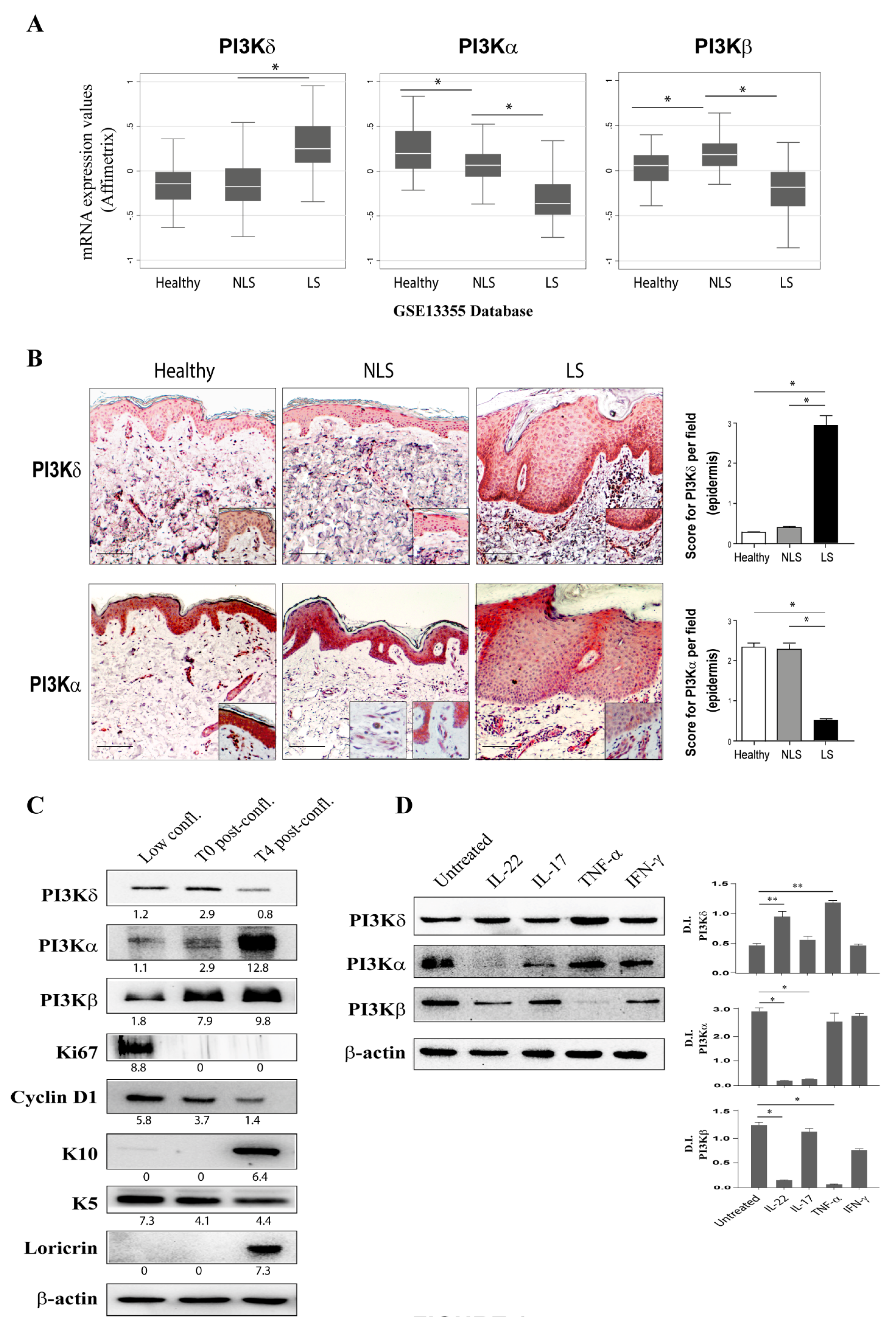
3.1. PI3Kδ Is Highly Expressed in Psoriatic Skin Lesions and Is Induced by Inflammatory Cytokines in Proliferating Keratinocytes
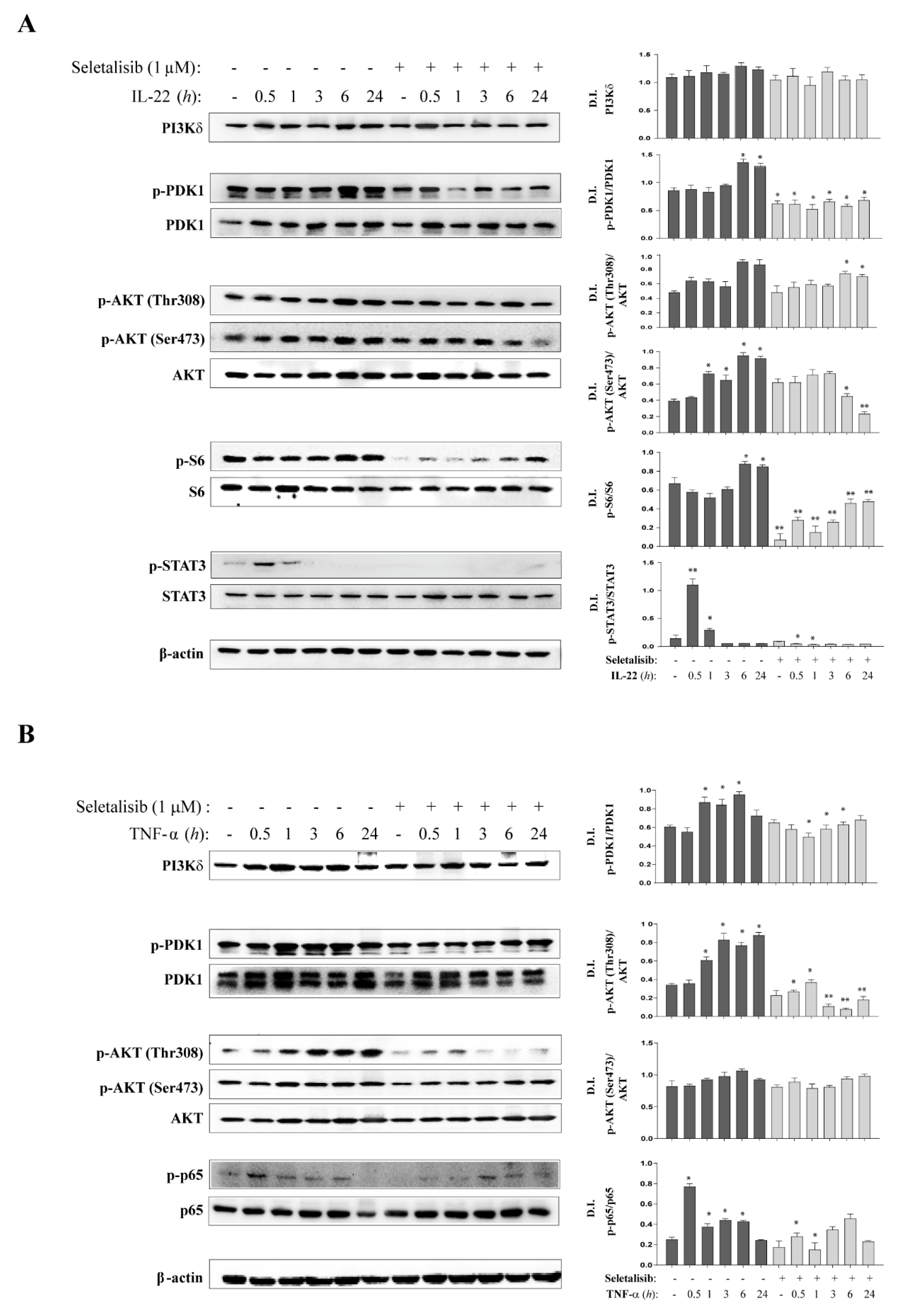
3.2. Selective PI3Kδ Inhibition Interferes with PI3K-Dependent Pathways Activated by IL-22- and TNF-α in Psoriatic Keratinocytes
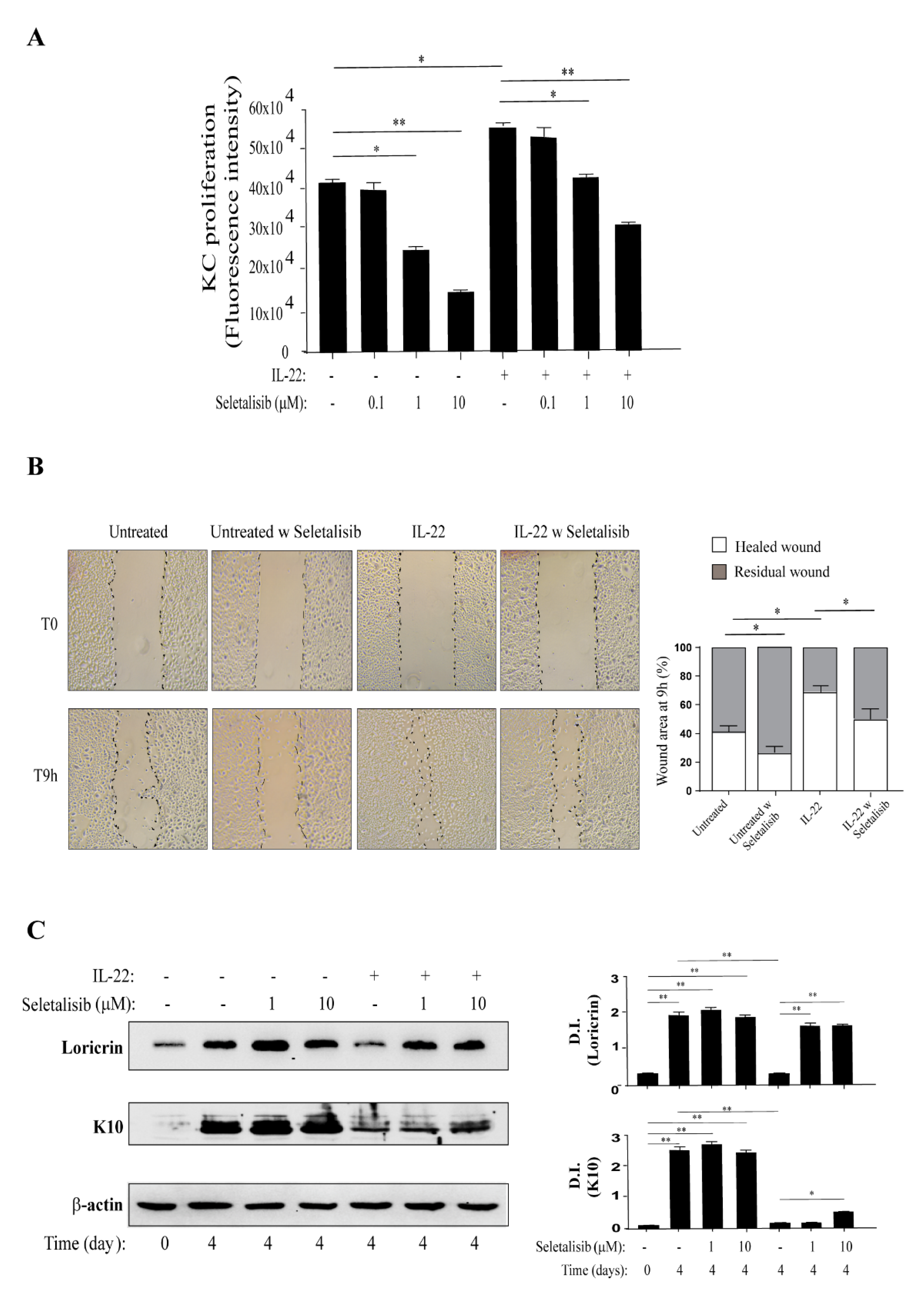
3.3. PI3Kδ Sustains Proliferation, Migration, and Differentiation of Human Keratinocytes Regulated by IL-22
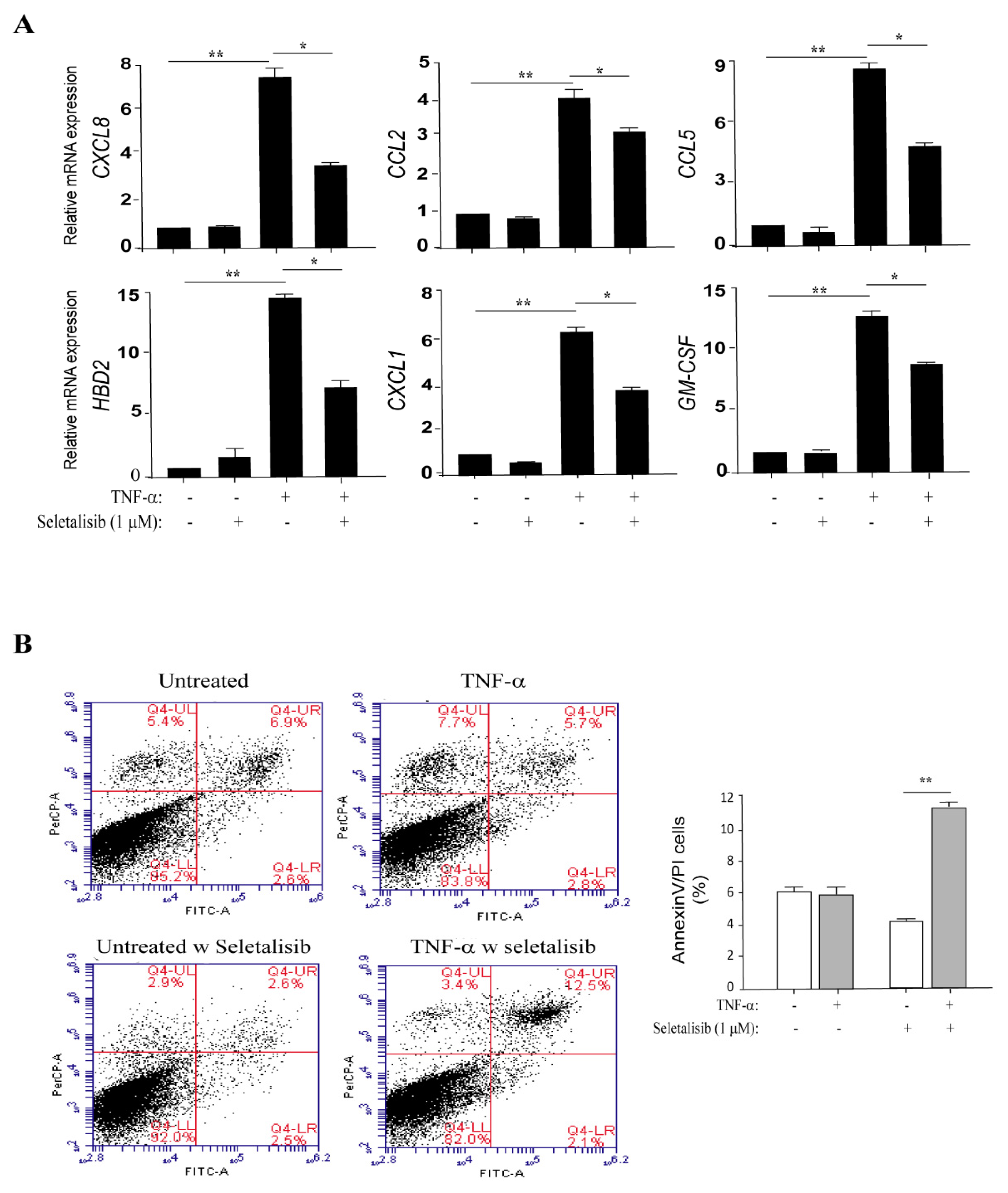
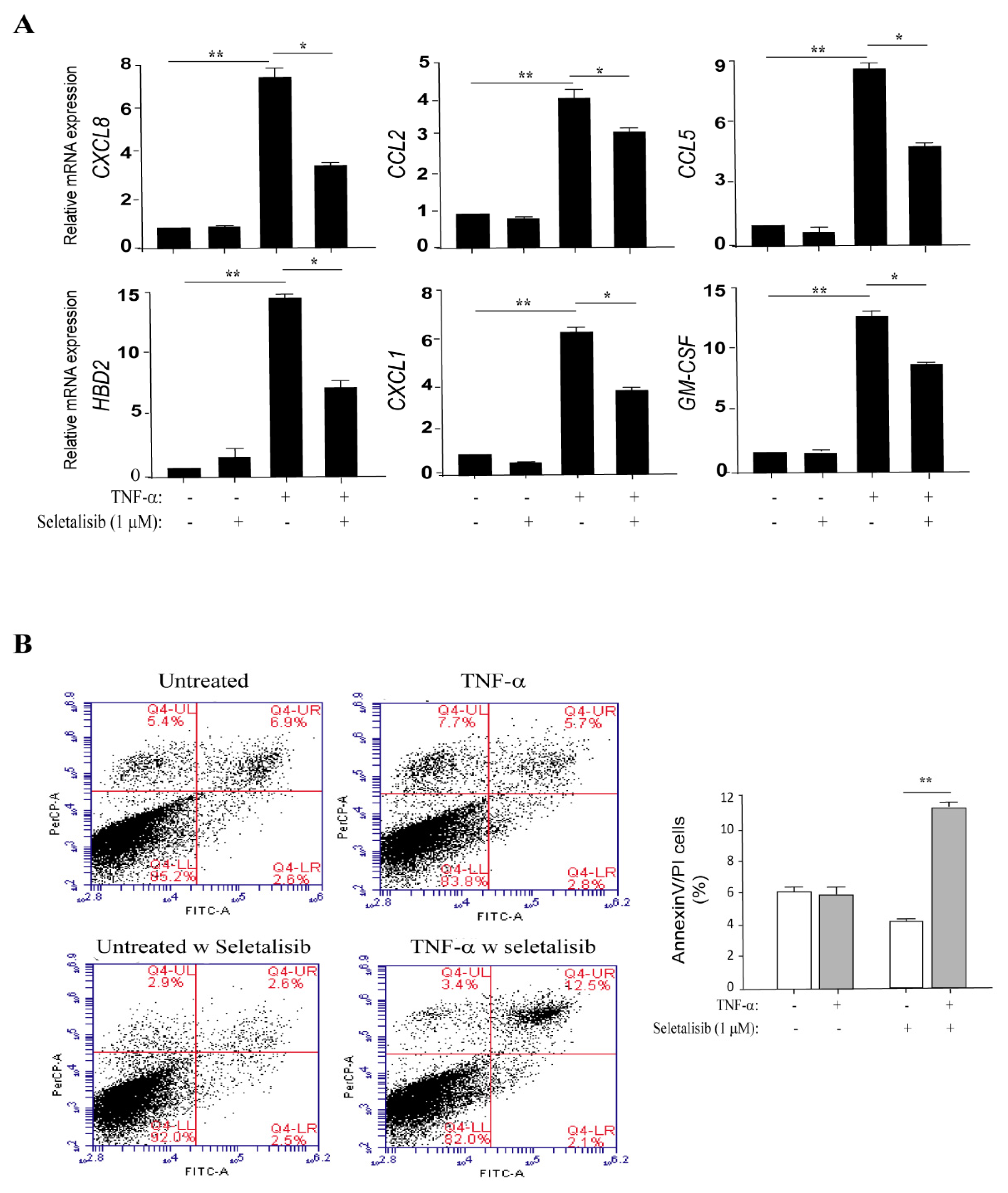
3.4. PI3Kδ Chemical Inhibition Reduces the Expression of Inflammatory Genes and Increases Apoptosis in TNF-α-Activated Keratinocytes
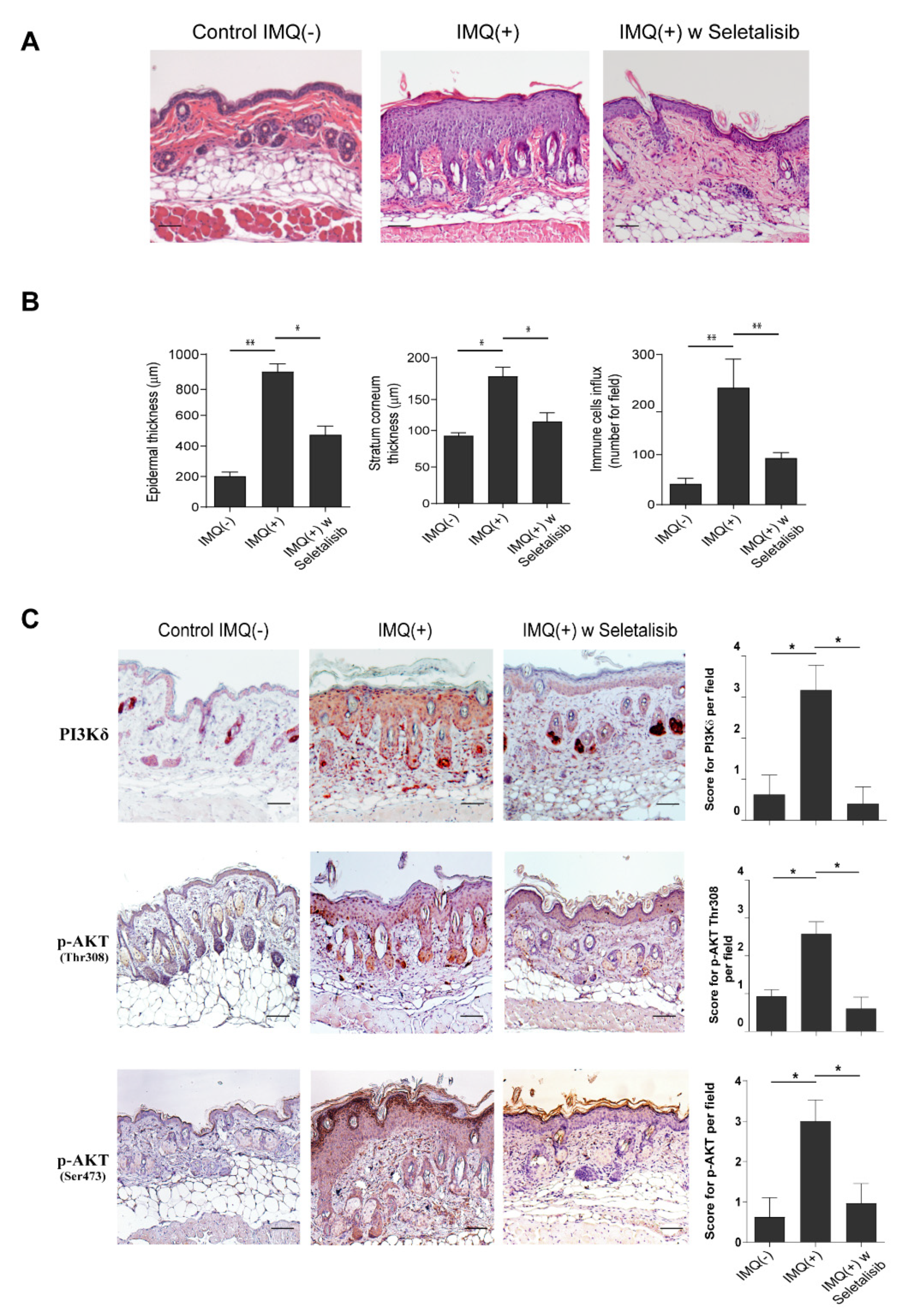
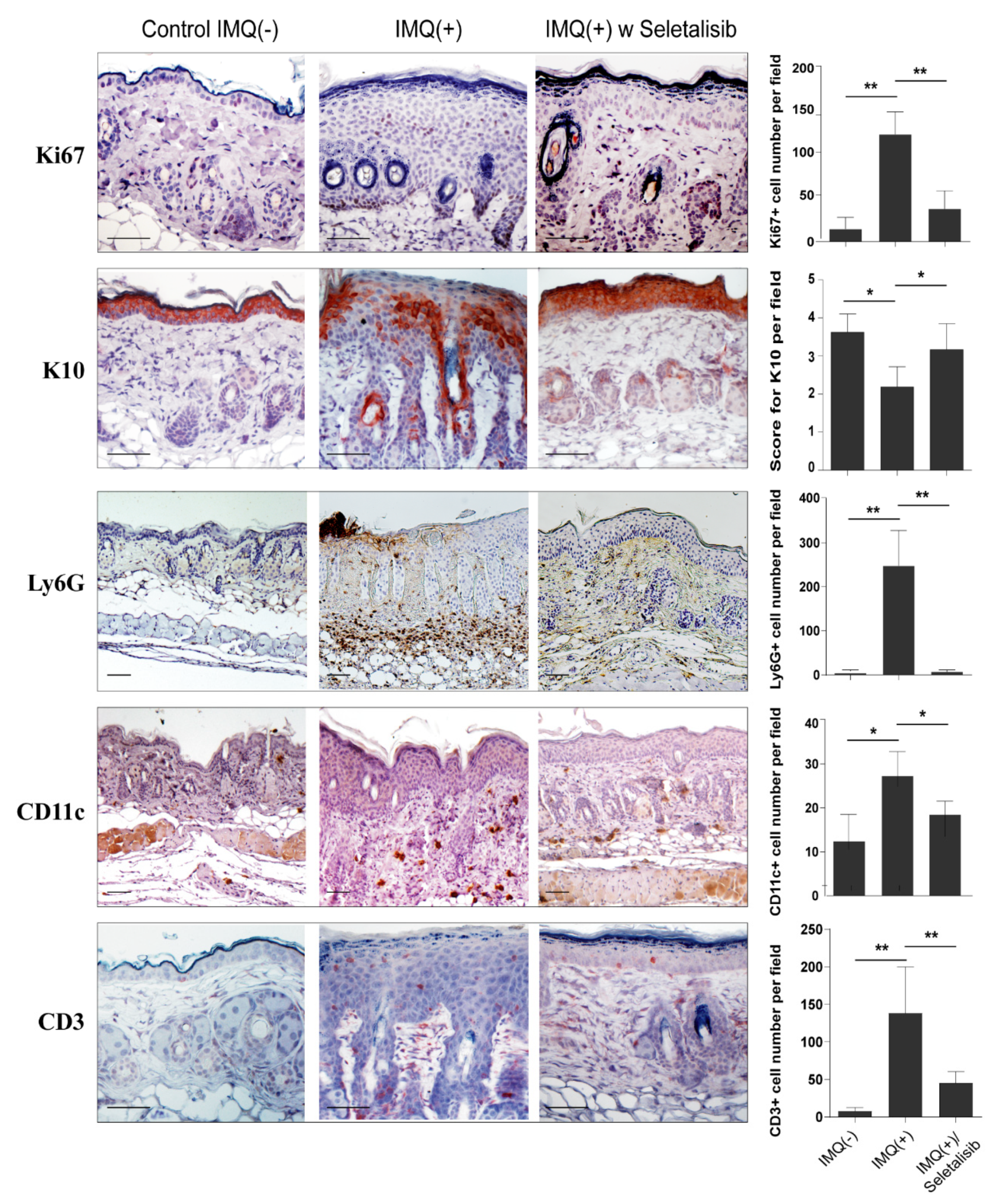
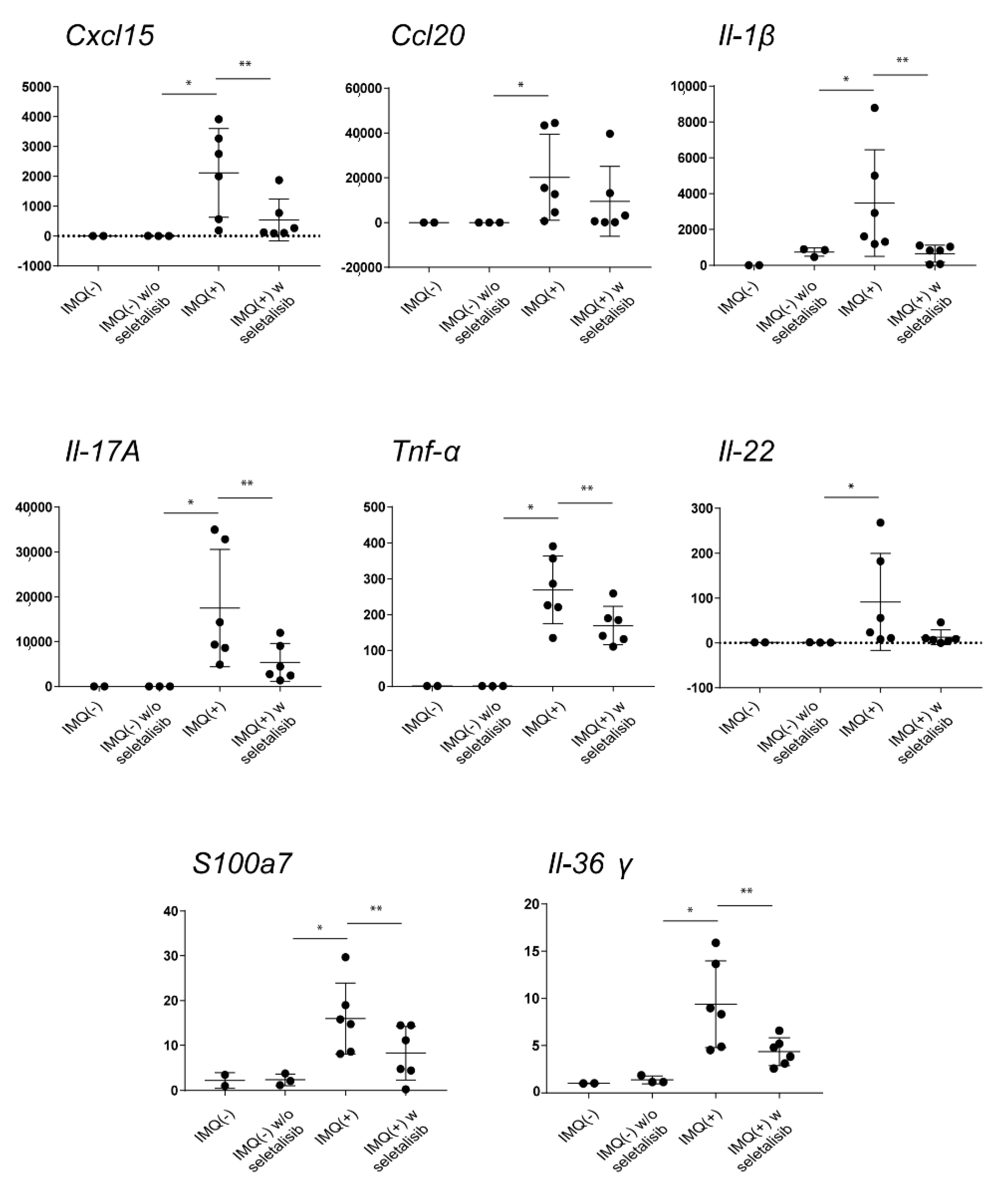
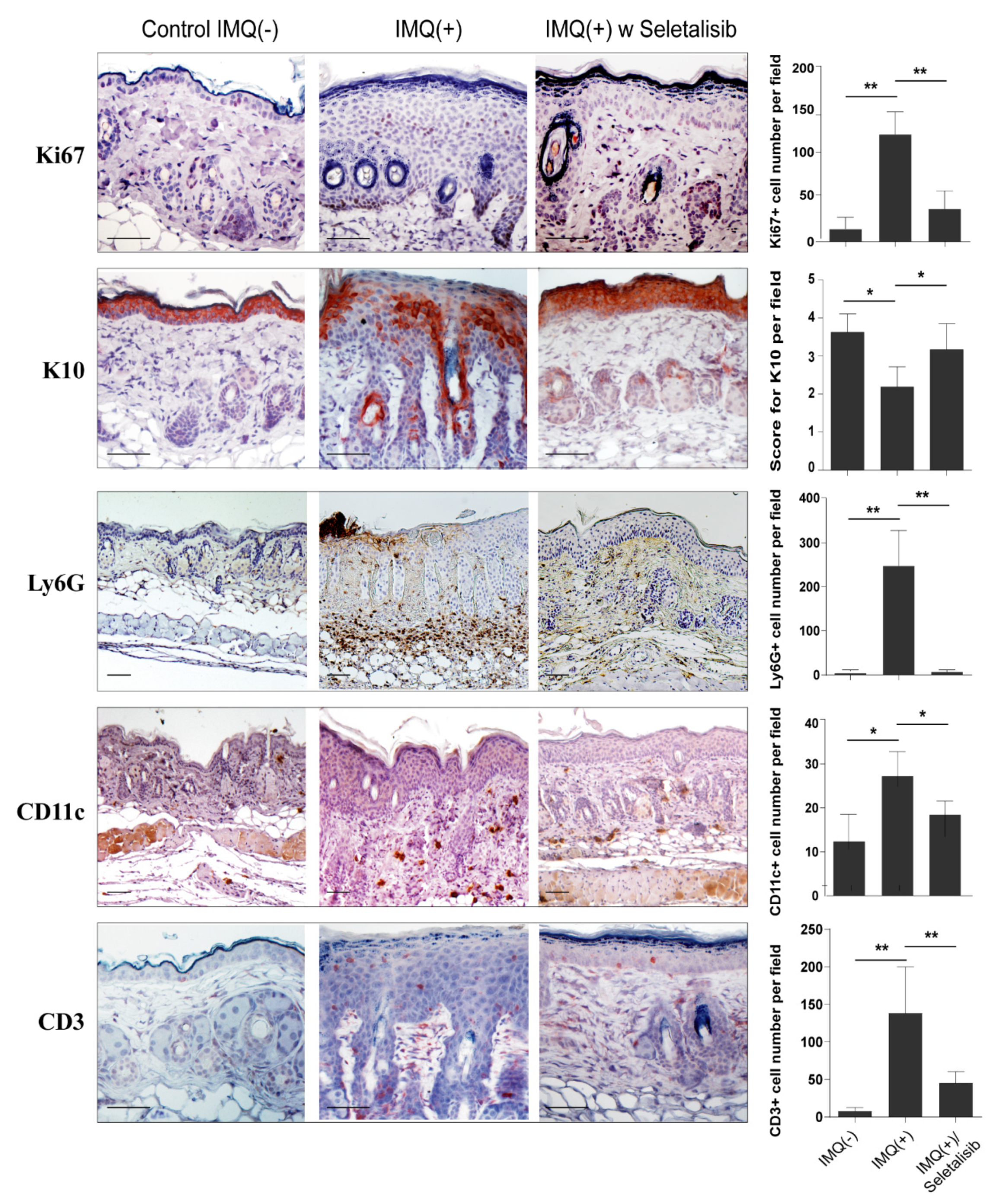
3.5. Topical Seletalisib Administration Ameliorates the Psoriasiform Phenotype of IMQ-Treated Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Mechanisms of disease: Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Di Meglio, P.; Perera, G.K.; Nestle, F.O. The Multitasking Organ: Recent Insights into Skin Immune Function. Immunity 2011, 35, 857–869. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Pastore, S. Pathobiology of Chronic Inflammatory Skin Diseases: Interplay Between Keratinocytes and Immune Cells as a Target for Anti-Inflammatory Drugs. Curr. Drug Metab. 2010, 11, 210–227. [Google Scholar] [CrossRef]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the Immunopathogenesis of Psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, E.; Sato, Y.; Minagawa, A.; Okuyama, R. Pathogenesis of psoriasis and development of treatment. J. Dermatol. 2018, 45, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madonna, S.; Scarponi, C.; Pallotta, S.; Cavani, A.; Albanesi, C. Anti-apoptotic effects of suppressor of cytokine signaling 3 and 1 in psoriasis. Cell Death Dis. 2012, 3, e334. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C.; Malisiewicz, B.; Eiser, A.; Hardt, K.; Boehncke, W.H. Mammalian target of rapamycin and its downstream signalling components are activated in psoriatic skin. Br. J. Dermatol. 2013, 169, 156–159. [Google Scholar] [CrossRef]
