Nucleic Acid-Based Approaches for Tumor Therapy




Abstract
1. Introduction
2. Nucleic Acid-Based Strategies to Induce Adaptive Anti-Tumor Responses
2.1. Clinical Trials Using Nucleic Acid-Based Vaccines for Tumor Therapy
2.1.1. pDNA Vaccines
2.1.2. mRNA Vaccines
2.2. Optimization Strategies for Nucleic Acid-Based Vaccines
2.2.1. Antigen
2.2.2. Adjuvant
2.2.3. Inhibition of Regulatory Proteins in APC
2.2.4. Structural Optimization of pDNA Vaccines
Expression Units
Size Reduction
Nuclear Transfer
Transcriptional Regulation
2.2.5. NPs for APC-Focused Delivery of Nucleic Acids
NP Size and Surface Characteristics Affecting Biodistribution
NP Types Suitable for APC Transfection
Administration Routes
Targeting of APC
3. Inhibition of Regulatory Immune Cells
3.1. Inhibition of Treg by RNA Interference
3.2. Strategies for MDSC Reprograming and Depletion
3.3. Inhibition of Treg and MDSC by Tumor-Directed Approaches
4. Generation of T Cells and NK Cells Expressing CARs for Tumor Therapy
5. Manipulating the TME Using Therapeutic Nucleic Acids
5.1. Modulation of Intratumoral Signaling by Nucleic Acids
5.2. Nucleic Acid-Mediated Immune Checkpoint Inhibition and T Cell Stimulation
5.3. Multi-Faceted Combat of Cancer by Oncolytic Virotherapy
5.4. Nucleic Acid-Based TLR Agonists to Boost Anti-Tumor Immune Response
5.5. Tumor Suppression by RNA Interference
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Think globally about cancer. Nat. Med. 2019, 25, 351. [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Rosenberg, S.A. A New Era for Cancer Immunotherapy Based on the Genes that Encode Cancer Antigens. Immunity 1999, 10, 281–287. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- Tariman, J.D. Changes in Cancer Treatment: Mabs, Mibs, Mids, Nabs, and Nibs. Nurs. Clin. North Am. 2017, 52, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Shojaosadati, S.A. Multifunctional nanoparticles for cancer immunotherapy. Hum. Vaccin. Immunother. 2016, 12, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Sami, H.; Ogris, M. Biopharmaceuticals and gene vectors opening new avenues in cancer immune therapy. Ther. Deliv. 2016, 7, 419–422. [Google Scholar] [CrossRef]
- Emens, L.A.; Ascierto, P.A.; Darcy, P.K.; Demaria, S.; Eggermont, A.M.M.; Redmond, W.L.; Seliger, B.; Marincola, F.M. Cancer immunotherapy: Opportunities and challenges in the rapidly evolving clinical landscape. Eur. J. Cancer 2017, 81, 116–129. [Google Scholar] [CrossRef]
- Rangel-Sosa, M.M.; Aguilar-Córdova, E.; Rojas-Martínez, A. Immunotherapy and gene therapy as novel treatments for cancer. Colomb. Med. (Cali) 2017, 48, 138–147. [Google Scholar] [CrossRef]
- Song, W.; Musetti, S.N.; Huang, L. Nanomaterials for cancer immunotherapy. Biomaterials 2017, 148, 16–30. [Google Scholar] [CrossRef]
- Musetti, S.; Huang, L. Nanoparticle-Mediated Remodeling of the Tumor Microenvironment to Enhance Immunotherapy. ACS Nano 2018, 12, 11740–11755. [Google Scholar] [CrossRef]
- Sau, S.; Alsaab, H.O.; Bhise, K.; Alzhrani, R.; Nabil, G.; Iyer, A.K. Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment. J. Controlled Release 2018, 274, 24–34. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, Y.; Zhang, X.; Fu, J.; Xing, X.; Wang, C.; Gao, L.; Liu, Y.; Shi, L. Potential applications of nanoparticles for tumor microenvironment remodeling to ameliorate cancer immunotherapy. Int. J. Pharm. 2019, 570, 118636. [Google Scholar] [CrossRef]
- Luo, Q.; Zhang, L.; Luo, C.; Jiang, M. Emerging strategies in cancer therapy combining chemotherapy with immunotherapy. Cancer Lett. 2019, 454, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Salvioni, L.; Rizzuto, A.M.; Bertolini, A.J.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty Years of Cancer Nanomedicine: Success, Frustration, and Hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lammers, T. Combining Nanomedicine and Immunotherapy. Acc. Chem. Res. 2019, 52, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar] [PubMed]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Wilky, B.A. Immune checkpoint inhibitors: The linchpins of modern immunotherapy. Immunol. Rev. 2019, 290, 6–23. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, C.; Chen, G.; Hu, Q.; Gu, Z. Delivery Strategies for Immune Checkpoint Blockade. Adv. Healthc. Mater. 2018, 7, e1800424. [Google Scholar] [CrossRef]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Hobernik, D.; Bros, M. DNA Vaccines—How Far From Clinical Use? Int. J. Mol. Sci. 2018, 19, 3605. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.J.; Nabel, E.G.; Yang, Z.Y.; Fox, B.A.; Plautz, G.E.; Gao, X.; Huang, L.; Shu, S.; Gordon, D.; Chang, A.E. Direct gene transfer with DNA-liposome complexes in melanoma: Expression, biologic activity, and lack of toxicity in humans. Proc. Natl. Acad. Sci. 1993, 90, 11307. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S. Improving cancer immunotherapy through nanotechnology. Nat. Rev. Cancer 2019, 19, 587–602. [Google Scholar] [CrossRef]
- Shae, D.; Baljon, J.J.; Wehbe, M.; Becker, K.W.; Sheehy, T.L.; Wilson, J.T. At the bench: Engineering the next generation of cancer vaccines. J. Leukoc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sadeghzadeh, M.; Bornehdeli, S.; Mohahammadrezakhani, H.; Abolghasemi, M.; Poursaei, E.; Asadi, M.; Zafari, V.; Aghebati-Maleki, L.; Shanehbandi, D. Dendritic cell therapy in cancer treatment; The state-of-the-art. Life Sci. 2020, 254, 117580. [Google Scholar] [CrossRef] [PubMed]
- Bastola, R.; Noh, G.; Keum, T.; Bashyal, S.; Seo, J.E.; Choi, J.; Oh, Y.; Cho, Y.; Lee, S. Vaccine adjuvants: Smart components to boost the immune system. Arch. Pharm. Res. 2017, 40, 1238–1248. [Google Scholar] [CrossRef]
- Marino, M.; Scuderi, F.; Provenzano, C.; Bartoccioni, E. Skeletal muscle cells: From local inflammatory response to active immunity. Gene ther. 2011, 18, 109–116. [Google Scholar] [CrossRef]
- Hengge, U.R.; Chan, E.F.; Foster, R.A.; Walker, P.S.; Vogel, J.C. Cytokine gene expression in epidermis with biological effects following injection of naked DNA. Nat. Genet. 1995, 10, 161–166. [Google Scholar] [CrossRef]
- Oh, S.; Kessler, J.A. Design, Assembly, Production, and Transfection of Synthetic Modified mRNA. Methods 2018, 133, 29–43. [Google Scholar] [CrossRef]
- Bai, H.; Lester, G.M.S.; Petishnok, L.C.; Dean, D.A. Cytoplasmic transport and nuclear import of plasmid DNA. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Lazzaro, S.; Giovani, C.; Mangiavacchi, S.; Magini, D.; Maione, D.; Baudner, B.; Geall, A.J.; De Gregorio, E.; D’Oro, U.; Buonsanti, C. CD8 T-cell priming upon mRNA vaccination is restricted to bone-marrow-derived antigen-presenting cells and may involve antigen transfer from myocytes. Immunology 2015, 146, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Sudowe, S.; Dominitzki, S.; Montermann, E.; Bros, M.; Grabbe, S.; Reske-Kunz, A.B. Uptake and presentation of exogenous antigen and presentation of endogenously produced antigen by skin dendritic cells represent equivalent pathways for the priming of cellular immune responses following biolistic DNA immunization. Immunology 2009, 128, e193–e205. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, H.; Peng, H.; Huyan, T.; Cacalano, N.A. Exosomes: Versatile Nano Mediators of Immune Regulation. Cancers 2019, 11, 1557. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Larregina, A.T.; Morelli, A.E. Impact of extracellular vesicles on innate immunity. Curr. Opin. Organ. Transplant. 2019, 24, 670–678. [Google Scholar] [CrossRef]
- Den Haan, J.M.; Arens, R.; van Zelm, M.C. The activation of the adaptive immune system: Cross-talk between antigen-presenting cells, T cells and B cells. Immunol. Lett. 2014, 162, 103–112. [Google Scholar] [CrossRef]
