Activation of Cdc42 GTPase upon CRY2-Induced Cortical Recruitment Is Antagonized by GAPs in Fission Yeast


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
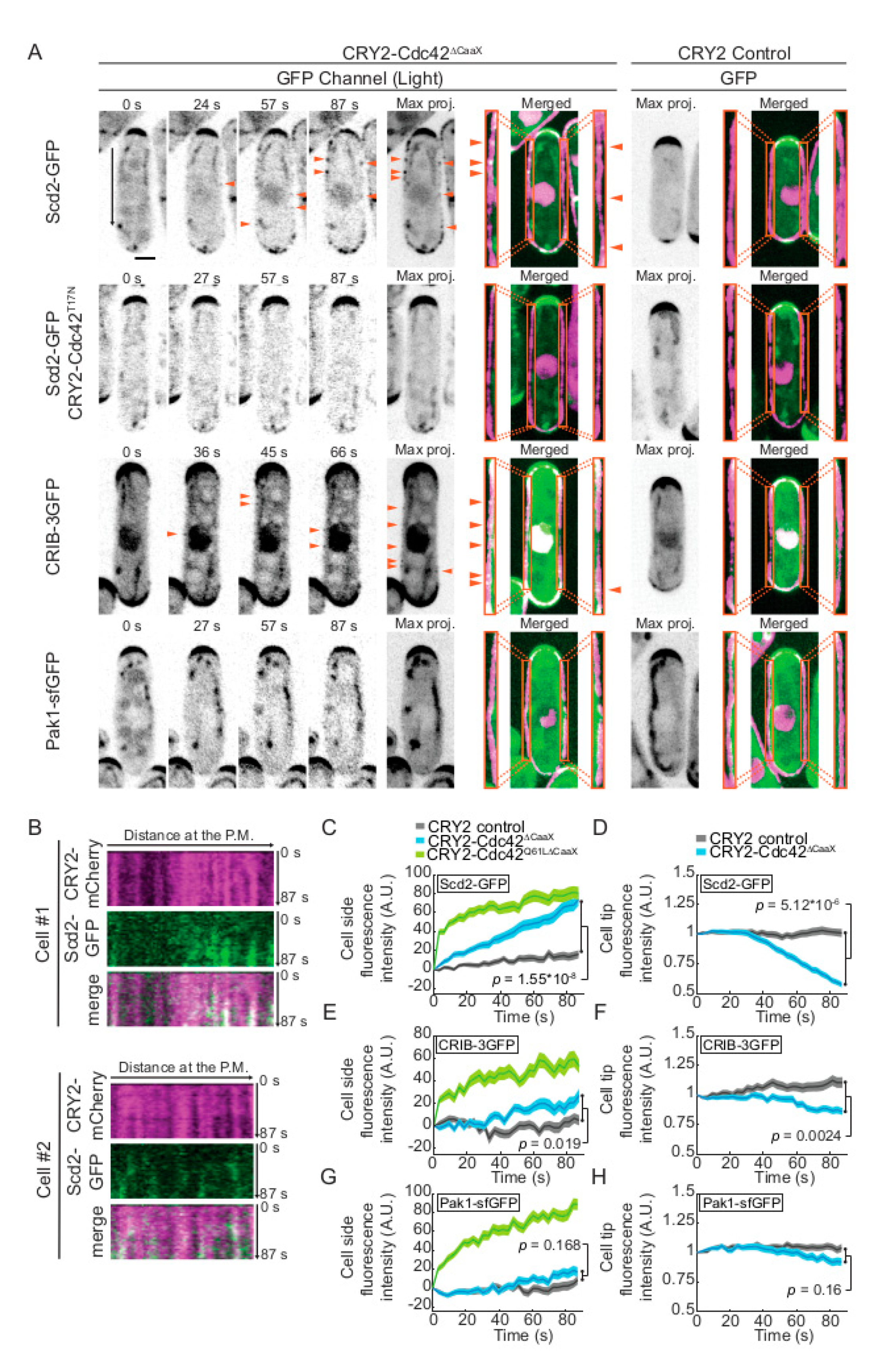
2.1. Weak Activation of CRY2-Cdc42 at the Cell Cortex
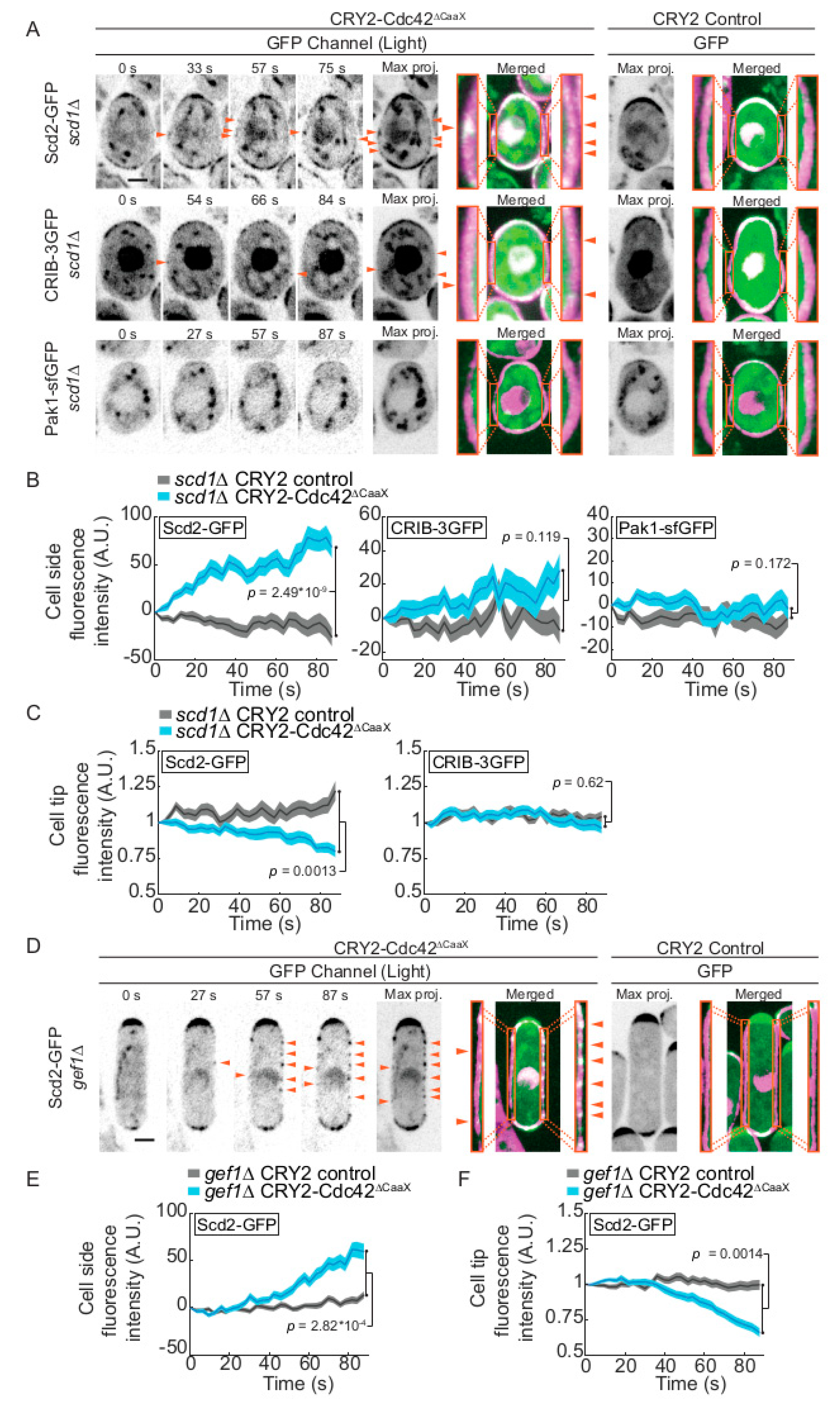
2.2. CRY2-Cdc42 Activation in Absence of Cdc42 GEFs