- Huang, T.; Lin, X.; Meng, X.; Lin, M. Phosphoinositide-3 Kinase/Protein Kinase-B/Mammalian Target of Rapamycin Pathway in Psoriasis Pathogenesis. A Potential Therapeutic Target? Acta Derm.-Venereol. 2014, 94, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Mercurio, L.; Albanesi, C.; Madonna, S. Recent Updates on the Involvement of PI3K/AKT/mTOR Molecular Cascade in the Pathogenesis of Hyperproliferative Skin Disorders. Front. Med. 2021, 8, 665647. [Google Scholar] [CrossRef]
- Rosenberger, C.; Solovan, C.; Rosenberger, A.D.; Jinping, L.; Treudler, R.; Frei, U.; Eckardt, K.U.; Brown, L.F. Upregulation of hypoxia-inducible factors in normal and psoriatic skin. J. Investig. Dermatol. 2007, 127, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2018, 48, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Adhami, V.M.; Siddiqui, A.; Wood, G.S.; Longley, B.J.; Mukhtar, H.; Rica, C. Upregulation of PI3K/AKT/mTOR, FABP5 and PPARβ/δ in Human Psoriasis and Imiquimod-induced Murine Psoriasiform Dermatitis Model. Acta Derm.-Venereol. 2016, 96, 854–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Fits, L.; Mourits, S.; Voerman, J.S.A.; Kant, M.; Boon, L.; Laman, J.D.; Cornelissen, F.; Mus, A.-M.; Florencia, E.; Prens, E.P.; et al. Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice Is Mediated via the IL-23/IL-17 Axis. J. Immunol. 2009, 182, 5836–5845. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Adhami, V.M.; Esnault, S.; Sechi, M.; Siddiqui, I.A.; Satyshur, K.A.; Syed, D.N.; Dodwad, S.J.M.; Chaves-Rodriquez, M.I.; Longley, B.J.; et al. Dual Inhibition of PI3K/Akt and mTOR by the Dietary Antioxidant, Delphinidin, Ameliorates Psoriatic Features in Vitro and in an Imiquimod-Induced Psoriasis-Like Disease in Mice. Antioxid. Redox Signal. 2017, 26, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Roller, A.; Perino, A.; Dapavo, P.; Soro, E.; Okkenhaug, K.; Hirsch, E.; Ji, H. Blockade of Phosphatidylinositol 3-Kinase (PI3K)δ or PI3Kγ Reduces IL-17 and Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis. J. Immunol. 2012, 189, 4612–4620. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Kriplani, N.; Hermida, M.A.; Brown, E.R.; Leslie, N.R. Class I PI 3-kinases: Function and evolution. Adv. Biol. Regul. 2015, 59, 53–64. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Benistant, C.; Chapuis, H.; Roche, S. A specific function for phosphatidylinositol 3-kinase alpha (p85alpha–p110alpha) in cell survival and for phosphatidylinositol 3-kinase beta (p85alpha–p110beta) in de novo DNA synthesis of human colon carcinoma cells. Oncogene 2000, 19, 5083–5090. [Google Scholar] [CrossRef] [Green Version]
