Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability

 ,
,  ,
, 
Abstract
1. Introduction
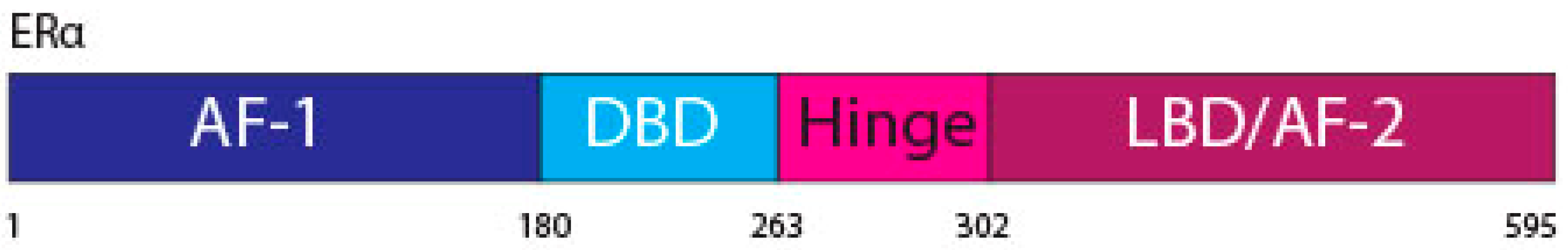
2. Estrogen Receptor Signaling
2.1. Estrogen Receptor Turnover
2.2. Degradation by the Ubiquitin Proteasome Pathway
3. Post Translational Modifications
3.1. Ubiquitylation
3.2. SUMOylation
3.3. Phosphorylation
3.4. Palmitoylation
3.5. Acetylation
3.6. Methylation
3.7. Glycosylation
4. Clinical Relevance
4.1. Estrogen Receptor Mutations and Resistance
4.1.1. S282C
4.1.2. K303R
4.1.3. Y537C/S/N
4.2. Other PTM Sites and Resistance
S167 and S118
4.3. Treatments
4.3.1. Targeting ESR1 Mutants
4.3.2. Clinical Proteasomal Inhibition
4.3.3. Targeting Kinases
4.3.4. Targeting Coactivators
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Boil. 2016, 16, 4–20. [Google Scholar] [CrossRef]
- Menasce, L.; White, G.; Harrison, C.J.; Boyle, J. Localization of the Estrogen Receptor Locus (ESR) to Chromosome 6q25.1 by FISH and a Simple Post-FISH Banding Technique. Genomics 1993, 17, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-A. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern. J. Clin. Endocr. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Gosden, J.; Middleton, P.; Rout, D. Localization of the human oestrogen receptor gene to chromosome 6q24→q27 by in situ hybridization. Cytogenet. Genome Res. 1986, 43, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Paech, K.; Webb, P.; Kuiper, G.G.J.M.; Nilsson, S.; Gustafsson, J.-Å.; Kushner, P.J.; Scanlan, T.S. Differential Ligand Activation of Estrogen Receptors ER and ER at AP1 Sites. Science 1997, 277, 1508–1510. [Google Scholar] [CrossRef]
- Kumar, V.; Green, S.; Stack, G.; Berry, M.; Jin, J.-R.; Chambon, P. Functional domains of the human estrogen receptor. Cell 1987, 51, 941–951. [Google Scholar] [CrossRef]
- Cowley, S.M.; Hoare, S.; Mosselman, S.; Parker, M.G. Estrogen Receptors α and β Form Heterodimers on DNA. J. Boil. Chem. 1997, 272, 19858–19862. [Google Scholar] [CrossRef]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef]
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V. Antitumour activity of the antiestrogen ICI 46,474 (Tamoxifen) in the dimethylbenzanthracene (DMBA)—Induced rat mammary carcinoma model. J. Steroid Biochem. 1974, 5, 354. [Google Scholar] [CrossRef]
- Jordan, V.C. Biochemical pharmacology of antiestrogen action. Pharmacol. Rev. 1984, 36, 245–276. [Google Scholar]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Casa, A.J.; Hochbaum, D.; Sreekumar, S.; Oesterreich, S.; Lee, A.V. The estrogen receptor alpha nuclear localization sequence is critical for fulvestrant-induced degradation of the receptor. Mol. Cell. Endocrinol. 2015, 415, 76–86. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef]
- Dauvois, S.; White, R.; Parker, M.G. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993, 106, 1377–1388. [Google Scholar]
- Dowsett, M.; Jones, A.; Johnston, S.R.; Jacobs, S.; Trunet, P.; Smith, I. In vivo measurement of aromatase inhibition by letrozole (CGS 20267) in postmenopausal patients with breast cancer. Clin. Cancer Res. 1995, 1, 1511–1515. [Google Scholar]
- Geisler, J.; King, N.; Anker, G.; Ornati, G.; Di Salle, E.; Lønning, P.E.; Dowsett, M. In vivo inhibition of aromatization by exemestane, a novel irreversible aromatase inhibitor, in postmenopausal breast cancer patients. Clin. Cancer Res. 1998, 4, 2089–2093. [Google Scholar]
- Geisler, J.; King, N.; Dowsett, M.; Ottestad, L.; Lundgren, S.; Walton, P.; Kormeset, P.; Lonning, P.E. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br. J. Cancer 1996, 74, 1286–1291. [Google Scholar] [CrossRef]
- Dowsett, M. Endocrine Resistance in Advanced Breast Cancer. Acta Oncol. 1996, 35, 91–95. [Google Scholar] [CrossRef]
- Haque, M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.W.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Weir, H.; Razavi, P.; Lawson, M.; Goeppert, A.U.; Mazzola, A.M.; Smith, A.; Wilson, J.; Morrow, C.; Wong, W.L.; et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 2016, 7, 277–287. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Martin, L.-A.; Farmer, I.; Johnston, S.R.D.; Ali, S.; Marshall, C.; Dowsett, M. Enhanced Estrogen Receptor (ER) α, ERBB2, and MAPK Signal Transduction Pathways Operate during the Adaptation of MCF-7 Cells to Long Term Estrogen Deprivation. J. Boil. Chem. 2003, 278, 30458–30468. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.G.; Ma, C.; Crowder, R.J.; Guintoli, T.; Phommaly, C.; Gao, F.; Lin, L.; Ellis, M.J. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011, 13, R21. [Google Scholar] [CrossRef]
