Targeted Activation of T Cells with IL-2-Coupled Nanoparticles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. IL-2 and Its Initial Use in Cancer Therapy
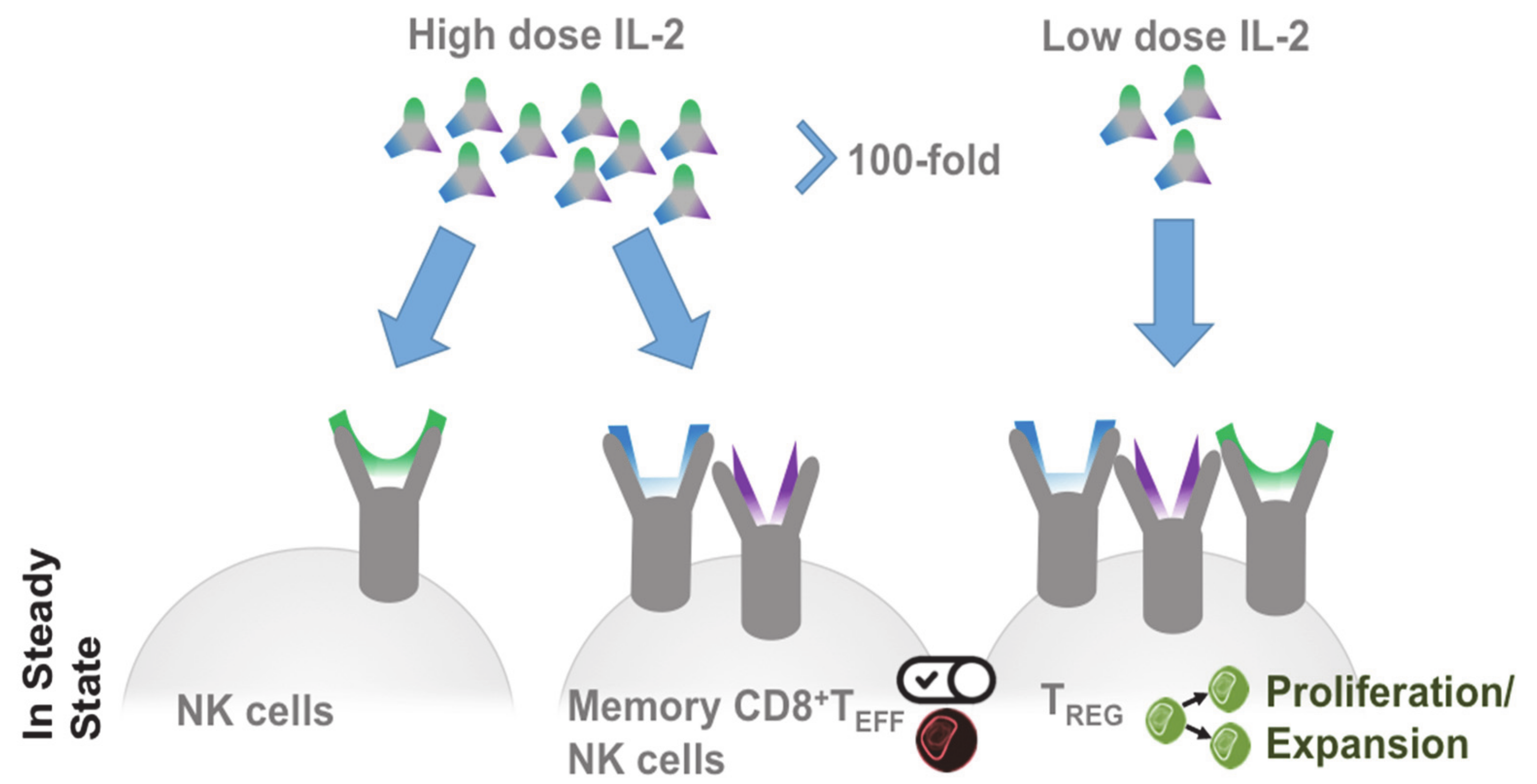
2.1. Producing and Responding Cells
2.2. Treg in Cancer
2.3. Targeted IL-2 Assignment
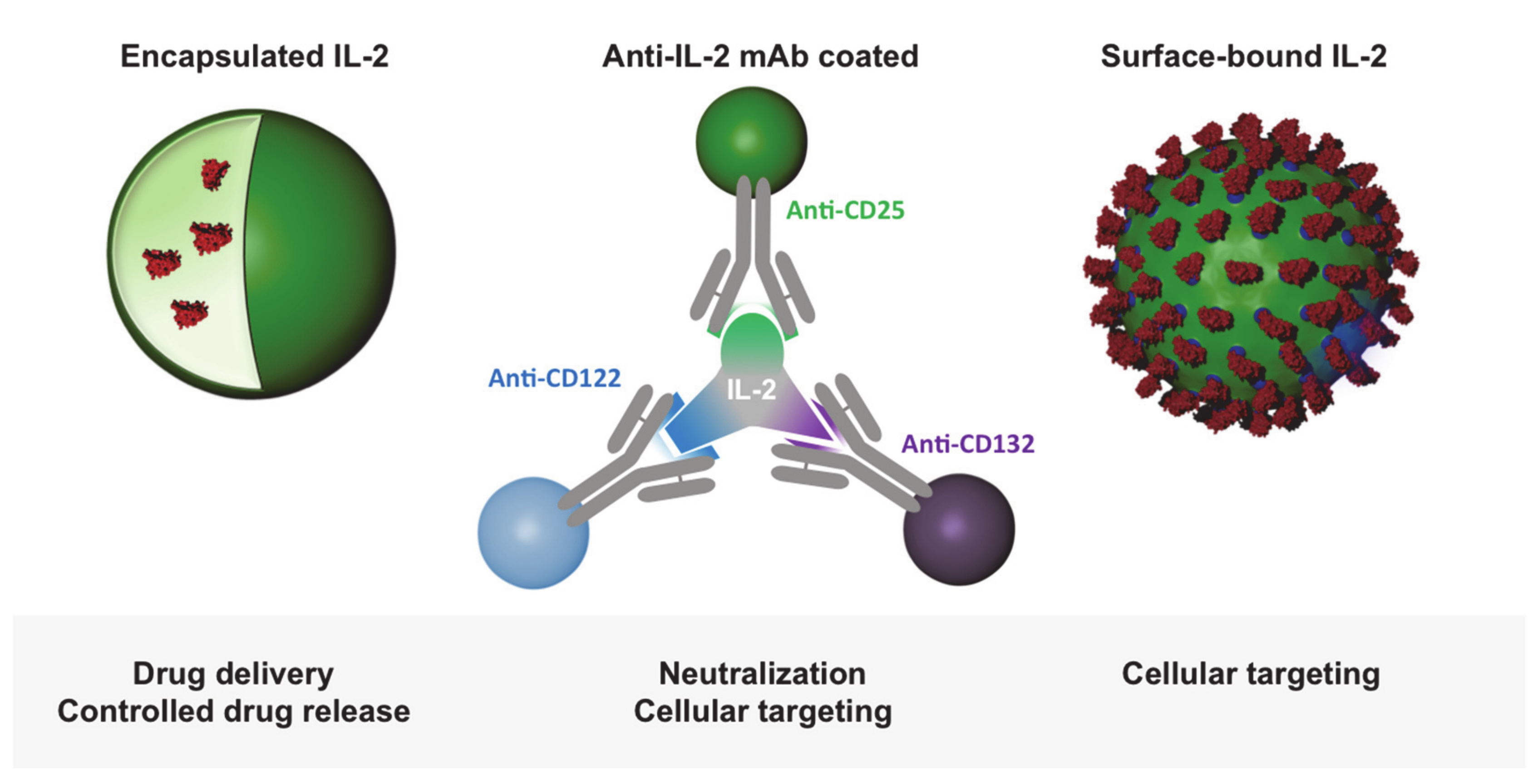
2.4. IL-2-Decorated Nanoparticles to Boost Melanoma Immunity—Potential Advantages and Pitfalls
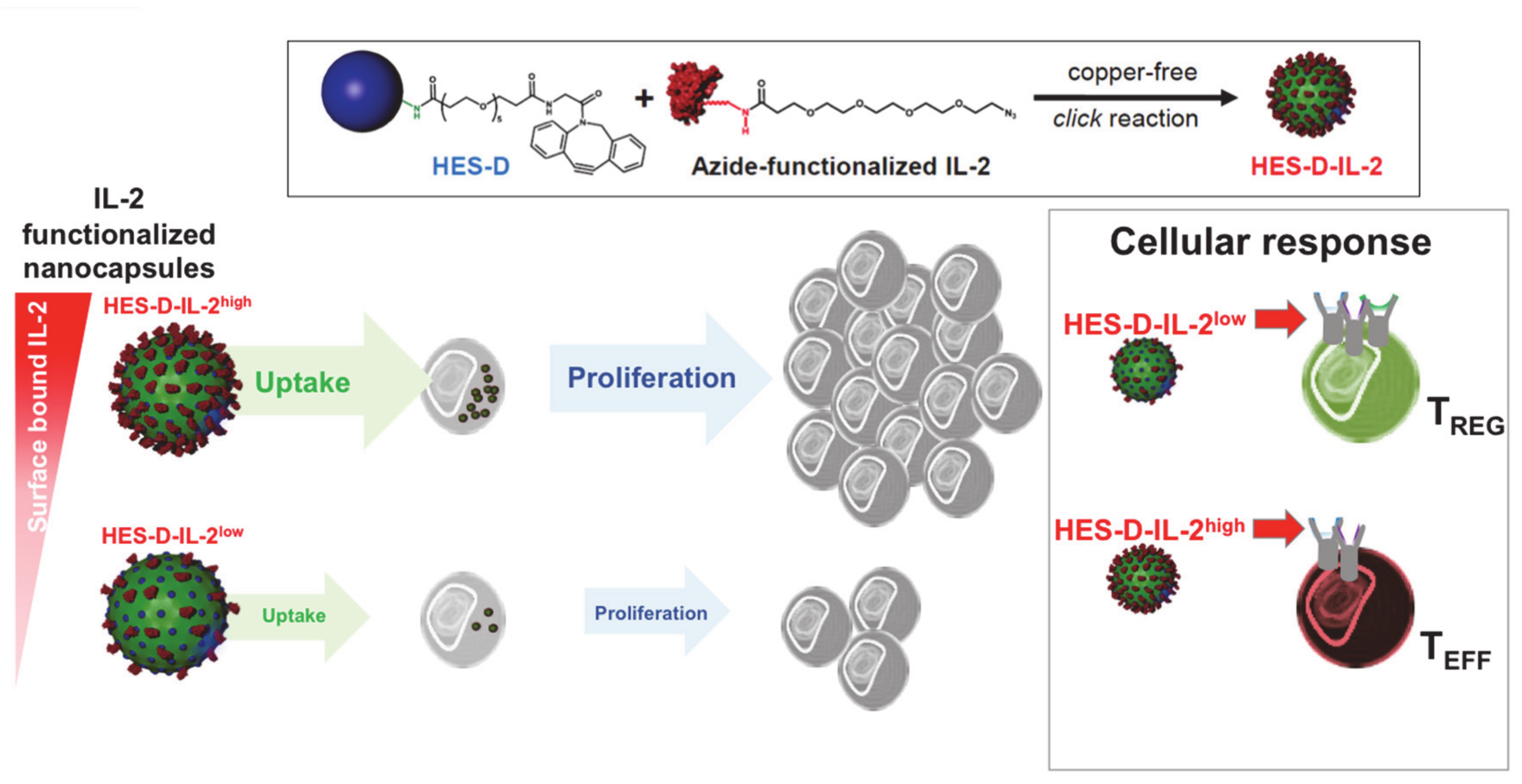
2.5. T Cell Activation through Delivery of Nanoparticle-Encapsulated IL-2
2.6. T Cell Targeting by IL-2-Functionalized Nanoparticles
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Al-Lawati, H.; Aliabadi, H.M.; Makhmalzadeh, B.S.; Lavasanifar, A. Nanomedicine for immunosuppressive therapy: Achievements in pre-clinical and clinical research. Expert Opin. Drug Deliv. 2018, 15, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.M.; Podojil, J.R.; Shea, L.D.; King, N.J.C.; Miller, S.D.; Getts, D.R. Overcoming challenges in treating autoimmuntity: Development of tolerogenic immune-modifying nanoparticles. Nanomedicine Nanotechnol. Biol. Med. 2019, 18, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Dane, E.L. Enhancing cancer immunotherapy with nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; White, D.E.; Steinberg, S.M. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: Identification of the antigens mediating response. Ann. Surg. 1998, 228, 307–319. [Google Scholar] [CrossRef]
- Rosenberg, S.A. IL-2: The first effective immunotherapy for human cancer. J. Immunol. 2014, 192, 5451–5458. [Google Scholar] [CrossRef]
- Klatzmann, D.; Abbas, A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015, 15, 283–294. [Google Scholar] [CrossRef]
- Tahvildari, M.; Dana, R. Low-Dose IL-2 Therapy in Transplantation, Autoimmunity, and Inflammatory Diseases. J. Immunol. 2019, 203, 2749–2755. [Google Scholar] [CrossRef]
- Arenas-Ramirez, N.; Woytschak, J.; Boyman, O. Interleukin-2: Biology, Design and Application. Trends Immunol. 2015, 36, 763–777. [Google Scholar] [CrossRef]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nature 1983, 302, 305–310. [Google Scholar] [CrossRef]
- Delorme, E.J.; Alexander, P. Treatment of primary fibrosarcoma in the rat with immune lymphocytes. Lancet Lond. Engl. 1964, 2, 117–120. [Google Scholar] [CrossRef]