- Macri, C.; Dumont, C.; Johnston, A.P.; Mintern, J.D. Targeting dendritic cells: A promising strategy to improve vaccine effectiveness. Clin. Transl. Immunol. 2016, 5, e66. [Google Scholar] [CrossRef]
- Porgador, A.; Irvine, K.R.; Iwasaki, A.; Barber, B.H.; Restifo, N.P.; Germain, R.N. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 1998, 188, 1075–1082. [Google Scholar] [CrossRef]
- Coban, C.; Kobiyama, K.; Jounai, N.; Tozuka, M.; Ishii, K.J. DNA vaccines: A simple DNA sensing matter? Hum. Vaccin. Immunother. 2013, 9, 2216–2221. [Google Scholar] [CrossRef]
- Joffre, O.; Nolte, M.A.; Sporri, R.; Reis e Sousa, C. Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol. Rev. 2009, 227, 234–247. [Google Scholar] [CrossRef]
- Maecker, H.T.; Umetsu, D.T.; DeKruyff, R.H.; Levy, S. Cytotoxic T cell responses to DNA vaccination: Dependence on antigen presentation via class II MHC. J. Immunol. (Baltimore, Md.: 1950) 1998, 161, 6532–6536. [Google Scholar]
- Aloulou, M.; Fazilleau, N. Regulation of B cell responses by distinct populations of CD4 T cells. Biomed. J. 2019, 42, 243–251. [Google Scholar] [CrossRef]
- Fu, S.H.; Chien, M.W.; Hsu, C.Y.; Liu, Y.W.; Sytwu, H.K. Interplay between Cytokine Circuitry and Transcriptional Regulation Shaping Helper T Cell Pathogenicity and Plasticity in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 3379. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Lee, P.; Snively, J.; Boswell, W.; Ounpraseuth, S.; Lee, S.; Hickingbottom, B.; Smith, J.; Johnson, D.; Weber, J.S. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer 2003, 98, 144–154. [Google Scholar] [CrossRef]
- Proudfoot, O.; Apostolopoulos, V.; Pietersz, G.A. Receptor-mediated delivery of antigens to dendritic cells: Anticancer applications. Mol. Pharm. 2007, 4, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.C.; Rao, A.; Marsden, J.R.; Armstrong, C.J.; Berhane, S.; Billingham, L.J.; Graham, N.; Roberts, C.; Ryan, G.; Uppal, H.; et al. Phase I/II trial of a dendritic cell vaccine transfected with DNA encoding melan A and gp100 for patients with metastatic melanoma. Gene Ther. 2011, 18, 584–593. [Google Scholar] [CrossRef]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J.Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef]
- McCann, K.J.; Mander, A.; Cazaly, A.; Chudley, L.; Stasakova, J.; Thirdborough, S.; King, A.; Lloyd-Evans, P.; Buxton, E.; Edwards, C.; et al. Targeting Carcinoembryonic Antigen with DNA Vaccination: On-Target Adverse Events Link with Immunologic and Clinical Outcomes. Clin. Cancer Res. 2016, 22, 4827–4836. [Google Scholar] [CrossRef]
- McNeel, D.G.; Eickhoff, J.C.; Wargowski, E.; Zahm, C.; Staab, M.J.; Straus, J.; Liu, G. Concurrent, but not sequential, PD-1 blockade with a DNA vaccine elicits anti-tumor responses in patients with metastatic, castration-resistant prostate cancer. Oncotarget 2018, 9, 25586–25596. [Google Scholar] [CrossRef] [PubMed]
- Tosch, C.; Bastien, B.; Barraud, L.; Grellier, B.; Nourtier, V.; Gantzer, M.; Limacher, J.M.; Quemeneur, E.; Bendjama, K.; Préville, X. Viral based vaccine TG4010 induces broadening of specific immune response and improves outcome in advanced NSCLC. J. Immunother. Cancer 2017, 5, 70. [Google Scholar] [CrossRef]
- Dörrie, J.; Schaft, N.; Schuler, G.; Schuler-Thurner, B. Therapeutic Cancer Vaccination with Ex Vivo RNA-Transfected Dendritic Cells-An Update. Pharmaceutics 2020, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Su, Z.; Vieweg, J.; Weizer, A.Z.; Dahm, P.; Yancey, D.; Turaga, V.; Higgins, J.; Boczkowski, D.; Gilboa, E.; Dannull, J. Enhanced induction of telomerase-specific CD4(+) T cells using dendritic cells transfected with RNA encoding a chimeric gene product. Cancer Res. 2002, 62, 5041–5048. [Google Scholar] [PubMed]
- Khoury, H.J.; Collins, R.H., Jr.; Blum, W.; Stiff, P.S.; Elias, L.; Lebkowski, J.S.; Reddy, A.; Nishimoto, K.P.; Sen, D.; Wirth, E.D., III; et al. Immune responses and long-term disease recurrence status after telomerase-based dendritic cell immunotherapy in patients with acute myeloid leukemia. Cancer 2017, 123, 3061–3072. [Google Scholar] [CrossRef]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Rahman, M.; Dastmalchi, F.; Karachi, A.; Mitchell, D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. Oncoimmunology 2019, 8, e1514921. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Van Nuffel, A.M.; Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Neyns, B. Long-term clinical outcome of melanoma patients treated with messenger RNA-electroporated dendritic cell therapy following complete resection of metastases. Cancer Immunol. Immunother. 2015, 64, 381–388. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, S.; Haas, H.; Diken, M.; Kranz, L.M.; Langguth, P.; Sahin, U. Translating nanoparticulate-personalized cancer vaccines into clinical applications: Case study with RNA-lipoplexes for the treatment of melanoma. Nanomedicine (London, England) 2016, 11, 2723–2734. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Nazarkina Zh, K.; Khar’kova, M.V.; Antonets, D.V.; Morozkin, E.S.; Bazhan, S.I.; Karpenko, L.I.; Vlasov, V.V.; Ilyichev, A.A.; Laktionov, P.P. Design of Polyepitope DNA Vaccine against Breast Carcinoma Cells and Analysis of Its Expression in Dendritic Cells. Bull. Exp. Biol. Med. 2016, 160, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Nishimura, Y. The present status and future prospects of peptide-based cancer vaccines. Int. Immunol. 2016, 28, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Brennick, C.A.; George, M.M.; Corwin, W.L.; Srivastava, P.K.; Ebrahimi-Nik, H. Neoepitopes as cancer immunotherapy targets: Key challenges and opportunities. Immunotherapy 2017, 9, 361–371. [Google Scholar] [CrossRef]
- Kiyotani, K.; Chan, H.T.; Nakamura, Y. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci. 2018, 109, 542–549. [Google Scholar] [CrossRef]
- Sultan, H.; Trillo-Tinoco, J.; Rodriguez, P.; Celis, E. Effective antitumor peptide vaccines can induce severe autoimmune pathology. Oncotarget 2017, 8, 70317–70331. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Bins, A.D.; Wolkers, M.C.; van den Boom, M.D.; Haanen, J.B.; Schumacher, T.N. In vivo antigen stability affects DNA vaccine immunogenicity. J. Immunol. (Baltimore, Md.: 1950) 2007, 179, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Hoppes, R.; Oostvogels, R.; Luimstra, J.J.; Wals, K.; Toebes, M.; Bies, L.; Ekkebus, R.; Rijal, P.; Celie, P.H.; Huang, J.H.; et al. Altered peptide ligands revisited: Vaccine design through chemically modified HLA-A2-restricted T cell epitopes. J. Immunol. 2014, 193, 4803–4813. [Google Scholar] [CrossRef] [PubMed]
- Seledtsova, G.V.; Shishkov, A.A.; Kaschenko, E.A.; Goncharov, A.G.; Gazatova, N.D.; Seledtsov, V.I. Xenogeneic cell-based vaccine therapy for stage III melanoma: Safety, immune-mediated responses and survival benefits. Eur. J. Dermatol. 2016, 26, 138–143. [Google Scholar] [CrossRef]
- Sponaas, A.; Carstens, C.; Koch, N. C-terminal extension of the MHC class II-associated invariant chain by an antigenic sequence triggers activation of naive T cells. Gene Ther. 1999, 6, 1826–1834. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tudor, D.; Dubuquoy, C.; Gaboriau, V.; Lefevre, F.; Charley, B.; Riffault, S. TLR9 pathway is involved in adjuvant effects of plasmid DNA-based vaccines. Vaccine 2005, 23, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Suschak, J.J.; Wang, S.; Fitzgerald, K.A.; Lu, S. A cGAS-Independent STING/IRF7 Pathway Mediates the Immunogenicity of DNA Vaccines. J. Immunol. 2016, 196, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Larregina, A.T.; Watkins, S.C.; Erdos, G.; Spencer, L.A.; Storkus, W.J.; Beer Stolz, D.; Falo, L.D., Jr. Direct transfection and activation of human cutaneous dendritic cells. Gene Ther. 2001, 8, 608–617. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Scheiermann, J.; Klinman, D.M. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014, 32, 6377–6389. [Google Scholar] [CrossRef]