- Park, S.; Song, J.; Joe, C.O.; Shin, I. Akt stabilizes estrogen receptor α with the concomitant reduction in its transcriptional activity. Cell. Signal. 2008, 20, 1368–1374. [Google Scholar] [CrossRef]
- Thomas, R.S.; Sarwar, N.; Phoenix, F.; Coombes, R.C.; Ali, S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J. Mol. Endocrinol. 2008, 40, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Cui, Y.; Herynk, M.H.; Rodriguez, C.-A.; Giordano, C.; Selever, J.; Beyer, A.; Andò, S.; Fuqua, A.W.S. Expression of the K303R estrogen receptor-α breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res. 2009, 69, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Iacopetta, D.; Covington, K.R.; Cui, Y.; Tsimelzon, A.; Beyer, A.; Andò, S.; Fuqua, S.A.; Anna, T. Phosphorylation of the mutant K303R estrogen receptor α at serine 305 affects aromatase inhibitor sensitivity. Oncogene 2010, 29, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.; Kok, M.; Michalides, R.; Fles, R.; Koornstra, R.; Wesseling, J.; Hauptmann, M.; Neefjes, J.; Peterse, J.; Stål, O.; et al. Phosphorylation of the oestrogen receptor α at serine 305 and prediction of tamoxifen resistance in breast cancer. J. Pathol. 2009, 217, 372–379. [Google Scholar] [CrossRef]
- Michalides, R.; Griekspoor, A.C.; Balkenende, A.; Verwoerd, D.; Janssen, L.; Jalink, K.; Floore, A.; Velds, A.; Veer, L.V.; Neefjes, J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell 2004, 5, 597–605. [Google Scholar] [CrossRef]
- Skliris, G.P.; Nugent, Z.; Watson, P.H.; Murphy, L.C. Estrogen Receptor Alpha Phosphorylated at Tyrosine 537 is Associated with Poor Clinical Outcome in Breast Cancer Patients Treated with Tamoxifen. Horm. Cancer 2010, 1, 215–221. [Google Scholar] [CrossRef]
- Skliris, G.P.; Nugent, Z.J.; Rowan, B.G.; Penner, C.R.; Watson, P.H.; Murphy, L.C. A phosphorylation code for oestrogen receptor-α predicts clinical outcome to endocrine therapy in breast cancer. Endocr. Relat. Cancer 2010, 17, 589–597. [Google Scholar] [CrossRef]
- Cheskis, B.; Greger, J.; Cooch, N.; McNally, C.; McLarney, S.; Lam, H.; Rutledge, S.; Mekonnen, B.; Hauze, D.; Nagpal, S. MNAR plays an important role in ERa activation of Src/MAPK and PI3K/Akt signaling pathways. Steroids 2008, 73, 901–905. [Google Scholar] [CrossRef]
- Zheng, F.F.; Wu, R.-C.; Smith, C.L.; O’Malley, B.W. Rapid Estrogen-Induced Phosphorylation of the SRC-3 Coactivator Occurs in an Extranuclear Complex Containing Estrogen Receptor. Mol. Cell. Boil. 2005, 25, 8273–8284. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef]
- Deng, B.; Tarhan, Y.E.; Ueda, K.; Ren, L.; Katagiri, T.; Park, J.-H.; Nakamura, Y. Critical Role of Estrogen Receptor Alpha O-Glycosylation by N-Acetylgalactosaminyltransferase 6 (GALNT6) in Its Nuclear Localization in Breast Cancer Cells. Neoplasia 2018, 20, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Faus, H.; Haendler, B. Post-translational modifications of steroid receptors. Biomed. Pharmacother. 2006, 60, 520–528. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Pesiri, V.; Leclercq, G.; Marino, M.; Acconcia, F. Palmitoylation Regulates 17β-Estradiol-Induced Estrogen Receptor-α Degradation and Transcriptional Activity. Mol. Endocrinol. 2012, 26, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tanaka, K.; Yan, J.; Li, J.; Peng, D.; Jiang, Y.; Yang, Z.; Barton, M.C.; Wen, H.; Shi, X. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 17284–17289. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Ramírez-Jarquín, J.O. Polyubiquitination inhibition of estrogen receptor alpha and its implications in breast cancer. World J. Clin. Oncol. 2018, 9, 60–70. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Boil. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Fan, M.; Nakshatri, H.; Nephew, K.P. Inhibiting Proteasomal Proteolysis Sustains Estrogen Receptor-α Activation. Mol. Endocrinol. 2004, 18, 2603–2615. [Google Scholar] [CrossRef][Green Version]
- Valley, C.C.; Solodin, N.M.; Powers, G.L.; Ellison, S.J.; Alarid, E.T. Temporal variation in estrogen receptor-α protein turnover in the presence of estrogen. J. Mol. Endocrinol. 2007, 40, 23–34. [Google Scholar] [CrossRef][Green Version]
- Kinyamu, H.K.; Collins, J.B.; Grissom, S.F.; Hebbar, P.B.; Archer, T.K. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol. Carcinog. 2008, 47, 845–885. [Google Scholar] [CrossRef]
- Powers, G.L.; Ellison-Zelski, S.J.; Casa, A.J.; Lee, A.V.; Alarid, E.T. Proteasome inhibition represses ERα gene expression in ER+ cells: A new link between proteasome activity and estrogen signaling in breast cancer. Oncogene 2009, 29, 1509–1518. [Google Scholar] [CrossRef]