- Utermark, T.; Rao, T.; Cheng, H.; Wang, Q.; Lee, S.H.; Wang, Z.C.; Dirk Iglehart, J.; Roberts, T.M.; Muller, W.J.; Zhao, J.J. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 2012, 26, 1573–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, F.M.; Traer, C.J.; Abraham, S.M.; Fry, M.J. The phosphoinositide (PI) 3-kinase family. J. Cell Sci. 2003, 116, 3037–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, C.; Sturge, J.; Bennett, D.C.; O’Hare, M.J.; Allen, W.E.; Bain, J.; Jones, G.E.; Vanhaesebroeck, B. Regulation of breast cancer cell chemotaxis by the phosphoinositide 3-kinase p110δ. Cancer Res. 2003, 63, 1667–1675. [Google Scholar] [PubMed]
- Eickholt, B.J.; Ahmed, A.I.; Davies, M.; Papakonstanti, E.A.; Pearce, W.; Starkey, M.L.; Bilancio, A.; Need, A.C.; Smith, A.J.H.; Hall, S.M.; et al. Control of axonal growth and regeneration of sensory neurons by the p110δ PI 3-kinase. PLoS ONE 2007, 2, e17846664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, E.; Fruciano, M.; Fagone, E.; Gili, E.; Caraci, F.; Iemmolo, M.; Crimi, N.; Vancheri, C. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class I P110 isoforms. PLoS ONE 2011, 6, e24663. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, M.A.; Bombardieri, M.; Pitzalis, C.; Vanhaesebroeck, B. Isoform-selective induction of human p110δ PI3K expression by TNFα: Identification of a new and inducible PIK3CD promoter. Biochem. J. 2012, 443, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Buerger, C. Epidermal mTORC1 signaling contributes to the pathogenesis of psoriasis and could serve as a therapeutic target. Front. Immunol. 2018, 9, 2786. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; Tsilioni, I.; Weng, Z.; Theoharides, T.C. TNF stimulates IL-6, CXCL8 and VEGF secretion from human keratinocytes via activation of mTOR, inhibited by tetramethoxyluteolin. Exp. Dermatol. 2018, 27, 135–143. [Google Scholar] [CrossRef]