- Garg, R.; Kaur, M.; Saxena, A.; Prasad, R.; Bhatnagar, R. Alum adjuvanted rabies DNA vaccine confers 80% protection against lethal 50 LD50 rabies challenge virus standard strain. Mol. Immunol. 2017, 85, 166–173. [Google Scholar] [CrossRef]
- Shedlock, D.J.; Tingey, C.; Mahadevan, L.; Hutnick, N.; Reuschel, E.L.; Kudchodkar, S.; Flingai, S.; Yan, J.; Kim, J.J.; Ugen, K.E.; et al. Co-Administration of Molecular Adjuvants Expressing NF-Kappa B Subunit p65/RelA or Type-1 Transactivator T-bet Enhance Antigen Specific DNA Vaccine-Induced Immunity. Vaccines 2014, 2, 196–215. [Google Scholar] [CrossRef]
- Pfeiffer, I.A.; Hoyer, S.; Gerer, K.F.; Voll, R.E.; Knippertz, I.; Gückel, E.; Schuler, G.; Schaft, N.; Dörrie, J. Triggering of NF-κB in cytokine-matured human DCs generates superior DCs for T-cell priming in cancer immunotherapy. Eur. J. Immunol. 2014, 44, 3413–3428. [Google Scholar] [CrossRef] [PubMed]
- Bosch, N.C.; Voll, R.E.; Voskens, C.J.; Gross, S.; Seliger, B.; Schuler, G.; Schaft, N.; Dörrie, J. NF-κB activation triggers NK-cell stimulation by monocyte-derived dendritic cells. Ther. Adv. Med. Oncol. 2019, 11, 1758835919891622. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Amara, R.R.; Yeow, W.S.; Pitha, P.M.; Robinson, H.L. Regulation of DNA-raised immune responses by cotransfected interferon regulatory factors. J. Virol. 2002, 76, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Bontkes, H.J.; Kramer, D.; Ruizendaal, J.J.; Meijer, C.J.; Hooijberg, E. Tumor associated antigen and interleukin-12 mRNA transfected dendritic cells enhance effector function of natural killer cells and antigen specific T-cells. Clin. Immunol. 2008, 127, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Kochar, N.K.; Elizaga, M.; Hay, C.M.; Wilson, G.J.; Cohen, K.W.; De Rosa, S.C.; Xu, R.; Ota-Setlik, A.; Morris, D.; et al. DNA Priming Increases Frequency of T-Cell Responses to a Vesicular Stomatitis Virus HIV Vaccine with Specific Enhancement of CD8(+) T-Cell Responses by Interleukin-12 Plasmid DNA. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef]
- Sun, L.; Yuan, Q.; Xu, T.; Yao, L.; Feng, J.; Ma, J.; Wang, L.; Lv, C.; Wang, D. Novel adjuvant for immunization against tuberculosis: DNA vaccine expressing Mycobacterium tuberculosis antigen 85A and interleukin-15 fusion product elicits strong immune responses in mice. Biotechnol. Lett. 2017, 39, 1159–1166. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, S.; Li, X.; Wang, L.; Zhang, J.; Xu, J.; Huo, S.; Cao, X.; Zhong, Z.; Zhong, F. Mutual enhancement of IL-2 and IL-7 on DNA vaccine immunogenicity mainly involves regulations on their receptor expression and receptor-expressing lymphocyte generation. Vaccine 2015, 33, 3480–3487. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, C.; Yi, H.; Li, P.; Pan, H.; Liu, L.; Cai, L.; Ma, Y. Nanovaccine loaded with poly I:C and STAT3 siRNA robustly elicits anti-tumor immune responses through modulating tumor-associated dendritic cells in vivo. Biomaterials 2015, 38, 50–60. [Google Scholar] [CrossRef]
- Luo, X.; Peng, X.; Hou, J.; Wu, S.; Shen, J.; Wang, L. Folic acid-functionalized polyethylenimine superparamagnetic iron oxide nanoparticles as theranostic agents for magnetic resonance imaging and PD-L1 siRNA delivery for gastric cancer. Int. J. Nanomed. 2017, 12, 5331–5343. [Google Scholar] [CrossRef]
- Self-Fordham, J.B.; Naqvi, A.R.; Uttamani, J.R.; Kulkarni, V.; Nares, S. MicroRNA: Dynamic Regulators of Macrophage Polarization and Plasticity. Front. Immunol. 2017, 8, 1062. [Google Scholar] [CrossRef]
- Migault, M.; Donnou-Fournet, E.; Galibert, M.D.; Gilot, D. Definition and identification of small RNA sponges: Focus on miRNA sequestration. Methods 2017, 117, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Tumor-targeted Delivery of Anti-microRNA for Cancer Therapy: pHLIP is Key. Angew. Chem. Int. Ed. Engl. 2015, 54, 5824–5826. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.E.; Salton, G.; Sodre de Castro Laino, A.; Pires, T.D.; Bonamino, M.; Lenz, G.; Delgado-Canedo, A. pLR: A lentiviral backbone series to stable transduction of bicistronic genes and exchange of promoters. Plasmid 2012, 68, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Terenin, I.M.; Smirnova, V.V.; Andreev, D.E.; Dmitriev, S.E.; Shatsky, I.N. A researcher’s guide to the galaxy of IRESs. Cell. Mol. Life Sci. 2017, 74, 1431–1455. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.L.; Park, H.J.; Kim, J.; Kim, H.; Youn, H.; Nam, J.H. Development of an RNA Expression Platform Controlled by Viral Internal Ribosome Entry Sites. J. Microbiol. Biotechnol. 2019, 29, 127–140. [Google Scholar] [CrossRef]
- Chng, J.; Wang, T.; Nian, R.; Lau, A.; Hoi, K.M.; Ho, S.C.; Gagnon, P.; Bi, X.; Yang, Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. mAbs 2015, 7, 403–412. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, H.J.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PloS ONE 2011, 6, e18556. [Google Scholar] [CrossRef]
- Gracey Maniar, L.E.; Maniar, J.M.; Chen, Z.Y.; Lu, J.; Fire, A.Z.; Kay, M.A. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol. Ther. 2013, 21, 131–138. [Google Scholar] [CrossRef]
- Stenler, S.; Blomberg, P.; Smith, C.I. Safety and efficacy of DNA vaccines: Plasmids vs. minicircles. Hum. Vaccin. Immunother. 2014, 10, 1306–1308. [Google Scholar] [CrossRef]
- Lechardeur, D.; Lukacs, G.L. Nucleocytoplasmic transport of plasmid DNA: A perilous journey from the cytoplasm to the nucleus. Hum. Gene Ther. 2006, 17, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A.; Dean, B.S.; Muller, S.; Smith, L.C. Sequence requirements for plasmid nuclear import. Exp. Cell. Res. 1999, 253, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Yamazaki, M.; Fukuda, T.; Takashima, Y.; Okada, H. Versatile nuclear localization signal-based oligopeptide as a gene vector. Biol. Pharm. Bull. 2015, 38, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Grubor-Bauk, B.; Yu, W.; Wijesundara, D.; Gummow, J.; Garrod, T.; Brennan, A.J.; Voskoboinik, I.; Gowans, E.J. Intradermal delivery of DNA encoding HCV NS3 and perforin elicits robust cell-mediated immunity in mice and pigs. Gene Ther. 2016, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Krinner, S.; Heitzer, A.; Asbach, B.; Wagner, R. Interplay of Promoter Usage and Intragenic CpG Content: Impact on GFP Reporter Gene Expression. Hum. Gene Ther. 2015, 26, 826–840. [Google Scholar] [CrossRef]
- Yagi, M.; Miyamoto, T.; Toyama, Y.; Suda, T. Role of DC-STAMP in cellular fusion of osteoclasts and macrophage giant cells. J. Bone Miner. Metab. 2006, 24, 355–358. [Google Scholar] [CrossRef]
- Dresch, C.; Edelmann, S.L.; Marconi, P.; Brocker, T. Lentiviral-mediated transcriptional targeting of dendritic cells for induction of T cell tolerance in vivo. J. Immunol. 2008, 181, 4495–4506. [Google Scholar] [CrossRef]
- Bonkobara, M.; Zukas, P.K.; Shikano, S.; Nakamura, S.; Cruz, P.D., Jr.; Ariizumi, K. Epidermal Langerhans cell-targeted gene expression by a dectin-2 promoter. J. Immunol. 2001, 167, 6893–6900. [Google Scholar] [CrossRef]
- Lopes, A.; Vanvarenberg, K.; Preat, V.; Vandermeulen, G. Codon-Optimized P1A-Encoding DNA Vaccine: Toward a Therapeutic Vaccination against P815 Mastocytoma. Mol. Ther. Nucleic Acids 2017, 8, 404–415. [Google Scholar] [CrossRef]
- Ross, R.; Sudowe, S.; Beisner, J.; Ross, X.L.; Ludwig-Portugall, I.; Steitz, J.; Tuting, T.; Knop, J.; Reske-Kunz, A.B. Transcriptional targeting of dendritic cells for gene therapy using the promoter of the cytoskeletal protein fascin. Gene Ther. 2003, 10, 1035–1040. [Google Scholar] [CrossRef]