- Mulé, J.J.; Yang, J.; Shu, S.; Rosenberg, S.A. The anti-tumor efficacy of lymphokine-activated killer cells and recombinant interleukin 2 in vivo: Direct correlation between reduction of established metastases and cytolytic activity of lymphokine-activated killer cells. J. Immunol. 1986, 136, 3899–3909. [Google Scholar] [PubMed]
- Lotze, M.T.; Frana, L.W.; Sharrow, S.O.; Robb, R.J.; Rosenberg, S.A. In vivo administration of purified human interleukin 2. I. Half-life and immunologic effects of the Jurkat cell line-derived interleukin 2. J. Immunol. 1985, 134, 157–166. [Google Scholar]
- Weiss, G.R.; Margolin, K.A.; Aronson, F.R.; Sznol, M.; Atkins, M.B.; Dutcher, J.P.; Gaynor, E.R.; Boldt, D.H.; Doroshow, J.H.; Bar, M.H. A randomized phase II trial of continuous infusion interleukin-2 or bolus injection interleukin-2 plus lymphokine-activated killer cells for advanced renal cell carcinoma. J. Clin. Oncol. 1992, 10, 275–281. [Google Scholar] [CrossRef]
- Krieg, C.; Létourneau, S.; Pantaleo, G.; Boyman, O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11906–11911. [Google Scholar] [CrossRef] [PubMed]
- Lentsch, A.B.; Miller, F.N.; Edwards, M.J. Mechanisms of leukocyte-mediated tissue injury induced by interleukin-2. Cancer Immunol. Immunother. CII 1999, 47, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Sherry, R.M.; Steinberg, S.M.; Topalian, S.L.; Schwartzentruber, D.J.; Hwu, P.; Seipp, C.A.; Rogers-Freezer, L.; Morton, K.E.; White, D.E.; et al. Randomized Study of High - Dose and Low - Dose Inter leukin - 2 in Patients With Metastatic Renal Cancer. J. Clin. Oncol. 2003, 21, 3127–3132. [Google Scholar] [CrossRef]
- Granucci, F.; Vizzardelli, C.; Pavelka, N.; Feau, S.; Persico, M.; Virzi, E.; Rescigno, M.; Moro, G.; Ricciardi-Castagnoli, P. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat. Immunol. 2001, 2, 882–888. [Google Scholar] [CrossRef]
- Wojciechowski, W.; Harris, D.P.; Sprague, F.; Mousseau, B.; Makris, M.; Kusser, K.; Honjo, T.; Mohrs, K.; Mohrs, M.; Randall, T.; et al. Cytokine-Producing Effector B Cells Regulate Type 2 Immunity to H. polygyrus. Immunity 2009, 30, 421–433. [Google Scholar] [CrossRef]