- Buerger, C.; Shirsath, N.; Lang, V.; Berard, A.; Diehl, S.; Kaufmann, R.; Boehncke, W.H.; Wolf, P. Inflammation dependent mTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS ONE 2017, 12, e0180853. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Chan, T.O.; Ahmed, N.N.; Datta, K.; Malstrom, S.; Stokoe, D.; McCormick, F.; Feng, J.; Tsichlis, P. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene 1998, 17, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.A.; Brookings, D.C.; Powell, M.J.; Delgado, J.; Shuttleworth, L.K.; Merriman, M.; Fahy, I.J.; Tewari, R.; Silva, J.P.; Healy, L.J.; et al. Seletalisib: Characterization of a Novel, Potent, and Selective Inhibitor of PI3Kd s. J. Pharmacol. Exp. Ther. 2017, 361, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Helmer, E.; Watling, M.; Jones, E.; Tytgat, D.; Jones, M.; Allen, R.; Payne, A.; Koch, A.; Healy, E. First-in-human studies of seletalisib, an orally bioavailable small-molecule PI3Kδ inhibitor for the treatment of immune and inflammatory diseases. Eur. J. Clin. Pharmacol. 2017, 73, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudjonsson, J.E.; Ding, J.; Li, X.; Nair, R.P.; Tejasvi, T.; Qin, Z.S.; Ghosh, D.; Aphale, A.; Gumucio, D.L.; Voorhees, J.J.; et al. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J. Investig. Dermatol. 2009, 129, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigler, J.; Rand, H.A.; Kerkof, K.; Timour, M.; Russell, C.B. Cross-Study Homogeneity of Psoriasis Gene Expression in Skin across a Large Expression Range. PLoS ONE 2013, 8, e52242. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Morelli, M.; Scarponi, C.; Eisenmesser, E.Z.; Doti, N.; Pagnanelli, G.; Gubinelli, E.; Mazzanti, C.; Cavani, A.; Ruvo, M.; et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018, 9, 1104. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Scarponi, C.; Mercurio, L.; Facchiano, F.; Pallotta, S.; Madonna, S.; Girolomoni, G.; Albanesi, C. Selective Immunomodulation of Inflammatory Pathways in Keratinocytes by the Janus Kinase (JAK) Inhibitor Tofacitinib: Implications for the Employment of JAK-Targeting Drugs in Psoriasis. J. Immunol. Res. 2018, 2018, 7897263. [Google Scholar] [CrossRef] [PubMed]
- Madonna, S.; Scarponi, C.; Sestito, R.; Pallotta, S.; Cavani, A.; Albanesi, C. The IFN-γ–Dependent Suppressor of Cytokine Signaling 1 Promoter Activity Is Positively Regulated by IFN Regulatory Factor-1 and Sp1 but Repressed by Growth Factor Independence-1b and Krüppel-Like Factor-4, and It Is Dysregulated in Psoriatic Keratinocytes. J. Immunol. 