- Powers, G.L.; Rajbhandari, P.; Solodin, N.M.; Bickford, B.; Alarid, E.T. The Proteasome Inhibitor Bortezomib Induces an Inhibitory Chromatin Environment at a Distal Enhancer of the Estrogen Receptor-α Gene. PLoS ONE 2013, 8, e81110. [Google Scholar] [CrossRef] [PubMed]
- Berry, N.B.; Fan, M.; Nephew, K.P. Estrogen receptor-alpha hinge-region lysines 302 and 303 regulate receptor degradation by the proteasome. Mol. Endocrinol. 2008, 22, 1535–1551. [Google Scholar] [CrossRef]
- Tecalco-Cruz, A.C.; Ramirez-Jarquin, J.O.; Cruz-Ramos, E. Estrogen Receptor Alpha and its Ubiquitination in Breast Cancer Cells. Curr. Drug Targets 2019, 20, 690–704. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zheng, Z.; Song, T.; Wei, C.; Ma, H.; Ma, Q.; Zhang, Y.; Xu, Y.; Shi, W.; Ye, Q.; et al. c-Abl regulates estrogen receptor α transcription activity through its stabilization by phosphorylation. Oncogene 2010, 29, 2238–2251. [Google Scholar] [CrossRef]
- Medunjanin, S.; Hermani, A.; De Servi, B.; Grisouard, J.; Rincke, G.; Mayer, D. Glycogen Synthase Kinase-3 Interacts with and Phosphorylates Estrogen Receptor α and Is Involved in the Regulation of Receptor Activity. J. Boil. Chem. 2005, 280, 33006–33014. [Google Scholar] [CrossRef]
- Hilmi, K.; Hussein, N.; Mendoza-Sanchez, R.; El-Ezzy, M.; Ismail, H.; Durette, C.; Bail, M.; Rozendaal, M.J.; Bouvier, M.; Thibault, P.; et al. Role of SUMOylation in Full Antiestrogenicity. Mol. Cell. Boil. 2012, 32, 3823–3837. [Google Scholar] [CrossRef] [PubMed]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.-C.; Corbo, L. Sumoylation of the Estrogen Receptor α Hinge Region Regulates Its Transcriptional Activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Murphy, L.C.; Seekallu, S.V.; Watson, P.H. Clinical significance of estrogen receptor phosphorylation. Endocr. Relat. Cancer 2010, 18, R1–R14. [Google Scholar] [CrossRef]
- Subramanian, K.; Jia, D.; Kapoor-Vazirani, P.; Powell, D.R.; Collins, R.E.; Sharma, D.; Peng, J.; Cheng, X.; Vertino, P.M. Regulation of Estrogen Receptor α by the SET7 Lysine Methyltransferase. Mol. Cell 2008, 30, 336–347. [Google Scholar] [CrossRef]
- Kim, M.Y.; Woo, E.M.; Chong, Y.T.E.; Homenko, D.R.; Kraus, W.L. Acetylation of Estrogen Receptor α by p300 at Lysines 266 and 268 Enhances the Deoxyribonucleic Acid Binding and Transactivation Activities of the Receptor. Mol. Endocrinol. 2006, 20, 1479–1493. [Google Scholar] [CrossRef]
- Williams, C.C.; Basu, A.; El-Gharbawy, A.; Carrier, L.; Smith, C.L.; Rowan, B.G. Identification of four novel phosphorylation sites in estrogen receptor α: Impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. 2009, 10, 36. [Google Scholar] [CrossRef]
- Xie, Y.; Li, G.; Chen, M.; Guo, X.; Tang, L.; Luo, X.; Wang, S.; Yi, W.; Dai, L.; Wang, J. Mutation screening of 10 cancer susceptibility genes in unselected breast cancer patients. Clin. Genet. 2017, 93, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct Acetylation of the Estrogen Receptor α Hinge Region by p300 Regulates Transactivation and Hormone Sensitivity. J. Boil. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef]
- Eakin, C.M.; MacCoss, M.J.; Finney, G.L.; Klevit, R.E. Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc. Nat. Acad. Sci. USA 2007, 104, 5794–5799. [Google Scholar] [CrossRef]
- Giordano, C.; Cui, Y.; Barone, I.; Ando, S.; Mancini, M.A.; Berno, V.; Fuqua, S.A.W. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor α and its phosphorylation at serine. Brea. Canc. Res. Treat. 2010, 119, 71–85. [Google Scholar] [CrossRef]
- Herynk, M.H.; Parra, I.; Cui, Y.; Beyer, A.; Wu, M.-F.; Hilsenbeck, S.G.; Fuqua, S.A. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin. Cancer Res. 2007, 13, 3235–3243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zwart, W.; Griekspoor, A.C.; Berno, V.; Lakeman, K.; Jalink, K.; Mancini, M.; Neefjes, J.; Michalides, R. PKA-induced resistance to tamoxifen is associated with an altered orientation of ERα towards co-activator SRC-1. EMBO J. 2007, 26, 3534–3544. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Deschenes, R.J.; Levin, E.R. DHHC-7 and -21 are palmitoylacyltransferases for sex steroid receptors. Mol. Boil. Cell 2012, 23, 188–199. [Google Scholar] [CrossRef]