2.3. CRY2-Cdc42 Promotes Recruitment of Its GEF Scd1 in Scd2 Scaffold-Dependent Manner
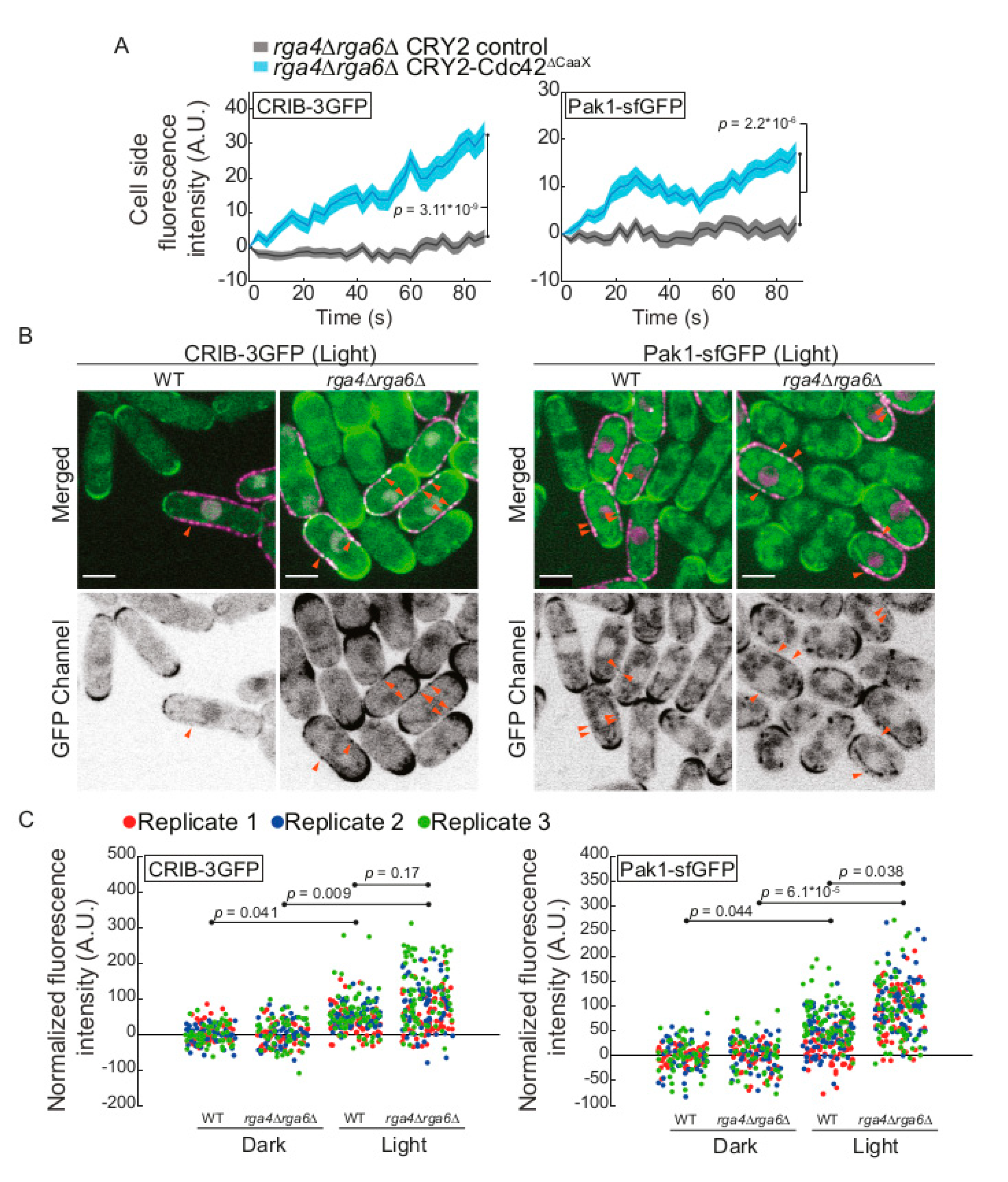
2.4. The Cdc42 GAP Rga4 Prevents Isotropic Growth of CRY2-Cdc42∆CaaX Cells
3. Materials and Methods
3.1. Strains, Media, and Growth Conditions
3.2. Cell Length and Width Measurements
3.3. Microscopy
- (1)
- RFP control: An RFP bleaching correction strain, expressing cytosolic CRY2PHR-mCherry.
- (2)
- GFP control: A wild type strain expressing the same GFP-tagged protein as the strain of interest but without the optogenetic system. This strain was used both as negative control for cell side re-localization experiments and as GFP bleaching correction strain (in Figure 1, Figure 2, Figure 3, Figure 5 and related Figures S2, S5, S6 and S8).