- Bros, M.; Ross, X.L.; Pautz, A.; Reske-Kunz, A.B.; Ross, R. The human fascin gene promoter is highly active in mature dendritic cells due to a stage-specific enhancer. J. Immunol. 2003, 171, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Raker, V.; Maxeiner, J.; Reske-Kunz, A.B.; Sudowe, S. Efficiency of biolistic DNA vaccination in experimental type I allergy. In Biolistic DNA Delivery: Methods in Molecular Biology; Sudowe, S., Reske-Kunz, A., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 357–370. [Google Scholar] [CrossRef]
- Castor, T.; Yogev, N.; Blank, T.; Barwig, C.; Prinz, M.; Waisman, A.; Bros, M.; Reske-Kunz, A.B. Inhibition of experimental autoimmune encephalomyelitis by tolerance-promoting DNA vaccination focused to dendritic cells. PloS ONE 2018, 13, e0191927. [Google Scholar] [CrossRef] [PubMed]
- Sudowe, S.; Höhn, Y.; Renzing, A.; Maxeiner, J.; Montermann, E.; Habermeier, A.; Closs, E.; Bros, M.; Reske-Kunz, A.B. Inhibition of antigen-specific immune responses by co-application of an indoleamine 2,3-dioxygenase (IDO)-encoding vector requires antigen transgene expression focused on dendritic cells. Amino Acids 2020, 52, 411–424. [Google Scholar] [CrossRef]
- Angell, C.; Xie, S.; Zhang, L.; Chen, Y. DNA Nanotechnology for Precise Control over Drug Delivery and Gene Therapy. Small (Weinheim an der Bergstrasse, Germany) 2016, 12, 1117–1132. [Google Scholar] [CrossRef]
- Das, S.K.; Menezes, M.E.; Bhatia, S.; Wang, X.Y.; Emdad, L.; Sarkar, D.; Fisher, P.B. Gene Therapies for Cancer: Strategies, Challenges and Successes. J. Cell. Physiol. 2015, 230, 259–271. [Google Scholar] [CrossRef]
- Cai, P.; Zhang, X.; Wang, M.; Wu, Y.L.; Chen, X. Combinatorial Nano-Bio Interfaces. ACS Nano 2018. [Google Scholar] [CrossRef] [PubMed]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef]
- Li, B.; Zhang, X.; Dong, Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1530. [Google Scholar] [CrossRef]
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert. Rev. Vaccines 2015, 14, 221–234. [Google Scholar] [CrossRef]
- Foged, C.; Brodin, B.; Frokjaer, S.; Sundblad, A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm. 2005, 298, 315–322. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Niikura, K.; Matsunaga, T.; Suzuki, T.; Kobayashi, S.; Yamaguchi, H.; Orba, Y.; Kawaguchi, A.; Hasegawa, H.; Kajino, K.; Ninomiya, T.; et al. Gold nanoparticles as a vaccine platform: Influence of size and shape on immunological responses in vitro and in vivo. ACS Nano 2013, 7, 3926–3938. [Google Scholar] [CrossRef] [PubMed]
- Radis-Baptista, G.; Campelo, I.S.; Morlighem, J.R.L.; Melo, L.M.; Freitas, V.J.F. Cell-penetrating peptides (CPPs): From delivery of nucleic acids and antigens to transduction of engineered nucleases for application in transgenesis. J. Biotechnol. 2017, 252, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Galdiero, S. Peptide chemistry encounters nanomedicine: Recent applications and upcoming scenarios in cancer. Future. Med. Chem. 2018, 10, 1877–1880. [Google Scholar] [CrossRef]
- Dane, K.Y.; Nembrini, C.; Tomei, A.A.; Eby, J.K.; O’Neil, C.P.; Velluto, D.; Swartz, M.A.; Inverardi, L.; Hubbell, J.A. Nano-sized drug-loaded micelles deliver payload to lymph node immune cells and prolong allograft survival. J. Controlled Release 2011, 156, 154–160. [Google Scholar] [CrossRef]
- Allen, T.M.; Hansen, C.B.; Guo, L.S. Subcutaneous administration of liposomes: A comparison with the intravenous and intraperitoneal routes of injection. Biochim. Biophys. Acta 1993, 1150, 9–16. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Nanomedicine (London, England) 2008, 3, 703–717. [Google Scholar] [CrossRef]
- Hu, J.; Sheng, Y.; Shi, J.; Yu, B.; Yu, Z.; Liao, G. Long circulating polymeric nanoparticles for gene/drug delivery. Curr. Drug Metab. 2018, 19, 723–738. [Google Scholar] [CrossRef]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef]
- Fogli, S.; Montis, C.; Paccosi, S.; Silvano, A.; Michelucci, E.; Berti, D.; Bosi, A.; Parenti, A.; Romagnoli, P. Inorganic nanoparticles as potential regulators of immune response in dendritic cells. Nanomedicine (London, England) 2017, 12, 1647–1660. [Google Scholar] [CrossRef]
- Svoboda, O.; Fohlerova, Z.; Baiazitova, L.; Mlynek, P.; Samouylov, K.; Provaznik, I.; Hubalek, J. Transfection by Polyethyleneimine-Coated Magnetic Nanoparticles: Fine-Tuning the Condition for Electrophysiological Experiments. J. Biomed. Nanotechnol. 2018, 14, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Qiao, S.Z. A mesoporous organosilica nano-bowl with high DNA loading capacity - a potential gene delivery carrier. Nanoscale 2016, 8, 17446–17450. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.P.; Herrera, C.E.; Singh, B.; Singh, S.; Singh, R.K.; Kumar, R. Graphene oxide: An efficient material and recent approach for biotechnological and biomedical applications. Mater. Sci. Eng. C Mater. Biol. App. 2018, 86, 173–197. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.; Lee, M.; Choi, H.C.; Kim, W.J. Stimuli-Regulated Enzymatically Degradable Smart Graphene-Oxide-Polymer Nanocarrier Facilitating Photothermal Gene Delivery. Adv. Healthc. Mater. 2016, 5, 1918–1930. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Zhou, X.; Cheng, M.; Xing, D. Graphene oxide-mediated Cas9/sgRNA delivery for efficient genome editing. Nanoscale 2018, 10, 1063–1071. [Google Scholar] [CrossRef]
- Quader, S.; Kataoka, K. Nanomaterial-Enabled Cancer Therapy. Mol. Ther. 2017, 25, 1501–1513. [Google Scholar] [CrossRef]
- Jang, H.J.; Jeong, E.J.; Lee, K.Y. Carbon Dioxide-Generating PLG Nanoparticles for Controlled Anti-Cancer Drug Delivery. Pharm. Res. 2018, 35, 59. [Google Scholar] [CrossRef]
- Li, Z.; Xiong, F.; He, J.; Dai, X.; Wang, G. Surface-functionalized, pH-responsive poly(lactic-co-glycolic acid)-based microparticles for intranasal vaccine delivery: Effect of surface modification with chitosan and mannan. Eur. J. Pharm. Biopharm. 2016, 109, 24–34. [Google Scholar] [CrossRef]
- Itaka, K.; Harada, A.; Yamasaki, Y.; Nakamura, K.; Kawaguchi, H.; Kataoka, K. In situ single cell observation by fluorescence resonance energy transfer reveals fast intra-cytoplasmic delivery and easy release of plasmid DNA complexed with linear polyethylenimine. J. Gene Med. 2004, 6, 76–84. [Google Scholar] [CrossRef]
- Zhu, J.; Qiao, M.; Wang, Q.; Ye, Y.; Ba, S.; Ma, J.; Hu, H.; Zhao, X.; Chen, D. Dual-responsive polyplexes with enhanced disassembly and endosomal escape for efficient delivery of siRNA. Biomaterials 2018, 162, 47–59. [Google Scholar] [CrossRef]
- Hao, F.; Li, Y.; Zhu, J.; Sun, J.; Marshall, B.; Lee, R.J.; Teng, L.; Yang, Z.; Xie, J. Polyethylenimine-based Formulations for Delivery of Oligonucleotides. Curr. Med. Chem. 2019, 26, 2264–2284. [Google Scholar] [CrossRef] [PubMed]
- Erbacher, P.; Zou, S.; Bettinger, T.; Steffan, A.M.; Remy, J.S. Chitosan-based vector/DNA complexes for gene delivery: Biophysical characteristics and transfection ability. Pharm. Res. 1998, 15, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chen, X.; Jia, J.; Zhang, W.; Yang, T.; Wang, L.; Ma, G. pH-Responsive Poly(D,L-lactic-co-glycolic acid) Nanoparticles with Rapid Antigen Release Behavior Promote Immune Response. ACS Nano 2015, 9, 4925–4938. [Google Scholar] [CrossRef] [PubMed]
- Slutter, B.; Plapied, L.; Fievez, V.; Sande, M.A.; des Rieux, A.; Schneider, Y.J.; Van Riet, E.; Jiskoot, W.; Preat, V. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J. Controlled Release 2009, 138, 113–121. [Google Scholar] [CrossRef]
- Lohcharoenkal, W.; Wang, L.; Chen, Y.C.; Rojanasakul, Y. Protein nanoparticles as drug delivery carriers for cancer therapy. BioMed Res. Int. 2014, 2014, 180549. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.C.; Rosell, N.; Ruano, G.; Busquets, M.A.; Vinardell, M.P. Gelatin-based nanoparticles as DNA delivery systems: Synthesis, physicochemical and biocompatible characterization. Colloids Surf. B. 2015, 134, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.; Liu, C.H.; Wu, W.C. Efficient gene delivery by oligochitosan conjugated serum albumin: Facile synthesis, polyplex stability, and transfection. Carbohydr. Polym. 2018, 183, 37–49. [Google Scholar] [CrossRef]