- Traboulsi, T.; El Ezzy, M.; Dumeaux, V.; Audemard, E.; Mader, S. Role of SUMOylation in differential ERα transcriptional repression by tamoxifen and fulvestrant in breast cancer cells. Oncogene 2018, 38, 1019–1037. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, W.; Kaliappan, K.; Nawaz, Z.; Slingerland, J. ERα phosphorylation at Y537 by Src triggers E6-AP-ERα binding, ERα ubiquitylation, promoter occupancy, and target gene expression. Mol. Endocrinol. 2012, 26, 1567–1577. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.M.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gomez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Lupini, L.; Moretti, A.; Bassi, C.; Schirone, A.; Pedriali, M.; Querzoli, P.; Roncarati, R.; Frassoldati, A.; Negrini, M. High-sensitivity assay for monitoring ESR1 mutations in circulating cell-free DNA of breast cancer patients receiving endocrine therapy. Sci. Rep. 2018, 8, 4371. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Fan, S.; Hu, C.; Meng, Q.; Fuqua, S.A.; Pestell, R.G.; Tomita, Y.A.; Rosen, E.M. BRCA1 Regulates Acetylation and Ubiquitination of Estrogen Receptor-α. Molec. Endocr. 2010, 24, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.C.; Skliris, G.P.; Rowan, B.G.; Al-Dhaheri, M.; Williams, C.; Penner, C.; Troup, S.; Begic, S.; Parisien, M.; Watson, P.H. The relevance of phosphorylated forms of estrogen receptor in human breast cancer in vivo. J. Steroid Biochem. Mol. Boil. 2009, 114, 90–95. [Google Scholar] [CrossRef]
- Huderson, B.P.; Duplessis, T.T.; Williams, C.C.; Seger, H.C.; Marsden, C.G.; Pouey, K.J.; Hill, S.M.; Rowan, B.G. Stable inhibition of specific estrogen receptor α (ERα) phosphorylation confers increased growth, migration/invasion, and disruption of estradiol signaling in MCF-7 breast cancer cells. Endocrinology 2012, 153, 4144–4159. [Google Scholar] [CrossRef]
- Valley, C.C.; Métivier, R.; Solodin, N.M.; Fowler, A.M.; Mashek, M.T.; Hill, L.; Alarid, E.T. Differential Regulation of Estrogen-Inducible Proteolysis and Transcription by the Estrogen Receptor α N Terminus. Mol. Cell. Boil. 2005, 25, 5417–5428. [Google Scholar] [CrossRef]
- Arnott, J.A.; Martinkovich, S.; Planey, S.L.; Shah, D. Selective estrogen receptor modulators: Tissue specificity and clinical utility. Clin. Interv. Aging 2014, 9, 1437–1452. [Google Scholar] [CrossRef]
- Rayala, S.K.; Talukder, A.H.; Balasenthil, S.; Tharakan, R.; Barnes, C.J.; Wang, R.-A.; Aldaz, C.M.; Khan, S.A.; Kumar, R. P21-Activated Kinase 1 Regulation of Estrogen Receptor- Activation Involves Serine 305 Activation Linked with Serine 118 Phosphorylation. Cancer Res. 2006, 66, 1694–1701. [Google Scholar] [CrossRef]
- Zheng, X.Q.; Guo, J.P.; Yang, H.; Kanai, M.; He, L.L.; Li, Y.Y.; Koomen, J.M.; Minton, S.; Gao, M.; Ren, X.B.; et al. Aurora-A is a determinant of tamoxifen sensitivity through phosphorylation of ERα in breast cancer. Oncogene 2013, 33, 4985–4996. [Google Scholar] [CrossRef]
- Bostner, J.; Skoog, L.; Fornander, T.; Nordenskjold, B.; Stål, O. Estrogen Receptor- Phosphorylation at Serine 305, Nuclear p21-Activated Kinase 1 Expression, and Response to Tamoxifen in Postmenopausal Breast Cancer. Clin. Cancer Res. 2010, 16, 1624–1633. [Google Scholar] [CrossRef]
- Castoria, G.; Giovannelli, P.; Lombardi, M.; De Rosa, C.; Giraldi, T.; De Falco, A.; Barone, M.V.; Abbondanza, C.; Migliaccio, A.; Auricchio, F. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene 2012, 31, 4868–4877. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Miedel, M.T.; Ngo, M.; Hessenius, R.; Chen, N.; Wang, P.; Bahreini, A.; Li, Z.; Ding, Z.; Shun, T.Y.; et al. Clinically Observed Estrogen Receptor Alpha Mutations within the Ligand-Binding Domain Confer Distinguishable Phenotypes. Oncology 2018, 94, 176–189. [Google Scholar] [CrossRef]
- Sun, M.; Paciga, J.E.; Feldman, R.I.; Yuan, Z.-Q.; Coppola, D.; Lu, Y.Y.; Shelley, S.A.; Nicosia, S.V.; Cheng, J.Q. Phosphatidylinositol-3-OH kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor α (ERα) via interaction between ERα and PI3K. Cancer Res. 2001, 61, 5985–5991. [Google Scholar]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent Estrogen Receptor α Membrane Localization: Regulation by 17β-Estradiol. Mol. Boil. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, M.; Pestell, R.; Curran, E.M.; Welshons, W.V.; Fuqua, S.A. Phosphorylation of Estrogen Receptor α Blocks Its Acetylation and Regulates Estrogen Sensitivity. Cancer Res. 2004, 64, 9199–9208. [Google Scholar] [CrossRef]
- Fuqua, S.A.; Wiltschke, C.; Zhang, Q.X.; Borg, Å.; Castles, C.G.; Friedrichs, W.E.; Hopp, T.; Hilsenbeck, S.; Mohsin, S.; O’Connell, P.; et al. A hypersensitive estrogen receptor-α mutation in premalignant breast lesions. Cancer Res. 2000, 60, 4026–4029. [Google Scholar]