- (1)
- 50 ms: RFP channel (200 ms), GFP channel (50 ms). This constitutes one cycle (≈ 0.5 s). 30 time points were acquired (≈ 0.5 s * 30 = 15.1 s).
- (2)
- 250 ms: RFP channel (200 ms), GFP channel (250 ms). This constitutes one cycle (≈ 0.7 s). 22 time points were acquired (≈ 0.7 s * 22 = 15.1 s).
- (3)
- 500 ms: RFP channel (200 ms), GFP channel (500 ms). This constitutes one cycle (≈ 0.9 s). 17 time points were acquired (0.9 s * 17 = 15.5 s).
3.4. Image Analysis
3.4.1. CRY2-Cdc42∆CaaX and CRY2 Quantifications
3.4.2. Quantifications of the Re-Localization of GFP-Tagged Proteins to Cell Sides
- (1)
- Calculation of the GFP bleaching correction coefficient (see below).
- (2)
- Negative control of the experiment: These strains carry the same endogenous GFP-tagged protein as the sample strain of the experiment, however lacking the optogenetic system. This controlled that GFP fluorescence changes were due to the optogenetic system and not caused by imaging per se. Control GFP strains were imaged in the same pad and analysed in the same way as optogenetic cells.
3.4.3. Quantifications of the Re-Localization of GFP-Tagged Proteins from Cell Tips
3.4.4. Quantifications of CRIB-3GFP and Cdc42-mChSW Relative Expression and Distribution Profiles
3.4.5. Quantifications of CRIB-3GFP and Pak1-sfGFP at the Cell Sides of WT and rga4∆rga6∆ Mutants
3.4.6. Cell Size Measurements, Aspect Ratios and Index of Ratios
3.4.7. Cell Images and Kymographs
3.4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goryachev, A.B.; Leda, M. Autoactivation of small GTPases by the GEF-effector positive feedback modules. F1000Research 2019, 8, 1676. [Google Scholar] [CrossRef] [PubMed]
- Woods, B.; Lew, D.J. Polarity establishment by Cdc42: Key roles for positive feedback and differential mobility. Small GTPases 2019, 10, 1301–1337. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.G. Spontaneous cell polarization: Feedback control of Cdc42 GTPase breaks cellular symmetry. Bioessays 2015, 37, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Bendezu, F.O.; Vincenzetti, V.; Vavylonis, D.; Wyss, R.; Vogel, H.; Martin, S.G. Spontaneous Cdc42 polarization independent of GDI-mediated extraction and actin-based trafficking. PLoS Biol. 2015, 13, e1002097. [Google Scholar] [CrossRef]
- Kelly, F.D.; Nurse, P. Spatial control of Cdc42 activation determines cell width in fission yeast. Mol. Biol. Cell 2011, 22, 38013–38811. [Google Scholar] [CrossRef]
- Lamas, I.; Merlini, L.; Vjestica, A.; Vincenzetti, V.; Martin, S.G. Optogenetics reveals Cdc42 local activation by scaffold-mediated positive feedback and Ras GTPase. PLoS Biol. 2020, 18, e3000600. [Google Scholar] [CrossRef]
- Endo, M.; Shirouzu, M.; Yokoyama, S. The Cdc42 binding and scaffolding activities of the fission yeast adaptor protein Scd2. J. Biol. Chem. 2003, 278, 8438–8452. [Google Scholar] [CrossRef]
- Chang, E.C.; Barr, M.; Wang, Y.; Jung, V.; Xu, H.P.; Wigler, M.H. Cooperative interaction of S. pombe proteins required for mating and morphogenesis. Cell 1994, 79, 1311–1341. [Google Scholar] [CrossRef]
- Tay, Y.D.; Leda, M.; Goryachev, A.B.; Sawin, K.E. Local and global Cdc42 GEFs for fission yeast cell polarity are coordinated by microtubules and the Tea1/Tea4/Pom1 axis. J. Cell Sci. 2018. [Google Scholar] [CrossRef]
- Vjestica, A.; Zhang, D.; Liu, J.; Oliferenko, S. Hsp70-Hsp40 chaperone complex functions in controlling polarized growth by repressing Hsf1-driven heat stress-associated transcription. PLoS Genet. 2013, 9, e100. [Google Scholar] [CrossRef]