- Han, J.; Wang, Q.; Zhang, Z.; Gong, T.; Sun, X. Cationic bovine serum albumin based self-assembled nanoparticles as siRNA delivery vector for treating lung metastatic cancer. Small (Weinheim an der Bergstrasse, Germany) 2014, 10, 524–535. [Google Scholar] [CrossRef]
- Rezaee, M.; Oskuee, R.K.; Nassirli, H.; Malaekeh-Nikouei, B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Controlled Release 2016, 236, 1–14. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Mevel, M.; Haudebourg, T.; Colombani, T.; Peuziat, P.; Dallet, L.; Chatin, B.; Lambert, O.; Berchel, M.; Montier, T.; Jaffres, P.A.; et al. Important role of phosphoramido linkage in imidazole-based dioleyl helper lipids for liposome stability and primary cell transfection. J. Gene Med. 2016, 18, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bahreman, A.; Daudey, G.; Bussmann, J.; Olsthoorn, R.C.; Kros, A. Drug Delivery via Cell Membrane Fusion Using Lipopeptide Modified Liposomes. ACS Cent. Sci. 2016, 2, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Glass, J.J.; Kent, S.J.; De Rose, R. Enhancing dendritic cell activation and HIV vaccine effectiveness through nanoparticle vaccination. Expert Rev. Vaccines 2016, 15, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Wagener, K.; Bros, M.; Krumb, M.; Langhanki, J.; Pektor, S.; Worm, M.; Schinnerer, M.; Montermann, E.; Miederer, M.; Frey, H.; et al. Targeting of Immune Cells with Trimannosylated Liposomes. Adv. Ther. 2020, 3, 1900185. [Google Scholar] [CrossRef]
- Lindén, M. Biodistribution and Excretion of Intravenously Injected Mesoporous Silica Nanoparticles: Implications for Drug Delivery Efficiency and Safety. Enzymes 2018, 43, 155–180. [Google Scholar] [CrossRef]
- Guo, X.; Zhuang, Q.; Ji, T.; Zhang, Y.; Li, C.; Wang, Y.; Li, H.; Jia, H.; Liu, Y.; Du, L. Multi-functionalized chitosan nanoparticles for enhanced chemotherapy in lung cancer. Carbohydr. Polym. 2018, 195, 311–320. [Google Scholar] [CrossRef]
- Meng, H.; Leong, W.; Leong, K.W.; Chen, C.; Zhao, Y. Walking the line: The fate of nanomaterials at biological barriers. Biomaterials 2018, 174, 41–53. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J. Controlled Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Sago, C.D.; Krupczak, B.R.; Lokugamage, M.P.; Gan, Z.; Dahlman, J.E. Cell Subtypes Within the Liver Microenvironment Differentially Interact with Lipid Nanoparticles. Cell. Mol. Bioeng. 2019, 12, 389–397. [Google Scholar] [CrossRef]
- Pustylnikov, S.; Sagar, D.; Jain, P.; Khan, Z.K. Targeting the C-type lectins-mediated host-pathogen interactions with dextran. J. Pharm. Sci. 2014, 17, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Elvevold, K.; Simon-Santamaria, J.; Hasvold, H.; McCourt, P.; Smedsrød, B.; Sørensen, K.K. Liver sinusoidal endothelial cells depend on mannose receptor-mediated recruitment of lysosomal enzymes for normal degradation capacity. Hepatology 2008, 48, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Fraser, I.P.; Gordon, S. Murine macrophage scavenger receptor: In vivo expression and function as receptor for macrophage adhesion in lymphoid and non-lymphoid organs. Eur. J. Immunol. 1995, 25, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef]
- Gül, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bögels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Invest. 2014, 124, 812–823. [Google Scholar] [CrossRef]
- Ganesan, L.P.; Kim, J.; Wu, Y.; Mohanty, S.; Phillips, G.S.; Birmingham, D.J.; Robinson, J.M.; Anderson, C.L. FcγRIIb on liver sinusoidal endothelium clears small immune complexes. J. Immunol. 2012, 189, 4981–4988. [Google Scholar] [CrossRef]
- Hinglais, N.; Kazatchkine, M.D.; Mandet, C.; Appay, M.D.; Bariety, J. Human liver Kupffer cells express CR1, CR3, and CR4 complement receptor antigens. An immunohistochemical study. Lab. Invest. 1989, 61, 509–514. [Google Scholar]
- Bros, M.; Nuhn, L.; Simon, J.; Moll, L.; Mailander, V.; Landfester, K.; Grabbe, S. The Protein Corona as a Confounding Variable of Nanoparticle-Mediated Targeted Vaccine Delivery. Front. Immunol. 2018, 9, 1760. [Google Scholar] [CrossRef]
- Shen, L.; Tenzer, S.; Storck, W.; Hobernik, D.; Raker, V.K.; Fischer, K.; Decker, S.; Dzionek, A.; Krauthauser, S.; Diken, M.; et al. Protein corona-mediated targeting of nanocarriers to B cells allows redirection of allergic immune responses. J. Allergy Clin. Immunol. 2018. [Google Scholar] [CrossRef]
- Sun, X.; Wang, G.; Zhang, H.; Hu, S.; Liu, X.; Tang, J.; Shen, Y. The Blood Clearance Kinetics and Pathway of Polymeric Micelles in Cancer Drug Delivery. ACS Nano 2018, 12, 6179–6192. [Google Scholar] [CrossRef]
- Zhou, H.; Fan, Z.; Li, P.Y.; Deng, J.; Arhontoulis, D.C.; Li, C.Y.; Bowne, W.B.; Cheng, H. Dense and Dynamic Polyethylene Glycol Shells Cloak Nanoparticles from Uptake by Liver Endothelial Cells for Long Blood Circulation. ACS Nano 2018, 12, 10130–10141. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.M.G.; Jaafari, M.R.; Hatamipour, M.; Penson, P.E.; Sahebkar, A. Liposome Circulation Time is Prolonged by CD47 Coating. Protein Pept. Lett. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gulla, S.K.; Rao, B.R.; Moku, G.; Jinka, S.; Nimmu, N.V.; Khalid, S.; Patra, C.R.; Chaudhuri, A. In vivo targeting of DNA vaccines to dendritic cells using functionalized gold nanoparticles. Biomater. Sci. 2019, 7, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Wi, T.I.; Byeon, Y.; Won, J.E.; Lee, J.M.; Kang, T.H.; Lee, J.W.; Lee, Y.J.; Sood, A.K.; Han, H.D.; Park, Y.M. Selective Tumor-Specific Antigen Delivery to Dendritic Cells Using Mannose-Labeled Poly(d, l-lactide-co-glycolide) Nanoparticles for Cancer Immunotherapy. J. Biomed. Nanotechnol. 2020, 16, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yuan, J.; Wang, Y.; Silvers, W.K. Distribution of ATPase-positive Langerhans cells in normal adult human skin. Br. J. Dermatol. 1985, 113, 707–711. [Google Scholar] [CrossRef]
- Russo, E.; Nitschke, M.; Halin, C. Dendritic cell interactions with lymphatic endothelium. Lymphatic Research and Biology 2013, 11, 172–182. [Google Scholar] [CrossRef]
- Fernando, G.J.; Zhang, J.; Ng, H.I.; Haigh, O.L.; Yukiko, S.R.; Kendall, M.A. Influenza nucleoprotein DNA vaccination by a skin targeted, dry coated, densely packed microprojection array (Nanopatch) induces potent antibody and CD8(+) T cell responses. J. Controlled Release 2016, 237, 35–41. [Google Scholar] [CrossRef]
- Lambracht-Washington, D.; Fu, M.; Frost, P.; Rosenberg, R.N. Evaluation of a DNA Aβ42 vaccine in adult rhesus monkeys (Macaca mulatta): Antibody kinetics and immune profile after intradermal immunization with full-length DNA Aβ42 trimer. Alzheimers Res. Ther. 2017, 9, 30. [Google Scholar] [CrossRef]
- Alvarez, R.D.; Huh, W.K.; Bae, S.; Lamb, L.S., Jr.; Conner, M.G.; Boyer, J.; Wang, C.; Hung, C.F.; Sauter, E.; Paradis, M.; et al. A pilot study of pNGVL4a-CRT/E7(detox) for the treatment of patients with HPV16+ cervical intraepithelial neoplasia 2/3 (CIN2/3). Gynecol. Oncol. 2016, 140, 245–252. [Google Scholar] [CrossRef]
- Duong, H.T.T.; Yin, Y.; Thambi, T.; Nguyen, T.L.; Giang Phan, V.H.; Lee, M.S.; Lee, J.E.; Kim, J.; Jeong, J.H.; Lee, D.S. Smart vaccine delivery based on microneedle arrays decorated with ultra-pH-responsive copolymers for cancer immunotherapy. Biomaterials 2018, 185, 13–24. [Google Scholar] [CrossRef]