- Zhang, X.; Peng, D.; Xi, Y.; Yuan, C.; Sagum, C.A.; Klein, B.J.; Tanaka, K.; Wen, H.; Kutateladze, T.G.; Li, L.; et al. G9a-mediated methylation of ERα links the PHF20/MOF histone acetyltransferase complex to hormonal gene expression. Nat. Comm. 2016, 7, 10810. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.M.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Franken, A.; Honisch, E.; Reinhardt, F.; Meier-Stiegen, F.; Yang, L.; Jaschinski, S.; Esposito, I.; Alberter, B.; Polzer, B.; Huebner, H.; et al. Detection of ESR1 Mutations in Single Circulating Tumor Cells on Estrogen Deprivation Therapy but Not in Primary Tumors from Metastatic Luminal Breast Cancer Patients. J. Mol. Diagn. 2019, 22, 111–121. [Google Scholar] [CrossRef]
- Bostner, J.; Karlsson, E.; Pandiyan, M.J.; Westman, H.; Skoog, L.; Fornander, T.; Nordenskjöld, B.; Stål, O. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res. Treat. 2012, 137, 397–406. [Google Scholar] [CrossRef]
- Chen, M.; Cui, Y.-K.; Huang, W.-H.; Man, K.; Zhang, G.-J. Phosphorylation of estrogen receptor α at serine 118 is correlated with breast cancer resistance to tamoxifen. Oncol. Lett. 2013, 6, 118–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, J.; Sarwar, N.; Peston, D.; Kulinskaya, E.; Shousha, S.; Coombes, R.C.; Ali, S. Phosphorylation of Estrogen Receptor- at Ser167 Is Indicative of Longer Disease-Free and Overall Survival in Breast Cancer Patients. Clin. Cancer Res. 2007, 13, 5769–5776. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Nishio, M.; Toyama, T.; Sugiura, H.; Kondo, N.; Kobayashi, S.; Fujii, Y.; Iwase, H. Low phosphorylation of estrogen receptor α (ERα) serine 118 and high phosphorylation of ERα serine 167 improve survival in ER-positive breast cancer. Endocr. Rel. Canc. 2008, 15, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.C.; Niu, Y.; Snell, L.; Watson, P. Phospho-Serine-118 Estrogen Receptor- Expression Is Associated with Better Disease Outcome in Women Treated with Tamoxifen. Clin. Cancer Res. 2004, 10, 5902–5906. [Google Scholar] [CrossRef][Green Version]
- Sarwar, N.; Kim, J.; Jiang, J.; Peston, D.; Sinnett, H.; Madden, P.; Gee, J.M.; Nicholson, R.I.; Lykkesfeldt, A.E.; Shousha, S.; et al. Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endoc. Relat. Cancer 2006, 13, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Button, B.; Croessmann, S.; Chu, D.; Rosen, D.M.; Zabransky, D.J.; Dalton, W.B.; Cravero, K.; Kyker-Snowman, K.; Waters, I.; Karthikeyan, S.; et al. The estrogen receptor-alpha S118P variant does not affect breast cancer incidence or response to endocrine therapies. Breast Cancer Res. Treat. 2018, 174, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.; Zwart, W.; Holm, C.; Fles, R.; Hauptmann, M.; Veer, L.J.V.; Wessels, L.F.A.; Neefjes, J.; Stål, O.; Linn, S.C.; et al. PKA-induced phosphorylation of ERα at serine 305 and high PAK1 levels is associated with sensitivity to tamoxifen in ER-positive breast cancer. Breast Cancer Res. Treat. 2010, 125, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Toaldo, C.B.; Alexi, X.; Beelen, K.; Kok, M.; Hauptmann, M.; Jansen, M.P.H.M.; Berns, P.M.J.J.; Neefjes, J.; Linn, S.; Michalides, R.; et al. Protein Kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer 2015, 15, 588. [Google Scholar] [CrossRef]
- Busonero, C.; Leone, S.; Bartoloni, S.; Acconcia, F. Strategies to degrade estrogen receptor α in primary and ESR1 mutant-expressing metastatic breast cancer. Mol. Cell. Endocrinol. 2019, 480, 107–121. [Google Scholar] [CrossRef]
- Gao, J.J.; Cheng, J.; Bloomquist, E.; Sanchez, J.; Wedam, S.B.; Singh, H.; Amiri-Kordestani, L.; Ibrahim, A.; Sridhara, R.; Goldberg, K.B.; et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: A US Food and Drug Administration pooled analysis. Lancet Oncol. 2020, 21, 250–260. [Google Scholar] [CrossRef]
- Puyang, X.; Furman, C.; Zheng, G.Z.; Wu, Z.J.; Banka, D.; Aithal, K.; Agoulnik, S.; Bolduc, D.M.; Buonamici, S.; Caleb, B.; et al. Discovery of selective estrogen receptor covalent antagonists for the treatment of ERαWT and ERαMUT breast cancer. Cancer Discov. 2018, 8, 1176–1193. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.P.; Patel, M.R.; Armstrong, A.C.; Baird, R.D.; Jhaveri, K.; Hoch, M.; Klinowska, T.; Lindemann, J.P.O.; Morgan, S.R.; Schiavon, G.; et al. A first-in-human study of the new oral selective estrogen receptor degrader AZD9496 for ER+/HER2− advanced breast cancer. Clin. Cancer Res. 2018, 24, 3510–3518. [Google Scholar] [CrossRef]
- Robertson, J.F.R.; Evans, A.; Henschen, S.; Kirwan, C.C.; Jahan, A.; Kenny, L.M.; Dixon, J.M.; Schmid, P.; Kothari, A.; Mohamed, O.; et al. A randomized, open-label, pre-surgical, window of opportunity study comparing the pharmacodynamic effects of the novel oral SERD AZD9496 with fulvestrant in patients with newly diagnosed ER+ HER2-primary breast cancer. Clin. Cancer Res. 2020, 26, 4242–4249. [Google Scholar]