2010, 185, 2467–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denley, A.; Gymnopoulos, M.; Kang, S.; Mitchell, C.; Vogt, P.K. Requirement of phosphatidylinositol(3,4,5)trisphosphate in phosphatidylinositol 3-kinase-induced oncogenic transformation. Mol. Cancer Res. 2009, 7, 1132–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardinocchi, L.; Sonego, G.; Passarelli, F.; Avitabile, S.; Scarponi, C.; Failla, C.M.; Simoni, S.; Albanesi, C.; Cavani, A. Interleukin-17 and interleukin-22 promote tumor progression in human nonmelanoma skin cancer. Eur. J. Immunol. 2015, 45, 922–931. [Google Scholar] [CrossRef]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Sestito, R.; Madonna, S.; Scarponi, C.; Cianfarani, F.; Failla, C.M.; Cavani, A.; Girolomoni, G.; Albanesi, C. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 2011, 25, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.R.; Liao, L.; Yates, J.R.; Vogt, P.K. Essential role of Stat3 in PI3K-induced oncogenic transformation. Proc. Natl. Acad. Sci. USA 2011, 108, 13247–13252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling [see comments]. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef]
- Boniface, K.; Bernard, F.-X.; Garcia, M.; Gurney, A.L.; Lecron, J.-C.; Morel, F. IL-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J. Immunol. 2005, 174, 3695–3702. [Google Scholar] [CrossRef] [Green Version]
- Ekman, A.K.; Bivik Eding, C.; Rundquist, I.; Enerbäck, C. IL-17 and IL-22 Promote Keratinocyte Stemness in the Germinative Compartment in Psoriasis. J. Investig. Dermatol. 2019, 139, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Madonna, S.; Scarponi, C.; Morelli, M.; Sestito, R.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. SOCS3 inhibits the pathological effects of IL-22 in nonmelanoma skin tumor-derived keratinocytes. Oncotarget 2017, 8, 24652–24667. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.; Brash, D.E.; Grossman, D. Keratinocyte apoptosis in epidermal development and disease. J. Investig. Dermatol. 2006, 126, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Yager, N.; Haddadeen, C.; Powell, M.; Payne, A.; Allen, R.; Healy, E. Expression of PI3K Signaling Associated with T Cells in Psoriasis Is Inhibited by Seletalisib, a PI3Kδ Inhibitor, and Is Required for Functional Activity. J. Investig. Dermatol. 2018, 138, 1435–1439. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Paveley, R.; Kraal, L.; Sritharan, L.; Stevens, E.; Dedi, N.; Shock, A.; Shaw, S.; Juarez, M.; Yeremenko, N.; et al. Selective targeting of PI3Kδ suppresses human IL-17-producing T cells and innate-like lymphocytes and may be therapeutic for IL-17-mediated diseases. J. Autoimmun. 2020, 111, 102435. [Google Scholar] [CrossRef]