- Cole, G.; Ali, A.A.; McErlean, E.; Mulholland, E.J.; Short, A.; McCrudden, C.M.; McCaffrey, J.; Robson, T.; Kett, V.L.; Coulter, J.A.; et al. DNA vaccination via RALA nanoparticles in a microneedle delivery system induces a potent immune response against the endogenous prostate cancer stem cell antigen. Acta Biomater. 2019, 96, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Samuels, S.; Marijne Heeren, A.; Zijlmans, H.; Welters, M.J.P.; van den Berg, J.H.; Philips, D.; Kvistborg, P.; Ehsan, I.; Scholl, S.M.E.; Nuijen, B.; et al. HPV16 E7 DNA tattooing: Safety, immunogenicity, and clinical response in patients with HPV-positive vulvar intraepithelial neoplasia. Cancer Immunol. Immunother. 2017, 66, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Bernelin-Cottet, C.; Urien, C.; McCaffrey, J.; Collins, D.; Donadei, A.; McDaid, D.; Jakob, V.; Barnier-Quer, C.; Collin, N.; Bouguyon, E.; et al. Electroporation of a nanoparticle-associated DNA vaccine induces higher inflammation and immunity compared to its delivery with microneedle patches in pigs. J. Controlled Release 2019, 308, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, K.; Smith, T.R.F.; Kiosses, W.B.; Kraynyak, K.A.; Wong, A.; Oh, J.; Broderick, K.E. Delineating the Cellular Mechanisms Associated with Skin Electroporation. Hum. Gene. Ther. Methods 2018, 29, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Lamolinara, A.; Stramucci, L.; Hysi, A.; Iezzi, M.; Marchini, C.; Mariotti, M.; Amici, A.; Curcio, C. Intradermal DNA Electroporation Induces Cellular and Humoral Immune Response and Confers Protection against HER2/neu Tumor. J. Immunol. Res. 2015, 2015, 159145. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Danishmalik, S.N.; Sin, J.I. DNA vaccines, electroporation and their applications in cancer treatment. Hum. Vaccin. Immunother. 2015, 11, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Gubara, S.M.; Fish, K.; Weber, T.; Bridges, C.R.; Hajjar, R.J.; Ishikawa, K. Targeted Gene Delivery through the Respiratory System: Rationale for Intratracheal Gene Transfer. J. Cardiovasc. Dev. Dis. 2019, 6, 8. [Google Scholar] [CrossRef]
- Davies, L.A.; Nunez-Alonso, G.A.; McLachlan, G.; Hyde, S.C.; Gill, D.R. Aerosol delivery of DNA/liposomes to the lung for cystic fibrosis gene therapy. Human Gene Ther. Clinical Develop. 2014, 25, 97–107. [Google Scholar] [CrossRef]
- Zheng, Z.; Diaz-Arevalo, D.; Guan, H.; Zeng, M. Noninvasive vaccination against infectious diseases. Hum. Vaccin. Immunother. 2018, 14, 1717–1733. [Google Scholar] [CrossRef]
- Mortimer, G.M.; Butcher, N.J.; Musumeci, A.W.; Deng, Z.J.; Martin, D.J.; Minchin, R.F. Cryptic epitopes of albumin determine mononuclear phagocyte system clearance of nanomaterials. ACS Nano 2014, 8, 3357–3366. [Google Scholar] [CrossRef]
- Huang, G.; Huang, H. Application of dextran as nanoscale drug carriers. Nanomedicine (London, England) 2018, 13, 3149–3158. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Harashima, H. An efficient PEGylated gene delivery system with improved targeting: Synergism between octaarginine and a fusogenic peptide. Int. J. Pharm. 2018, 538, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, F.; Liu, S.; Jiang, S. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J. Controlled Release 2016, 244, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Frenz, T.; Grabski, E.; Duran, V.; Hozsa, C.; Stepczynska, A.; Furch, M.; Gieseler, R.K.; Kalinke, U. Antigen presenting cell-selective drug delivery by glycan-decorated nanocarriers. Eur. J. Pharm. Biopharm. 2015, 95, 13–17. [Google Scholar] [CrossRef]
- Burgdorf, S.; Lukacs-Kornek, V.; Kurts, C. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J. Immunol. 2006, 176, 6770–6776. [Google Scholar] [CrossRef]
- Appelmelk, B.J.; van Die, I.; van Vliet, S.J.; Vandenbroucke-Grauls, C.M.; Geijtenbeek, T.B.; van Kooyk, Y. Cutting edge: Carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J. Immunol. 2003, 170, 1635–1639. [Google Scholar] [CrossRef]
- Qiao, C.; Liu, J.; Yang, J.; Li, Y.; Weng, J.; Shao, Y.; Zhang, X. Enhanced non-inflammasome mediated immune responses by mannosylated zwitterionic-based cationic liposomes for HIV DNA vaccines. Biomaterials 2016, 85, 1–17. [Google Scholar] [CrossRef]
- Wang, Q.; Cao, W.; Yang, Z.G.; Zhao, G.F. DC targeting DNA vaccines induce protective and therapeutic antitumor immunity in mice. Int. J. Clin. Exp. Med. 2015, 8, 17565–17577. [Google Scholar]
- Shimizu, K.; Iyoda, T.; Okada, M.; Yamasaki, S.; Fujii, S.I. Immune suppression and reversal of the suppressive tumor microenvironment. Int. Immunol. 2018, 30, 445–454. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Ahrends, T.; Borst, J. The opposing roles of CD4(+) T cells in anti-tumour immunity. Immunology 2018. [Google Scholar] [CrossRef] [PubMed]
- Hippen, K.L.; Loschi, M.; Nicholls, J.; MacDonald, K.P.A.; Blazar, B.R. Effects of MicroRNA on Regulatory T Cells and Implications for Adoptive Cellular Therapy to Ameliorate Graft-versus-Host Disease. Front. Immunol. 2018, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, S.; Liu, Y.; Yang, C. Epigenetics in myeloid derived suppressor cells: A sheathed sword towards cancer. Oncotarget 2016, 7, 57452–57463. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liang, P.; Guo, G.; Huang, Z.; Niu, Y.; Dong, L.; Wang, C.; Zhang, J. Re-polarizing Myeloid-derived Suppressor Cells (MDSCs) with Cationic Polymers for Cancer Immunotherapy. Sci. Rep. 2016, 6, 24506. [Google Scholar] [CrossRef]
- Li, W.; Deng, C.; Yang, H.; Wang, G. The Regulatory T Cell in Active Systemic Lupus Erythematosus Patients: A Systemic Review and Meta-Analysis. Front. Immunol. 2019, 10, 159. [Google Scholar] [CrossRef]
- Bacher, P.; Scheffold, A. Antigen-specific regulatory T-cell responses against aeroantigens and their role in allergy. Mucosal Immunol. 2018, 11, 1537–1550. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef]
- Attias, M.; Al-Aubodah, T.; Piccirillo, C.A. Mechanisms of human FoxP3(+) T(reg) cell development and function in health and disease. Clin. Exp. Immunol. 2019, 197, 36–51. [Google Scholar] [CrossRef]
- Yang, S.; Xie, C.; Chen, Y.; Wang, J.; Chen, X.; Lu, Z.; June, R.R.; Zheng, S.G. Differential roles of TNFα-TNFR1 and TNFα-TNFR2 in the differentiation and function of CD4(+)Foxp3(+) induced Treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis. 2019, 10, 27. [Google Scholar] [CrossRef]
- Oh, J.; Wang, W.; Thomas, R.; Su, D.M. Capacity of tTreg generation is not impaired in the atrophied thymus. PLoS Biol. 2017, 15, e2003352. [Google Scholar] [CrossRef]
- Zhong, H.; Liu, Y.; Xu, Z.; Liang, P.; Yang, H.; Zhang, X.; Zhao, J.; Chen, J.; Fu, S.; Tang, Y.; et al. TGF-β-Induced CD8(+)CD103(+) Regulatory T Cells Show Potent Therapeutic Effect on Chronic Graft-versus-Host Disease Lupus by Suppressing B Cells. Front. Immunol. 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.S.; Anandasabapathy, N. The origin of DCs and capacity for immunologic tolerance in central and peripheral tissues. Semin. Immunopathol. 2017, 39, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Quintana, F.J. Tolerogenic dendritic cells. Semin. Immunopathol. 2017, 39, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Robinson, C.M.; Plain, K.M.; Verma, N.D.; Tran, G.T.; Nomura, M.; Carter, N.; Boyd, R.; Hodgkinson, S.J. Changes in Reactivity In Vitro of CD4(+)CD25(+) and CD4(+)CD25(-) T Cell Subsets in Transplant Tolerance. Front. Immunol. 2017, 8, 994. [Google Scholar] [CrossRef]
- Sun, X.; He, S.; Lv, C.; Sun, X.; Wang, J.; Zheng, W.; Wang, D. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 2893–2898. [Google Scholar] [CrossRef]
- Huang, Y.H.; Chang, C.Y.; Kuo, Y.Z.; Fang, W.Y.; Kao, H.Y.; Tsai, S.T.; Wu, L.W. Cancer-associated fibroblast-derived interleukin-1β activates protumor C-C motif chemokine ligand 22 signaling in head and neck cancer. Cancer Sci. 2019, 110, 2783–2793. [Google Scholar] [CrossRef]
- Siede, J.; Fröhlich, A.; Datsi, A.; Hegazy, A.N.; Varga, D.V.; Holecska, V.; Saito, H.; Nakae, S.; Löhning, M. IL-33 Receptor-Expressing Regulatory T Cells Are Highly Activated, Th2 Biased and Suppress CD4 T Cell Proliferation through IL-10 and TGFβ Release. PloS ONE 2016, 11, e0161507. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef]