- Weir, H.M.; Bradbury, R.H.; Lawson, M.; Rabow, A.A.; Buttar, D.; Callis, R.J.; Curwen, J.O.; de Almeida, C.; Ballard, P.; Hulse, M.; et al. AZD9496: An oral estrogen receptor inhibitor that blocks the growth of ER-positive and ESR1-mutant breast tumors in preclinical models. Cancer Res. 2016, 76, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, Y.; Arnold, M.; Nakajima, H.; Zalcberg, J.; Maruta, H. Signal therapy of breast cancers by the HDAC inhibitor FK228 that blocks the activation of PAK1 and abrogates the tamoxifen-resistance. Cancer Boil. Ther. 2005, 4, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.H.; Casbard, A.; Carucci, M.; Cox, C.; Butler, R.; Alchami, F.; Madden, T.-A.; Bale, C.; Bezecny, P.; Joffe, J.; et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): A multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2020, 21, 345–357. [Google Scholar] [CrossRef]
- Haddad, T.C.; D’Assoro, A.; Suman, V.; Opyrchal, M.; Peethambaram, P.; Liu, M.C.; Goetz, M.P.; Ingle, J.N. Phase I trial to evaluate the addition of alisertib to fulvestrant in women with endocrine-resistant, ER+ metastatic breast cancer. Breast Cancer Res. Treat. 2017, 168, 639–647. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Borakove, M.; Feng, Y.; Wuest, W.M.; Koval, A.B.; Nikonova, A.S.; Serebriiskii, I.; Chernoff, J.; Borges, V.F.; Golemis, E.A.; et al. Combined inhibition of Aurora A and p21-activated kinase 1 as a new treatment strategy in breast cancer. Breast Cancer Res. Treat. 2019, 177, 369–382. [Google Scholar] [CrossRef]
- Paul, D.; Vukelja, S.J.; Holmes, F.A.; Blum, J.L.; McIntyre, K.J.; Lindquist, D.L.; Osborne, C.R.; Sanchez, I.J.; Goldschmidt, J.H.; Wang, Y.; et al. Randomized phase-II evaluation of letrozole plus dasatinib in hormone receptor positive metastatic breast cancer patients. NPJ Breast Cancer 2019, 5, 36–37. [Google Scholar] [CrossRef]
- Chen, Y.; Alvarez, E.A.; Azzam, D.J.; Wander, S.A.; Guggisberg, N.; Jorda, M.; Ju, Z.; Hennessy, B.T.; Slingerland, J. Combined Src and ER blockade impairs human breast cancer proliferation in vitro and in vivo. Breast Cancer Res. Treat. 2010, 128, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guggisberg, N.; Jorda, M.; Gonzalez-Angulo, A.; Hennessy, B.; Mills, G.B.; Tan, C.-K.; Slingerland, J.M. Combined Src and Aromatase Inhibition Impairs Human Breast Cancer Growth In vivo and Bypass Pathways Are Activated in AZD0530-Resistant Tumors. Clin. Cancer Res. 2009, 15, 3396–3405. [Google Scholar] [CrossRef] [PubMed]
- Moy, B.; Neven, P.; Lebrun, F.; Bellet, M.; Xu, B.; Sarosiek, T.; Chow, L.; Goss, P.; Zacharchuk, C.; Leip, E.; et al. Bosutinib in Combination With the Aromatase Inhibitor Exemestane: A Phase II Trial in Postmenopausal Women With Previously Treated Locally Advanced or Metastatic Hormone Receptor-Positive/HER2-Negative Breast Cancer. Oncology 2014, 19, 346–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moy, B.; Neven, P.; Lebrun, F.; Bellet, M.; Xu, B.; Sarosiek, T.; Chow, L.; Goss, P.; Zacharchuk, C.; Leip, E.; et al. Bosutinib in Combination With the Aromatase Inhibitor Letrozole: A Phase II Trial in Postmenopausal Women Evaluating First-Line Endocrine Therapy in Locally Advanced or Metastatic Hormone Receptor-Positive/HER2-Negative Breast Cancer. Oncology 2014, 19, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Adler, J.; Myers, N.; Biran, A.; Reuven, N.; Shaul, Y. Oncogenic addiction to high 26S proteasome level. Cell Death Dis. 2018, 9, 773. [Google Scholar] [CrossRef]
- Wong, D.J.; Nuyten, D.S.A.; Regev, A.; Lin, M.; Adler, A.S.; Segal, E.; Van De Vijver, M.J.; Chang, H.Y. Revealing Targeted Therapy for Human Cancer by Gene Module Maps. Cancer Res. 2008, 68, 369–378. [Google Scholar] [CrossRef]
- Thaler, S.; Thiede, G.; Hengstler, J.G.; Schad, A.; Schmidt, M.; Sleeman, J.P. The proteasome inhibitor Bortezomib (Velcade) as potential inhibitor of estrogen receptor-positive breast cancer. Int. J. Cancer 2015, 137, 686–697. [Google Scholar] [CrossRef]
- Maynadier, M.; Basile, I.; Gallud, A.; Gary-Bobo, M.; Garcia, M. Combination treatment with proteasome inhibitors and antiestrogens has a synergistic effect mediated by p21WAF1 in estrogen receptor-positive breast cancer. Oncol. Rep. 2016, 36, 1127–1134. [Google Scholar] [CrossRef]
- Yang, C.H.; Gonzalez-Angulo, A.M.; Reuben, J.M.; Booser, D.J.; Pusztai, L.; Krishnamurthy, S.; Esseltine, D.; Stec, J.; Broglio, K.R.; Islam, R.; et al. Bortezomib (VELCADE®) in metastatic breast cancer: Pharmacodynamics, biological effects, and prediction of clinical benefits. Ann. Oncol. 2006, 17, 813–817. [Google Scholar] [CrossRef]