- Alessi, D.; Kozlowski, M.T.; Weng, Q.P.; Morrice, N.; Avruch, J. 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro. Curr. Biol. 1998, 8, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Rintelen, F.; Stocker, H.; Thomas, G.; Hafen, E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc. Natl. Acad. Sci. USA 2001, 98, 15020–15025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Blasio, L.; Gagliardi, P.; Puliafito, A.; Primo, L. Serine/Threonine Kinase 3-Phosphoinositide-Dependent Protein Kinase-1 (PDK1) as a Key Regulator of Cell Migration and Cancer Dissemination. Cancers 2017, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Chan, K.S.; Carbajal, S.; Clifford, J.; Peavey, M.; Kiguchi, K.; Itami, S.; Nickoloff, B.J.; DiGiovanni, J. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat. Med. 2005, 11, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kastelan, M.; Prpić-Massari, L.; Brajac, I. Apoptosis in psoriasis. Acta Dermatovenerol. Croat. 2009, 17, 182–186. [Google Scholar]
- Claerhout, S.; Decraene, D.; Van Laethem, A.; Van Kelst, S.; Agostinis, P.; Garmyn, M. AKT delays the early-activated apoptotic pathway in UVB-irradiated keratinocytes via BAD translocation. J. Investig. Dermatol. 2007, 127, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Ozes, O.N.; Ozes, O.N.; Mayo, L.D.; Mayo, L.D.; Gustin, J.A.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, S.R.; Pfeffer, L.M.; Pfeffer, L.M.; et al. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef]
- Yoshiki, R.; Kabashima, K.; Honda, T.; Nakamizo, S.; Sawada, Y.; Sugita, K.; Yoshioka, H.; Ohmori, S.; Malissen, B.; Tokura, Y.; et al. IL-23 from langerhans cells is required for the development of imiquimod-induced psoriasis-like dermatitis by induction of IL-17A-producing γδ T cells. J. Investig. Dermatol. 2014, 134, 1912–1921. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Shen, X.; Ding, C.; Qi, C.; Li, K.; Li, X.; Jala, V.R.; Zhang, H.; Wang, T.; Zheng, J.; et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity 2011, 35, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Kwon, M.