- Owen, D.L.; Mahmud, S.A.; Vang, K.B.; Kelly, R.M.; Blazar, B.R.; Smith, K.A.; Farrar, M.A. Identification of Cellular Sources of IL-2 Needed for Regulatory T Cell Development and Homeostasis. J. Immunol. 2018, 200, 3926–3933. [Google Scholar] [CrossRef]
- Naramura, M.; Hu, R.-J.; Gu, H. Mice with a Fluorescent Marker for Interleukin 2 Gene Activation. Immunity 1998, 9, 209–216. [Google Scholar] [CrossRef]
- Serfling, E.; Avots, A.; Neumann, M. The architecture of the interleukin-2 promoter: A reflection of T lymphocyte activation. Biochim. Biophys. Acta 1995, 1263, 181–200. [Google Scholar] [CrossRef]
- Kim, H.P.; Imbert, J.; Leonard, W.J. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev. 2006, 17, 349–366. [Google Scholar] [CrossRef]
- Leung, D.T.; Morefield, S.; Willerford, D.M. Regulation of lymphoid homeostasis by IL-2 receptor signals in vivo. J. Immunol. 2000, 164, 3527–3534. [Google Scholar] [CrossRef] [PubMed]
- Wuest, S.C.; Edwan, J.; Martin, J.F.; Han, S.; Perry, J.S.A.; Cartagena, C.M.; Matsuura, E.; Maric, D.; Waldmann, T.A.; Bielekova, B. A vital role for IL-2 trans-presentation in DC-mediated T cell activation in humans as revealed by daclizumab therapy. Nat. Med. 2011, 17, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Sadlack, B.; Merz, H.; Schorle, H.; Schimpl, A.; Feller, A.C.; Horak, I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993, 75, 253–261. [Google Scholar] [CrossRef]
- Suzuki, H.; Kündig, T.M.; Furlonger, C.; Wakeham, A.; Timms, E.; Matsuyama, T.; Schmits, R.; Simard, J.J.; Ohashi, P.S.; Griesser, H. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science 1995, 268, 1472–1476. [Google Scholar] [CrossRef]
- Willerford, D.M.; Chen, J.; Ferry, J.A.; Davidson, L.; Ma, A.; Alt, F.W. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 1995, 3, 521–530. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Ohkura, N.; Hamaguchi, M.; Morikawa, H.; Sugimura, K.; Tanaka, A.; Ito, Y.; Osaki, M.; Tanaka, Y.; Yamashita, R.; Nakano, N.; et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity 2012, 37, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Yu, A.; Vincek, V.; Scibelli, P.; Kong, L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity 2002, 17, 167–178. [Google Scholar] [CrossRef]