- Conroy, H.; Galvin, K.C.; Higgins, S.C.; Mills, K.H. Gene silencing of TGF-β1 enhances antitumor immunity induced with a dendritic cell vaccine by reducing tumor-associated regulatory T cells. Cancer Immunol. Immunother. 2012, 61, 425–431. [Google Scholar] [CrossRef]
- Masjedi, A.; Ahmadi, A.; Ghani, S.; Malakotikhah, F.; Nabi Afjadi, M.; Irandoust, M.; Karoon Kiani, F.; Heydarzadeh Asl, S.; Atyabi, F.; Hassannia, H.; et al. Silencing adenosine A2a receptor enhances dendritic cell-based cancer immunotherapy. Nanomedicine 2020, 29, 102240. [Google Scholar] [CrossRef]
- Zhang, H.H.; Fei, R.; Xie, X.W.; Wang, L.; Luo, H.; Wang, X.Y.; Wei, L.; Chen, H.S. Specific suppression in regulatory T cells by Foxp3 siRNA contributes to enhance the in vitro anti-tumor immune response in hepatocellular carcinoma patients. Beijing Da Xue Xue Bao Yi Xue Ban 2009, 41, 313–318. [Google Scholar] [PubMed]
- Kang, S.; Xie, J.; Ma, S.; Liao, W.; Zhang, J.; Luo, R. Targeted knock down of CCL22 and CCL17 by siRNA during DC differentiation and maturation affects the recruitment of T subsets. Immunobiology 2010, 215, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Jebbawi, F.; Fayyad-Kazan, H.; Merimi, M.; Lewalle, P.; Verougstraete, J.C.; Leo, O.; Romero, P.; Burny, A.; Badran, B.; Martiat, P.; et al. A microRNA profile of human CD8(+) regulatory T cells and characterization of the effects of microRNAs on Treg cell-associated genes. J. Transl. Med. 2014, 12, 218. [Google Scholar] [CrossRef] [PubMed]
- Jonuleit, H.; Bopp, T.; Becker, C. Treg cells as potential cellular targets for functionalized nanoparticles in cancer therapy. Nanomedicine (London, England) 2016, 11, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Naghavian, R.; Ghaedi, K.; Kiani-Esfahani, A.; Ganjalikhani-Hakemi, M.; Etemadifar, M.; Nasr-Esfahani, M.H. miR-141 and miR-200a, Revelation of New Possible Players in Modulation of Th17/Treg Differentiation and Pathogenesis of Multiple Sclerosis. PloS ONE 2015, 10, e0124555. [Google Scholar] [CrossRef]
- Zhou, J.; Li, X.; Wu, X.; Zhang, T.; Zhu, Q.; Wang, X.; Wang, H.; Wang, K.; Lin, Y.; Wang, X. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol. Res. 2018, 6, 1578–1592. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Bopp, T. Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front. Immunol. 2016, 7, 315. [Google Scholar] [CrossRef]
- Frick, S.U.; Domogalla, M.P.; Baier, G.; Wurm, F.R.; Mailänder, V.; Landfester, K.; Steinbrink, K. Interleukin-2 Functionalized Nanocapsules for T Cell-Based Immunotherapy. ACS Nano 2016, 10, 9216–9226. [Google Scholar] [CrossRef]
- Woller, N.; Knocke, S.; Mundt, B.; Gürlevik, E.; Strüver, N.; Kloos, A.; Boozari, B.; Schache, P.; Manns, M.P.; Malek, N.P.; et al. Virus-induced tumor inflammation facilitates effective DC cancer immunotherapy in a Treg-dependent manner in mice. J. Clin. Invest. 2011, 121, 2570–2582. [Google Scholar] [CrossRef]
- Al Sayed, M.F.; Amrein, M.A.; Bührer, E.D.; Huguenin, A.L.; Radpour, R.; Riether, C.; Ochsenbein, A.F. T-cell-Secreted TNFα Induces Emergency Myelopoiesis and Myeloid-Derived Suppressor Cell Differentiation in Cancer. Cancer Res. 2019, 79, 346–359. [Google Scholar] [CrossRef]
- Keskinov, A.A.; Shurin, M.R. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology 2015, 220, 236–242. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. (Berl.) 2019, 97, 1049–1064. [Google Scholar] [CrossRef]
- Bruger, A.M.; Dorhoi, A.; Esendagli, G.; Barczyk-Kahlert, K.; van der Bruggen, P.; Lipoldova, M.; Perecko, T.; Santibanez, J.; Saraiva, M.; Van Ginderachter, J.A.; et al. How to measure the immunosuppressive activity of MDSC: Assays, problems and potential solutions. Cancer Immunol. Immunother. 2019, 68, 631–644. [Google Scholar] [CrossRef]
- Zeng, Y.; Hahn, S.; Stokes, J.; Hoffman, E.A.; Schmelz, M.; Proytcheva, M.; Chernoff, J.; Katsanis, E. Pak2 regulates myeloid-derived suppressor cell development in mice. Blood Adv. 2017, 1, 1923–1933. [Google Scholar] [CrossRef]
- Fleet, J.C.; Burcham, G.N.; Calvert, R.D.; Elzey, B.D.; Ratliff, T.L. 1α, 25 Dihydroxyvitamin D (1,25(OH)(2)D) inhibits the T cell suppressive function of myeloid derived suppressor cells (MDSC). J. Steroid Biochem. Mol. Biol. 2020, 198, 105557. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J.; Ochoa, A.C. Editorial: Myeloid Derived Suppressor Cells as Disease Modulators. Front. Immunol. 2020, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Boros, P.; Ochando, J.; Zeher, M. Myeloid derived suppressor cells and autoimmunity. Hum. Immunol. 2016, 77, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.; Hartl, D. Myeloid-Derived Suppressor Cells in Infection: A General Overview. J. Innate Immun. 2018, 10, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Li, B.H.; Garstka, M.A.; Li, Z.F. Chemokines and their receptors promoting the recruitment of myeloid-derived suppressor cells into the tumor. Mol. Immunol. 2020, 117, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A.; et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, M.; Jiang, X.; Zhang, Z.; Dai, L.; Min, S.; Wu, X.; He, Q.; Liu, J.; Zhang, Y.; et al. miR-223 suppresses differentiation of tumor-induced CD11b+ Gr1+ myeloid-derived suppressor cells from bone marrow cells. Int. J. Cancer 2011, 129, 2662–2673. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Muroski, M.E.; Miska, J.; Lee-Chang, C.; Shen, Y.; Rashidi, A.; Zhang, P.; Xiao, T.; Han, Y.; Lopez-Rosas, A.; et al. Repolarization of myeloid derived suppressor cells via magnetic nanoparticles to promote radiotherapy for glioma treatment. Nanomedicine 2019, 16, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Shirota, H.; Tross, D.; Klinman, D.M. CpG Oligonucleotides as Cancer Vaccine Adjuvants. Vaccines 2015, 3, 390–407. [Google Scholar] [CrossRef]
- Lee, W.C.; Hsu, P.Y.; Hsu, H.Y. Stem cell factor produced by tumor cells expands myeloid-derived suppressor cells in mice. Sci. Rep. 2020, 10, 11257. [Google Scholar] [CrossRef]
- Kao, J.; Ko, E.C.; Eisenstein, S.; Sikora, A.G.; Fu, S.; Chen, S.H. Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit. Rev. Oncol. Hematol. 2011, 77, 12–19. [Google Scholar] [CrossRef]
- Shao, B.; Wei, X.; Luo, M.; Yu, J.; Tong, A.; Ma, X.; Ye, T.; Deng, H.; Sang, Y.; Liang, X.; et al. Inhibition of A20 expression in tumor microenvironment exerts anti-tumor effect through inducing myeloid-derived suppressor cells apoptosis. Sci. Rep. 2015, 5, 16437. [Google Scholar] [CrossRef]
- Fujii, H.; Shin-Ya, M.; Takeda, S.; Hashimoto, Y.; Mukai, S.A.; Sawada, S.; Adachi, T.; Akiyoshi, K.; Miki, T.; Mazda, O. Cycloamylose-nanogel drug delivery system-mediated intratumor silencing of the vascular endothelial growth factor regulates neovascularization in tumor microenvironment. Cancer Sci. 2014, 105, 1616–1625. [Google Scholar] [CrossRef]
- Ni, J.; Galani, I.E.; Cerwenka, A.; Schirrmacher, V.; Fournier, P. Antitumor vaccination by Newcastle Disease Virus Hemagglutinin-Neuraminidase plasmid DNA application: Changes in tumor microenvironment and activation of innate anti-tumor immunity. Vaccine 2011, 29, 1185–1193. [Google Scholar] [CrossRef]
- Principe, M.; Ceruti, P.; Shih, N.Y.; Chattaragada, M.S.; Rolla, S.; Conti, L.; Bestagno, M.; Zentilin, L.; Yang, S.H.; Migliorini, P.; et al. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget 2015, 6, 11098–11113. [Google Scholar] [CrossRef] [PubMed]