- Coll, P.M.; Trillo, Y.; Ametzazurra, A.; Perez, P. Gef1p, a New Guanine Nucleotide Exchange Factor for Cdc42p, Regulates Polarity in Schizosaccharomyces pombe. Mol. Biol. Cell 2003, 14, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Tanaka, K.; Ohta, K.; Yamamoto, M. Gef1p and Scd1p, the Two GDP-GTP exchange factors for Cdc42p, form a ring structure that shrinks during cytokinesis in Schizosaccharomyces pombe. Mol. Biol. Cell 2003, 14, 36173–36627. [Google Scholar] [CrossRef] [PubMed]
- Gallo Castro, D.; Martin, S.G. Differential GAP requirement for Cdc42-GTP polarization during proliferation and sexual reproduction. J. Cell Biol. 2018, 217, 42154–42229. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Wiley, D.J.; Medina, S.; Vincent, H.A.; Larrea, M.; Oriolo, A.; Verde, F. Regulation of cell diameter, For3p localization, and cell symmetry by fission yeast Rho-GAP Rga4p. Mol. Biol. Cell 2007, 18, 2090–2101. [Google Scholar] [CrossRef] [PubMed]
- Revilla-Guarinos, M.T.; Martin-Garcia, R.; Villar-Tajadura, M.A.; Estravis, M.; Coll, P.M.; Perez, P. Rga6 is a Fission Yeast Rho GAP Involved in Cdc42 Regulation of Polarized Growth. Mol. Biol. Cell 2016, 27, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Mutoh, T.; Arai, R.; Mabuchi, I. The small GTPase Rho4 is involved in controlling cell morphology and septation in fission yeast. Genes Cells 2003, 8, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Bugaj, L.J.; Choksi, A.T.; Mesuda, C.K.; Kane, R.S.; Schaffer, D.V. Optogenetic protein clustering and signaling activation in mammalian cells. Nat. Methods 2013, 10, 249–252. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Jang, H. Ras assemblies and signaling at the membrane. Curr. Opin. Struct. Biol. 2020, 62, 1401–1448. [Google Scholar] [CrossRef]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem. 2000, 275, 37373–37740. [Google Scholar] [CrossRef]
- Kang, P.J.; Beven, L.; Hariharan, S.; Park, H.O. The Rsr1/Bud1 GTPase interacts with itself and the Cdc42 GTPase during bud-site selection and polarity establishment in budding yeast. Mol. Biol. Cell 2010, 21, 3007–3016. [Google Scholar] [CrossRef]
- Zhang, B.; Gao, Y.; Moon, S.Y.; Zhang, Y.; Zheng, Y. Oligomerization of Rac1 gtpase mediated by the carboxyl-terminal polybasic domain. J. Biol. Chem. 2001, 276, 89588–89967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zheng, Y. Negative regulation of Rho family GTPases Cdc42 and Rac2 by homodimer formation. J. Biol. Chem. 1998, 273, 25728–25733. [Google Scholar] [CrossRef]
- Remorino, A.; De Beco, S.; Cayrac, F.; Di Federico, F.; Cornilleau, G.; Gautreau, A.; Parrini, M.C.; Masson, J.B.; Dahan, M.; Coppey, M. Gradients of Rac1 Nanoclusters Support Spatial Patterns of Rac1 Signaling. Cell Rep. 2017, 21, 19221–19935. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Zhou, Y.; Hancock, J.F. Rac1 Nanoscale Organization on the Plasma Membrane Is Driven by Lipid Binding Specificity Encoded in the Membrane Anchor. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Sartorel, E.; Unlu, C.; Jose, M.; Massoni-Laporte, A.; Meca, J.; Sibarita, J.B.; McCusker, D. Phosphatidylserine and GTPase activation control Cdc42 nanoclustering to counter dissipative diffusion. Mol. Biol. Cell 2018, 29, 1299–1310. [Google Scholar] [CrossRef]