- Dooms, H.; Wolslegel, K.; Lin, P.; Abbas, A.K. Interleukin-2 enhances CD4+ T cell memory by promoting the generation of IL-7R alpha-expressing cells. J. Exp. Med. 2007, 204, 547–557. [Google Scholar] [CrossRef]
- Teague, R.M.; Tempero, R.M.; Thomas, S.; Murali-Krishna, K.; Nelson, B.H. Proliferation and differentiation of CD8+ T cells in the absence of IL-2/15 receptor beta-chain expression or STAT5 activation. J. Immunol. 2004, 173, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Granucci, F.; Zanoni, I.; Feau, S.; Ricciardi-Castagnoli, P. Dendritic cell regulation of immune responses: A new role for interleukin 2 at the intersection of innate and adaptive immunity. EMBO J. 2003, 22, 2546–2551. [Google Scholar] [CrossRef]
- Malek, T.R. The biology of interleukin-2. Annu. Rev. Immunol. 2008, 26, 453–479. [Google Scholar] [CrossRef]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef]
- Koreth, J.; Matsuoka, K.; Kim, H.T.; McDonough, S.M.; Bindra, B.; Alyea, E.P.; Armand, P.; Cutler, C.; Ho, V.T.; Treister, N.S.; et al. Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease. N. Engl. J. Med. 2011, 365, 2055–2066. [Google Scholar] [CrossRef]
- Hartemann, A.; Bensimon, G.; Payan, C.A.; Jacqueminet, S.; Bourron, O.; Nicolas, N.; Fonfrede, M.; Rosenzwajg, M.; Bernard, C.; Klatzmann, D. Low-dose interleukin 2 in patients with type 1 diabetes: A phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2013, 1, 295–305. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Churlaud, G.; Mallone, R.; Six, A.; Dérian, N.; Chaara, W.; Lorenzon, R.; Long, S.A.; Buckner, J.H.; Afonso, G.; et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J. Autoimmun. 2015, 58, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Nasser, A.A.; Ku, S.; Castillo-Caro, P.; Hazrat, Y.; Wu, M.-F.; Liu, H.; Melenhorst, J.; Barrett, A.J.; Ito, S.; Foster, A.; et al. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin. Cancer. 2014, 20, 2215–2225. [Google Scholar] [CrossRef] [PubMed]
- Woo, E.Y.; Chu, C.S.; Goletz, T.J.; Schlienger, K.; Yeh, H.; Coukos, G.; Rubin, S.C.; Kaiser, L.R.; June, C.H. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001, 61, 4766–4772. [Google Scholar] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.-G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef]