-A.; Hwang, J.; Lee, S.J.; Lee, J.H.; Kim, H.J.; Lee, K.B.; Lee, S.J.; Jeong, E.M.; Chung, J.H.; et al. Keratinocyte transglutaminase 2 promotes CCR6+ γδT-cell recruitment by upregulating CCL20 in psoriatic inflammation. Cell Death Dis. 2020, 11, 301. [Google Scholar] [CrossRef]
- Li, Z.J.; Sohn, K.C.; Choi, D.K.; Shi, G.; Hong, D.; Lee, H.E.; Whang, K.U.; Lee, Y.H.; Im, M.; Lee, Y.; et al. Roles of TLR7 in Activation of NF-κB Signaling of Keratinocytes by Imiquimod. PLoS ONE 2013, 8, e77159. [Google Scholar] [CrossRef] [Green Version]
- Madonna; Girolomoni; Dinarello; Albanesi The Significance of IL-36 Hyperactivation and IL-36R Targeting in Psoriasis. Int. J. Mol. Sci. 2019, 20, 3318. [CrossRef] [PubMed] [Green Version]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-Independent Signaling Downstream of Oncogenic PIK3CA Mutations in Human Cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, P.A.; di Blasio, L.; Orso, F.; Seano, G.; Sessa, R.; Taverna, D.; Bussolino, F.; Primo, L. 3-Phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia 2012, 14, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, J.M. The Src, Syk, and Tec family kinases: Distinct types of molecular switches. Cell. Signal. 2010, 22, 1175–1184. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human | Sequence | |
| CXCL8 | Fw | 5′-CCC CTA AGA GCA GTA ACA GTT CCT-3′ |
| Rv | 5′-GGT GAA GAT AAG CCA GCC AATC-3′ | |
| CCL2 | Fw | 5′-CAC CAG CAG CAA GTG TCCC-3′ |
| Rv | 5′-CCA TGG AAT CCT GAA CCC AC-3′ | |
| HBD-2 | Fw | 5′-TCC TCT TCT CGT TCC TCT TCA TATT-3′ |
| Rv | 5′-TTA AGG CAG GTA ACA GGA TCGC-3′ | |
| HBD-2 probe | Fw | 5′-ACC ACC AAA AAC ACC TGG AAG AGG CA-3′ |
| GM-CSF | Fw | 5′-GCG TCT CCT GAA CCT GAG TAG-3′ |
| Rv | 5′-TCG GCT CCT GGA GGT CAA AC-3′ | |
| CCL5 | Fw | 5′-CTACTGCCCTCTGCGCTCC-3′ |
| Rv | 5′-TGGTGTCCGAGGAATATGGG-3′ | |
| CXCL1 | Fw | 5′-CCTCAATCCTGCATCCC-3′ |
| Rv | 5′-AGTTGGATTTGTCACTGT-3′ | |
| HPRT1 | Fw | 5′-TGACACTGGCAAAACAATGCA-3′ |
| Rv | 5′-GGTCCTTTTCACCAGCAAGCT′-3′ | |
| Mouse | Sequence | |
| Ccl20 | Fw | 5′-AGG CAG AAG CAG CAA GCA AC-3′ |
| Rv | 5′-ACA AGC TTC ATC GGC CAT CT-3′ | |
| Il-36γ | Fw | 5′-AGCAGGTGTGGSTCTTTCGT-3′ |
| Rv | 5′-ACGCTGACTGGGGTTACTCT-3′ | |
| S100a7 | Fw | 5′-AGC AAC AGA CTC TCC GCT G-3′ |