- Adelson, K.; Ramaswamy, B.; Sparano, J.A.; Christos, P.J.; Wright, J.J.; Raptis, G.; Han, G.; Villalona-Calero, M.; Ma, C.X.; Hershman, D.; et al. Randomized phase II trial of fulvestrant alone or in combination with bortezomib in hormone receptor-positive metastatic breast cancer resistant to aromatase inhibitors: A New York Cancer Consortium trial. NPJ Breast Cancer 2016, 2, 16037. [Google Scholar] [CrossRef][Green Version]
- Ishii, Y.; Papa, L.; Bahadur, U.; Yue, Z.; Aguirre-Ghiso, J.; Shioda, T.; Waxman, S.; Germain, D. Bortezomib enhances the efficacy of fulvestrant by amplifying the aggregation of the estrogen receptor, which leads to a proapoptotic unfolded protein response. Clin. Cancer Res. 2011, 17, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Gu, G.; Chen, Y.; Rohira, A.D.; Lei, J.T.; Hamilton, R.A.; Yu, Y.; Lonard, D.M.; Wang, J.; Wang, S.-P.; et al. Proteomic profiling identifies key coactivators utilized by mutant ERα proteins as potential new therapeutic targets. Oncogene 2018, 37, 4581–4598. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Site | Posttranslational Modification (Mediator) | Effect | ESR1 Mutations # | ET Resistant | Frequency | Functionally Tested |
|---|---|---|---|---|---|---|
| Y52 | Phosphorylation (c-Abl) | Increased gene transactivation, increased DNA binding and dimerisation, rapid cell growth and increased invasive capacity [54] | None | N/A | N/A | N/A |
| S104 | Phosphorylation (Cyclin A/CDK2, MAPK ERK1/2, GSK3β) | Increased gene transactivation, tamoxifen sensitivity, ER stability in absence of E2 [31,55] | None | N/A | N/A | N/A |
| S106 | Phosphorylation (MAPK ERK1/2, GSK3β) | Increased gene transactivation, tamoxifen sensitivity, ER stability in absence of E2 [31,55] | None | N/A | N/A | N/A |
| S118 | Phosphorylation (CDK7, ERK1/2, IKKα, GSK3β, ILK, EGFR, IGF1R, DNA-PK, RET) | Increased stability [48] | None | N/A | N/A | N/A |
| S167 | Phosphorylation (p90, RSK1, S6 K1, Akt, IKKƐ, CK2) | Inhibits proteasomal degradation [30] | None | N/A | N/A | N/A |
| K171 | SUMOylation (SUMO3) | Repressed gene transactivation, antiestrogen sensitivity [56] | None | N/A | N/A | N/A |
| K180 | SUMOylation (SUMO3) | Repressed gene transactivation, antiestrogen sensitivity [56] | None | N/A | N/A | N/A |
| Y219 | Phosphorylation (c-Abl) | Enhanced gene transactivation, increased DNA binding and dimerisation, rapid cell growth and increased invasive capacity [54] | None | N/A | N/A | N/A |
| K235 | Methylation (SMYND2) | Repressed gene transactivation [44] | None | N/A | N/A | N/A |
| K266 | SUMOylation (SUMO-1) | Repressed gene transactivation [57] | None | N/A | N/A | N/A |
| Acetylation (p300) | Promotes DNA binding and transactivation capacity [58] | |||||
| Methylation (SET7, SMYND2) | Repressed gene transactivation [44] | |||||
| K268 | SUMOylation (SUMO-1) | Repressed gene transactivation [59] | None | N/A | N/A | N/A |
| Acetylation (p300) | Enhances DNA binding and gene transactivation [60] | |||||
| S282 | Phosphorylation (CK2) | Tamoxifen sensitivity, suppression of gene transactivation, ER stability [37,61] | S282C | Unknown | 0.3% (1/292) [62] | N/A |
| K299 | SUMOylation (SUMO1, SUMO2/3) | Repressed gene transactivation, antiestrogen sensitivity [56,57] | None | N/A | N/A | N/A |
| Acetylation (p300) | Not major target of acetylation, hinge lysines are preferentially acetylated [60,63] | |||||
| K302 | Ubiquitylation (BRCA1/BARD1) | ER degradation, induced by estrogen or fulvestrant [52,64] | None | N/A | N/A | N/A |
| SUMOylation (SUMO-1) | Repressed gene transactivation [59] | |||||
| Methylation (SET7/9, SMYD2) | ER stability, recruitment to promoter [59] | |||||
| K303 | Ubiquitylation (BRCA1/BARD1?) | ER degradation, induced by estrogen or fulvestrant [52,64] | K303R | Yes, resistant to AIs and tamoxifen [33,65] | 49.81% (133/267) of invasive BCa [66] | In combination with S305, promotes crosstalk with growth factor pathways, and confers resistance to AIs and tamoxifen [32,33,65] |
| SUMOylation (SUMO-1) | Enhances estrogen induced DNA binding and transcription [52] Repressed gene transactivation [57] | |||||
| Acetylation (p300) | Represses ER transactivation activity [63] | |||||
| S305 | Phosphorylation (PAK1, PKA) | Tamoxifen resistance In the presence of the K303R mutation, enhances crosstalk with IGF-1/IRS/Akt pathway and aromatase inhibitor resistance [33,34,35,61,67] | None | N/A | N/A | N/A |
| C447 | Palmitoylation (DHHC-7, DHHC-21) | Membrane localisation [68] | None | N/A | N/A | N/A |
| C451 | SUMOylation (SUMO3) | Sensitivity to fulvestrant and tamoxifen, through gene repression [69] | None | N/A | N/A | N/A |
| K472 | SUMOylation (SUMO3) | Repressed gene transactivation, antiestrogen sensitivity [56] | None | N/A | N/A | N/A |