- Klages, K.; Mayer, C.T.; Lahl, K.; Loddenkemper, C.; Teng, M.W.L.; Ngiow, S.F.; Smyth, M.J.; Hamann, A.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res. 2010, 70, 7788–7799. [Google Scholar] [CrossRef]
- Li, X.; Kostareli, E.; Suffner, J.; Garbi, N.; Hämmerling, G.J. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur. J. Immunol. 2010, 40, 3325–3335. [Google Scholar] [CrossRef]
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar] [CrossRef]
- Erdman, S.E.; Poutahidis, T. Cancer inflammation and regulatory T cells. Int. J. Cancer 2010, 127, 768–779. [Google Scholar] [CrossRef]
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: The paradox of colorectal cancer. Cancer Immunol. Immunother. CII 2011, 60, 909–918. [Google Scholar] [CrossRef]
- Frey, D.M.; Droeser, R.A.; Viehl, C.T.; Zlobec, I.; Lugli, A.; Zingg, U.; Oertli, D.; Kettelhack, C.; Terracciano, L.; Tornillo, L. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int. J. Cancer 2010, 126, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Droeser, R.; Zlobec, I.; Kilic, E.; Güth, U.; Heberer, M.; Spagnoli, G.; Oertli, D.; Tapia, C. Differential pattern and prognostic significance of CD4+, FOXP3+ and IL-17+ tumor infiltrating lymphocytes in ductal and lobular breast cancers. BMC Cancer 2012, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef] [PubMed]
- Létourneau, S.; van Leeuwen, E.M.M.; Krieg, C.; Martin, C.; Pantaleo, G.; Sprent, J.; Surh, C.D.; Boyman, O. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc. Natl. Acad. Sci. USA 2010, 107, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Ramirez, N.; Zou, C.; Popp, S.; Zingg, D.; Brannetti, B.; Wirth, E.; Calzascia, T.; Kovarik, J.; Sommer, L.; Zenke, G.; et al. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci. Transl. Med. 2016, 8, 367ra166. [Google Scholar] [CrossRef]
- Margolin, K.; Atkins, M.B.; Dutcher, J.P.; Ernstoff, M.S.; Smith, J.W.; Clark, J.I.; Baar, J.; Sosman, J.; Weber, J.; Lathia, C.; et al. Phase I trial of BAY 50-4798, an interleukin-2-specific agonist in advanced melanoma and renal cancer. Clin. Cancer Res. 2007, 13, 3312–3319. [Google Scholar] [CrossRef]
- Pretto, F.; Elia, G.; Castioni, N.; Neri, D. Preclinical evaluation of IL2-based immunocytokines supports their use in combination with dacarbazine, paclitaxel and TNF-based immunotherapy. Cancer Immunol. Immunother. CII 2014, 63, 901–910. [Google Scholar] [CrossRef]