| Rv | 5′-CTG GCA TGA CTG ATG GAC CC-3′ | |
| Il-22 | Fw | 5′-TGT TCC GAG GAG TCA GTG CTA-3′ |
| Rv | 5′-CAG AAC GTC TTC CAG GGT GA-3′ | |
| Tnf-α | Fw | 5′-GTC CCC AAA GGG ATG AGA AGT T-3′ |
| Rv | 5′-GGG TCT GGG CCA TAG AAC TG-3′ | |
| Il-17a | Fw | 5′-AGA AGG CCC TCA GAC TAC CT-3′ |
| Rv | 5′-CTT CAT TGC GGT GGA GAG TC-3′ | |
| Hprt1 | Fw | 5′-TCC TCA GAC CGC TTT TTG CC-3′ |
| Rv | 5′-ATC GCT AAT CAC GAC GCT GG-3′ |
| Inflammatory Molecules | IMQ(−) | IMQ(+) | IMQ(+) w Ly294002 | IMQ(+) w MK2206 | IMQ(+) w Seletalisib |
|---|---|---|---|---|---|
| Cxcl15 | 5.21 ± 1.8 | 232.25 ± 52.3 | 9.42 ± 3.45 ** | 2.16 ± 0.8 ** | 1.00 ± 0.14 ** |
| Ccl20 | 1.00 ± 0.12 | 7859.84 ± 78.22 | 3311.33 ± 43.56 * | 8450.58 ± 143.22 | 3325.83 ± 54.12 ** |
| Il-1β | 1.00 ± 0.02 | 161.37 ± 43.23 | 171.18 ± 38.92 | 177.99 ± 41.65 | 57.34 ± 18.76 * |
| Il-17a | 1.00 ± 0.18 | 465.80 ± 56.87 | 552.09 ± 48.82 | 492.95 ± 35.98 | 215.65 ± 40.53 * |
| Tnf-α | 1.00 ± 0.08 | 43.83 ± 12.43 | 50.96 ± 9.87 | 49.33 ± 12.32 | 17.32 ± 8.98 * |
| Il-22 | 1.00 ± 0.14 | 513.02 ± 29.81 | 83.60 ± 12.56 * | 578.00 ± 39.45 | 84.12 ± 30.45 * |
| S100a7 | 1.00 ± 0.09 | 2384.84 ± 98.65 | 2193.30 ± 87.45 | 2065.05 ± 79.65 | 609.66 ± 35.56 ** |
| Il-36γ | 1.00 ± 0.03 | 11.17 ± 1.21 | 11.41 ± 1.80 | 12.49 ± 2.01 | 7.51 ± 1.98 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercurio, L.; Morelli, M.; Scarponi, C.; Scaglione, G.L.; Pallotta, S.; Albanesi, C.; Madonna, S. PI3Kδ Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis. Cells 2021, 10, 2636. https://doi.org/10.3390/cells10102636
Mercurio L, Morelli M, Scarponi C, Scaglione GL, Pallotta S, Albanesi C, Madonna S. PI3Kδ Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis. Cells. 2021; 10(10):2636. https://doi.org/10.3390/cells10102636
Chicago/Turabian StyleMercurio, Laura, Martina Morelli, Claudia Scarponi, Giovanni Luca Scaglione, Sabatino Pallotta, Cristina Albanesi, and Stefania Madonna. 2021. "PI3Kδ Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis" Cells 10, no. 10: 2636. https://doi.org/10.3390/cells10102636
APA StyleMercurio, L., Morelli, M., Scarponi, C., Scaglione, G. L., Pallotta, S., Albanesi, C., & Madonna, S. (2021). PI3Kδ Sustains Keratinocyte Hyperproliferation and Epithelial Inflammation: Implications for a Topically Druggable Target in Psoriasis. Cells, 10(10), 2636. https://doi.org/10.3390/cells10102636