| V534 | Ubiquitylation (E6-AP ligase) | ER degradation, induced by estrogen Subcellular localisation, gene transactivation and the degradation of the ER [70] | V534E | Unlikely | 1/616 (0.16%) metastatic BCa [25] | No effect, neither constitutively active nor inactivating [25] |
| Y537 | Ubiquitylation (E6-AP ligase) | ER degradation, induced by estrogen Subcellular localisation, gene transactivation and the degradation of the ER [70] | Y537C | Yes | 6/616 (0.97%) metastatic BCa [25] | Ligand independent, somewhat resistant to tamoxifen, fulvestrant and estrogen deprivation [25,71] |
| Phosphorylation (SRC) | Regulation of subcellular localisation, transcriptional activity and degradation of the ER [25,45] | Y537D | Unknown | 1/616 (0.16%) metastatic BCa [25] | Estrogen independent, increased activation of progesterone receptor [25] | |
| Y537H | Unknown | 1/56 (1.79%) cfDNA [72] | N/A | |||
| Y537N | Yes | 5/616 (0.81%) metastatic BCa [25] | Ligand independent, relatively resistant to tamoxifen, fulvestrant and estrogen deprivation [25,71] | |||
| Y537S | Yes | 13/616 (2.11%) metastatic BCa [25] | Constitutively active, adopts conformation that enhances coactivator binding, requires much higher concentrations of fulvestrant for inhibition of ER activities than WT [25] | |||
| S573 | Glycosylation (GALNT6) | Nuclear localisation, gene transactivation [41] | None | N/A | N/A | N/A |
| Treatment | Target | Experimental/Clinical Trial/Current Treatment | Outcome/Conclusion |
|---|---|---|---|
| Aromatase Inhibitors (Anastrozole, Exemestane, Letrozole) | Aromatase | Current | The proliferation marker, Ki67, was significantly suppressed by anastrozole (76%) after 2 weeks of treatment compared to tamoxifen (62%) and the combination (64%) [12] |
| Tamoxifen | ER (SERD) | Current | 5-yr adjuvant tamoxifen use results 47% reduction in recurrence [12] |
| Fulvestrant | ER (SERM | Current | Greater suppression of ER, PgR and Ki-67 was observed in the higher dose fulvestrant [12] |
| Cyclin-dependent kinase 4/6 inhibitors (CDKIs) | Cyclin-dependent kinase 4/6 | Current | CDKI treatment in combination with ET extends PFS by a median of 8.8 months [100] |
| H3B-5942 | ER (SERCA) | Experimental | Greater antiproliferative effect than fulvestrant. Has synergistic effect when combined with CDK4/6 and mTOR inhibitors [101] |
| AZD9496 | ER | Trial | Disease stabilisation [25,102,103,104] |
| Bortezomib | Proteasome inhibitor | Experimental | Inhibit cell growth of both ER+ and ER- cells [49,50,51] |
| FK228 | Histone deacetylase inhibitor | Trial | Combined with vorinostat and tamoxifen results in tumour regression [105,106] |
| Vorinostat | Histone deacetylase inhibitor | Trial | Show tumour regression with FK228 [106] |
| Capivasertib | AKT inhibitor | Trial | Patients who received capivasertib plus fulvestrant had a median PFS of 10.3 months compared to 4.8 months in patients who received a placebo plus fulvestrant, warranting further investigation, in a phase III trial [107] |
| Alisertib | Aurora Kinase A inhibitor | Trial | In combination with fulvestrant demonstrated anti-tumour activity [108] |
| FRAX1036 | PAK1 inhibitor | Experimental | Acts synergistically with alisertib with greater efficacy [109] |
| Dasatinib | SRC and AbI inhibitor | Experimental | In combination with letrozole was promising in 71% of patients [110] |
| Saracatinib (AZD0530) | SRC inhibitor | Experimental | In combination with fulvestrant had a greater effect at reducing proliferation than alone [111,112] |
| Bosutinib | SRC inhibitor | Experimental | Had an unfavourable risk-benefit ratio [113,114] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeffreys, S.A.; Powter, B.; Balakrishnar, B.; Mok, K.; Soon, P.; Franken, A.; Neubauer, H.; de Souza, P.; Becker, T.M. Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells 2020, 9, 2077. https://doi.org/10.3390/cells9092077
Jeffreys SA, Powter B, Balakrishnar B, Mok K, Soon P, Franken A, Neubauer H, de Souza P, Becker TM. Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells. 2020; 9(9):2077. https://doi.org/10.3390/cells9092077
Chicago/Turabian StyleJeffreys, Sarah A., Branka Powter, Bavanthi Balakrishnar, Kelly Mok, Patsy Soon, André Franken, Hans Neubauer, Paul de Souza, and Therese M. Becker. 2020. "Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability" Cells 9, no. 9: 2077. https://doi.org/10.3390/cells9092077
APA StyleJeffreys, S. A., Powter, B., Balakrishnar, B., Mok, K., Soon, P., Franken, A., Neubauer, H., de Souza, P., & Becker, T. M. (2020). Endocrine Resistance in Breast Cancer: The Role of Estrogen Receptor Stability. Cells, 9(9), 2077. https://doi.org/10.3390/cells9092077