- Levin, A.M.; Bates, D.L.; Ring, A.M.; Krieg, C.; Lin, J.T.; Su, L.; Moraga, I.; Raeber, M.E.; Bowman, G.R.; Novick, P.; et al. Exploiting a natural conformational switch to engineer an interleukin-2 “superkine”. Nature 2012, 484, 529–533. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef]
- Ekladious, I.; Colson, Y.L.; Grinstaff, M.W. Polymer-drug conjugate therapeutics: Advances, insights and prospects. Nat. Rev. Drug Discov. 2019, 18, 273–294. [Google Scholar] [CrossRef]
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.-E.; Janssen, L.M.E.; Chesson, B.C.; Creasy, C.A.; Forget, M.-A.; Kahn, L.M.S.; Pazdrak, B.; et al. Bempegaldesleukin selectively depletes intratumoral Tregs and potentiates T cell-mediated cancer therapy. Nat. Commun. 2020, 11, 661. [Google Scholar] [CrossRef]
- Vazquez-Lombardi, R.; Loetsch, C.; Zinkl, D.; Jackson, J.; Schofield, P.; Deenick, E.K.; King, C.; Phan, T.G.; Webster, K.E.; Sprent, J.; et al. Potent antitumour activity of interleukin-2-Fc fusion proteins requires Fc-mediated depletion of regulatory T-cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, E.; Hong, S.-W.; Kim, D.; Eunju, O.; Sprent, J.; Im, S.-H.; Lee, Y.J.; Surh, C.D. TCB2, a new anti-human interleukin-2 antibody, facilitates heterodimeric IL-2 receptor signaling and improves anti-tumor immunity. OncoImmunology 2020, 9, 1681869. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Petschauer, J.S.; Madden, A.J.; Zamboni, W.C. Nanoparticles and the mononuclear phagocyte system: Pharmacokinetics and applications for inflammatory diseases. Curr. Rheumatol. Rev. 2014, 10, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, K.M.; MacParland, S.A.; Ma, X.-Z.; Spetzler, V.N.; Echeverri, J.; Ouyang, B.; Fadel, S.M.; Sykes, E.A.; Goldaracena, N.; Kaths, J.M.; et al. Mechanism of hard-nanomaterial clearance by the liver. Nat. Mater. 2016, 15, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Burdick, L.M.; Somani, N.; Somani, A.-K. Type I IFNs and their role in the development of autoimmune diseases. Expert Opin. Drug Saf. 2009, 8, 459–472. [Google Scholar] [CrossRef]
- Becker, C.; Bopp, T.; Steinbrink, K. Interferon α interferes with immunological tolerance. OncoImmunology 2013, 2. [Google Scholar] [CrossRef]