- Slaughter, B.D.; Unruh, J.R.; Das, A.; Smith, S.E.; Rubinstein, B.; Li, R. Non-uniform membrane diffusion enables steady-state cell polarization via vesicular trafficking. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Meca, J.; Massoni-Laporte, A.; Martinez, D.; Sartorel, E.; Loquet, A.; Habenstein, B.; McCusker, D. Avidity-driven polarity establishment via multivalent lipid-GTPase module interactions. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Kennedy, M.J.; Hughes, R.M.; Peteya, L.A.; Schwartz, J.W.; Ehlers, M.D.; Tucker, C.L. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 2010, 7, 9739–9775. [Google Scholar] [CrossRef]
- Wheatley, E.; Rittinger, K. Interactions between Cdc42 and the scaffold protein Scd2: Requirement of SH3 domains for GTPase binding. Biochem. J. 2005, 388, 1771–1784. [Google Scholar] [CrossRef]
- Tatebe, H.; Nakano, K.; Maximo, R.; Shiozaki, K. Pom1 DYRK regulates localization of the Rga4 GAP to ensure bipolar activation of Cdc42 in fission yeast. Curr. Biol. 2008, 18, 3223–3230. [Google Scholar] [CrossRef]
- Merlini, L.; Khalili, B.; Dudin, O.; Michon, L.; Vincenzetti, V.; Martin, S.G. Inhibition of Ras activity coordinates cell fusion with cell-cell contact during yeast mating. J. Cell Biol. 2018, 217, 1467–1483. [Google Scholar] [CrossRef]
- Merlini, L.; Khalili, B.; Bendezu, F.O.; Hurwitz, D.; Vincenzetti, V.; Vavylonis, D.; Martin, S.G. Local Pheromone Release from Dynamic Polarity Sites Underlies Cell-Cell Pairing during Yeast Mating. Curr. Biol. 2016, 26, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.E.; Magliozzi, J.O.; Picard, N.A.; Moseley, J. Sequestration of the exocytic SNARE Psy1 into multiprotein nodes reinforces polarized morphogenesis in fission yeast. BioRxiv 2020. [Google Scholar] [CrossRef]
- Dudin, O.; Bendezu, F.O.; Groux, R.; Laroche, T.; Seitz, A.; Martin, S.G. A formin-nucleated actin aster concentrates cell wall hydrolases for cell fusion in fission yeast. J. Cell Biol. 2015, 208, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Bähler, J.; Wu, J.Q.; Longtine, M.S.; Shah, N.G.; McKenzie, A., III; Steever, A.B.; Wach, A.; Philippsen, P.; Pringle, J.R. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 1998, 14, 9439–9451. [Google Scholar] [CrossRef]
- Vjestica, A.; Marek, M.; Nkosi, P.J.; Merlini, L.; Liu, G.; Berard, M.; Billault-Chaumartin, I.; Martin, S.G. A toolbox of Stable Integration Vectors (SIV) in the fission yeast Schizosaccharomyces pombe. J. Cell Sci. 2019. [Google Scholar] [CrossRef]
- Bendezu, F.O.; Martin, S.G. Actin cables and the exocyst form two independent morphogenesis pathways in the fission yeast. Mol. Biol. Cell 2011, 22, 44–53. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamas, I.; Weber, N.; Martin, S.G. Activation of Cdc42 GTPase upon CRY2-Induced Cortical Recruitment Is Antagonized by GAPs in Fission Yeast. Cells 2020, 9, 2089. https://doi.org/10.3390/cells9092089
Lamas I, Weber N, Martin SG. Activation of Cdc42 GTPase upon CRY2-Induced Cortical Recruitment Is Antagonized by GAPs in Fission Yeast. Cells. 2020; 9(9):2089. https://doi.org/10.3390/cells9092089
Chicago/Turabian StyleLamas, Iker, Nathalie Weber, and Sophie G. Martin. 2020. "Activation of Cdc42 GTPase upon CRY2-Induced Cortical Recruitment Is Antagonized by GAPs in Fission Yeast" Cells 9, no. 9: 2089. https://doi.org/10.3390/cells9092089
APA StyleLamas, I., Weber, N., & Martin, S. G. (2020). Activation of Cdc42 GTPase upon CRY2-Induced Cortical Recruitment Is Antagonized by GAPs in Fission Yeast. Cells, 9(9), 2089. https://doi.org/10.3390/cells9092089