- Khan, S.; Gerber, D.E. Autoimmunity, checkpoint inhibitor therapy and immune-related adverse events: A review. Semin. Cancer Biol. 2020, 64, 93–101. [Google Scholar] [CrossRef]
- Das, P.K.; Wijngaard, R.M.J.G.J.v.d.; Wankowicz-Kalinska, A.; Poole, I.C.L. A symbiotic concept of autoimmunity and tumour immunity: Lessons from vitiligo. Trends Immunol. 2001, 22, 130–136. [Google Scholar] [CrossRef]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- McHugh, M.D.; Park, J.; Uhrich, R.; Gao, W.; Horwitz, D.A.; Fahmy, T.M. Paracrine co-delivery of TGF-β and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials 2015, 59, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Bickerton, S.; Koss, M.; Fahmy, T.M.; La Cava, A. Suppression of Murine Lupus by CD4+ and CD8+ Treg Cells Induced by T Cell-Targeted Nanoparticles Loaded With Interleukin-2 and Transforming Growth Factor β. Arthritis Rheumatol. Hoboken NJ 2019, 71, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Votavova, P.; Tomala, J.; Subr, V.; Strohalm, J.; Ulbrich, K.; Rihova, B.; Kovar, M. Novel IL-2-Poly(HPMA)Nanoconjugate Based Immunotherapy. J. Biomed. Nanotechnol. 2015, 11, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tang, C.; Yin, C. Co-delivery of doxorubicin and interleukin-2 via chitosan based nanoparticles for enhanced antitumor efficacy. Acta Biomater. 2017, 47, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Fadel, T.R.; Sharp, F.A.; Vudattu, N.; Ragheb, R.; Garyu, J.; Kim, D.; Hong, E.; Li, N.; Haller, G.L.; Pfefferle, L.D.; et al. A carbon nanotube-polymer composite for T-cell therapy. Nat. Nanotechnol. 2014, 9, 639–647. [Google Scholar] [CrossRef]
- Liu, M.; Miao, T.; Zhu, H.; Symonds, A.L.J.; Li, L.; Schurich, A.; Maini, M.K.; Zhang, J.; Kennedy, P.T.F.; Li, S.; et al. IL-2-engineered nano-APC effectively activates viral antigen-mediated T cell responses from chronic hepatitis B virus-infected patients. J. Immunol. 2012, 188, 1534–1543. [Google Scholar] [CrossRef]
- Wojta-Stremayr, D.; Neunkirchner, A.; Srinivasan, B.; Trapin, D.; Schmetterer, K.G.; Pickl, W.F. CD8+ T Cell Fate and Function Influenced by Antigen-Specific Virus-Like Nanoparticles Co-Expressing Membrane Tethered IL-2. PloS One 2015, 10, e0126034. [Google Scholar] [CrossRef]
- Konigsberg, P.J.; Godtel, R.; Kissel, T.; Richer, L.L. The development of IL-2 conjugated liposomes for therapeutic purposes. Biochim. Biophys. Acta 1998, 1370, 243–251. [Google Scholar] [CrossRef]
- Zheng, Y.; Stephan, M.T.; Gai, S.A.; Abraham, W.; Shearer, A.; Irvine, D.J. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J. Control. 2013, 172, 426–435. [Google Scholar] [CrossRef]
- Frick, S.U.; Domogalla, M.P.; Baier, G.; Wurm, F.R.; Mailänder, V.; Landfester, K.; Steinbrink, K. Interleukin-2 Functionalized Nanocapsules for T Cell-Based Immunotherapy. ACS Nano 2016, 10, 9216–9226. [Google Scholar] [CrossRef]
- Baier, G.; Baumann, D.; Siebert, J.M.; Musyanovych, A.; Mailänder, V.; Landfester, K. Suppressing unspecific cell uptake for targeted delivery using hydroxyethyl starch nanocapsules. Biomacromolecules 2012, 13, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Duprez, V.; Ferrer, M.; Cornet, V.; Olive, D.; Dautry-Varsat, A. Modulation of interleukin 2 internalization and interleukin 2-dependent cell growth by antireceptor antibodies. J. Biol. Chem. 1991, 266, 1497–1501. [Google Scholar] [PubMed]
- Tonigold, M.; Simon, J.; Estupiñán, D.; Kokkinopoulou, M.; Reinholz, J.; Kintzel, U.; Kaltbeitzel, A.; Renz, P.; Domogalla, M.P.; Steinbrink, K.; et al. Pre-adsorption of antibodies enables targeting of nanocarriers despite a biomolecular corona. Nat. Nanotechnol. 2018, 13, 862–869. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raker, V.K.; Becker, C.; Landfester, K.; Steinbrink, K. Targeted Activation of T Cells with IL-2-Coupled Nanoparticles. Cells 2020, 9, 2063. https://doi.org/10.3390/cells9092063
Raker VK, Becker C, Landfester K, Steinbrink K. Targeted Activation of T Cells with IL-2-Coupled Nanoparticles. Cells. 2020; 9(9):2063. https://doi.org/10.3390/cells9092063
Chicago/Turabian StyleRaker, Verena K., Christian Becker, Katharina Landfester, and Kerstin Steinbrink. 2020. "Targeted Activation of T Cells with IL-2-Coupled Nanoparticles" Cells 9, no. 9: 2063. https://doi.org/10.3390/cells9092063
APA StyleRaker, V. K., Becker, C., Landfester, K., & Steinbrink, K. (2020). Targeted Activation of T Cells with IL-2-Coupled Nanoparticles. Cells, 9(9), 2063. https://doi.org/10.3390/cells9092063