- Cappello, P.; Rolla, S.; Chiarle, R.; Principe, M.; Cavallo, F.; Perconti, G.; Feo, S.; Giovarelli, M.; Novelli, F. Vaccination with ENO1 DNA prolongs survival of genetically engineered mice with pancreatic cancer. Gastroenterology 2013, 144, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Bachmann, M.; Bergmann, R.; Berndt, N.; Feldmann, A.; Koristka, S. Theranostic CAR T cell targeting: A brief review. J. Labelled Comp. Radiopharm. 2019, 62, 533–540. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Puré, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Darowski, D.; Jost, C.; Stubenrauch, K.; Wessels, U.; Benz, J.; Ehler, A.; Freimoser-Grundschober, A.; Brünker, P.; Mössner, E.; Umaña, P.; et al. P329G-CAR-J: A novel Jurkat-NFAT-based CAR-T reporter system recognizing the P329G Fc mutation. Protein Eng. Des. Sel. 2019, 32, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.H.; Hughes, G.; Koffman, B.; Turtle, C.J.; Maloney, D.G.; Acharya, U.H. Not so crystal clear: Observations from a case of crystalline arthritis with cytokine release syndrome (CRS) after chimeric antigen receptor (CAR)-T cell therapy. Bone Marrow Transplant. 2019, 54, 632–634. [Google Scholar] [CrossRef]
- Rohrs, J.A.; Siegler, E.L.; Wang, P.; Finley, S.D. ERK Activation in CAR T Cells Is Amplified by CD28-Mediated Increase in CD3ζ Phosphorylation. iScience 2020, 23, 101023. [Google Scholar] [CrossRef]
- Kintz, H.; Nylen, E.; Barber, A. Inclusion of Dap10 or 4–1BB costimulation domains in the chPD1 receptor enhances anti-tumor efficacy of T cells in murine models of lymphoma and melanoma. Cell Immunol. 2020, 351, 104069. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Oberschmidt, O.; Kloess, S.; Koehl, U. Redirected Primary Human Chimeric Antigen Receptor Natural Killer Cells As an “Off-the-Shelf Immunotherapy” for Improvement in Cancer Treatment. Front. Immunol. 2017, 8, 654. [Google Scholar] [CrossRef] [PubMed]
- Sievers, N.M.; Dörrie, J.; Schaft, N. CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int. J. Mol. Sci. 2020, 21, 3525. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Turtle, C.J. Toxicities of CD19 CAR-T cell immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef]
- Belay, Y.; Yirdaw, K.; Enawgaw, B. Tumor Lysis Syndrome in Patients with Hematological Malignancies. J. Oncol. 2017, 2017, 9684909. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Cervantes, E.V.; Boucher, J.C.; Lee, S.B.; Spitler, K.; Reid, K.; Davila, M.L. MDSC Suppression of CAR T Cells Can be Reduced By Targeted Signaling Disruption. Blood 2019, 134, 4438. [Google Scholar] [CrossRef]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Joseph Espat, N.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Zhang, Y.; Shang, Y.; Gao, Q. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget 2016, 7, 4760–4769. [Google Scholar] [CrossRef] [PubMed]
- Crotti, C.; Agape, E.; Becciolini, A.; Biggioggero, M.; Favalli, E.G. Targeting Granulocyte-Monocyte Colony-Stimulating Factor Signaling in Rheumatoid Arthritis: Future Prospects. Drugs 2019, 79, 1741–1755. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Fultang, L.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with Gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine 2019, 47, 235–246. [Google Scholar] [CrossRef]
- Wang, H.; Ye, X.; Ju, Y.; Cai, Z.; Wang, X.; Du, P.; Zhang, M.; Li, Y.; Cai, J. Minicircle DNA-Mediated CAR T Cells Targeting CD44 Suppressed Hepatocellular Carcinoma Both in vitro and in vivo. Onco Targets Ther. 2020, 13, 3703–3716. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687. [Google Scholar] [CrossRef] [PubMed]
- Östman, A. The tumor microenvironment controls drug sensitivity. Nat. Med. 2012, 18, 1332–1334. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Prenen, H.; Mazzone, M. Tumor-associated macrophages: A short compendium. Cell. Mol. Life Sci. 2019, 76, 1447–1458. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef]
- Badalamenti, G.; Fanale, D.; Incorvaia, L.; Barraco, N.; Listì, A.; Maragliano, R.; Vincenzi, B.; Calò, V.; Iovanna, J.L.; Bazan, V.; et al. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell Immunol. 2019, 343, 103753. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148. [Google Scholar] [CrossRef]
- Ferreira, L.M.R. Cancer metabolism: The Warburg effect today. Exp. Mol. Pathol. 2010, 89, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Kuppusamy, P.; Li, H.; Ilangovan, G.; Cardounel, A.J.; Zweier, J.L.; Yamada, K.; Krishna, M.C.; Mitchell, J.B. Noninvasive Imaging of Tumor Redox Status and Its Modification by Tissue Glutathione Levels. Cancer Res. 2002, 62, 307. [Google Scholar] [PubMed]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Hager, S.; Wagner, E. Bioresponsive polyplexes - chemically programmed for nucleic acid delivery. Expert Opin. Drug Deliv. 2018, 15, 1067–1083. [Google Scholar] [CrossRef]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef]
- Ruoslahti, E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater. 2012, 24, 3747–3756. [Google Scholar] [CrossRef]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef]
- Lächelt, U.; Wagner, E. Nucleic Acid Therapeutics Using Polyplexes: A Journey of 50 Years (and Beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Kulmburg, P.; Veelken, H.; Mackensen, A.; Mézes, B.; Lindemann, A.; Mertelsmann, R.; Rosenthal, F.M. Cytokine gene transfer in cancer therapy. Stem Cells 1998, 16, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Parmiani, G.; Rivoltini, L.; Andreola, G.; Carrabba, M. Cytokines in cancer therapy. Immunol. Lett. 2000, 74, 41–44. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Golomb, H.M.; Jacobs, A.; Fefer, A.; Ozer, H.; Thompson, J.; Portlock, C.; Ratain, M.; Golde, D.; Vardiman, J.; Burke, J.S. Alpha-2 interferon therapy of hairy-cell leukemia: A multicenter study of 64 patients. J. Clin. Oncol. 1986, 4, 900–905. [Google Scholar] [CrossRef]
- Antony, G.K.; Dudek, A.Z. Interleukin 2 in Cancer Therapy. Curr. Med. Chem. 2010, 17, 3297–3302. [Google Scholar] [CrossRef]
- Schreiber, S.; Kämpgen, E.; Wagner, E.; Pirkhammer, D.; Trcka, J.; Korschan, H.; Lindemann, A.; Dorffner, R.; Kittler, H.; Kasteliz, F.; et al. Immunotherapy of metastatic malignant melanoma by a vaccine consisting of autologous interleukin 2-transfected cancer cells: Outcome of a phase I study. Hum. Gene Ther. 1999, 10, 983–993. [Google Scholar] [CrossRef]
- Gansbacher, B.; Houghton, A.; Livingston, P.; Minasian, L.; Rosenthal, F.; Gilboa, E.; Oettgen, H.; Steffens, T.; Yang, S.Y.; Wong, G. A Pilot Study of Immunization with HLA-A2 Matched Allogeneic Melanoma Cells That Secrete Interleukin-2 in Patients with Metastatic Melanoma. Hum. Gene Ther. 1992, 3, 677–690. [Google Scholar] [CrossRef]
- Osanto, S.; Brouwenstÿn, N.; Vaessen, N.; Figdor, C.G.; Melief, C.J.; Schrier, P.I. Immunization with interleukin-2 transfected melanoma cells. A phase I-II study in patients with metastatic melanoma. Hum. Gene Ther. 1993, 4, 323–330. [Google Scholar] [CrossRef]
- Bowman, L.C.; Grossmann, M.; Rill, D.; Brown, M.; Zhong, W.Y.; Alexander, B.; Leimig, T.; Coustan-Smith, E.; Campana, D.; Jenkins, J.; et al. Interleukin-2 gene-modified allogeneic tumor cells for treatment of relapsed neuroblastoma. Hum. Gene Ther. 1998, 9, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Kali, A. TNFerade, an innovative cancer immunotherapeutic. Indian J. Pharmacol. 2015, 47, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Wagner, E. Technology evaluation: TNFerade, GenVec. Curr. Opin. Mol. Ther. 2003, 5, 437–447. [Google Scholar] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J., III; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Duplisea, J.J.; Mokkapati, S.; Plote, D.; Schluns, K.S.; McConkey, D.J.; Yla-Herttuala, S.; Parker, N.R.; Dinney, C.P. The development of interferon-based gene therapy for BCG unresponsive bladder cancer: From bench to bedside. World J. Urol. 2019, 37, 2041–2049. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Nemunaitis, M.; Senzer, N.; Snitz, P.; Bedell, C.; Kumar, P.; Pappen, B.; Maples, P.B.; Shawler, D.; Fakhrai, H. Phase II trial of Belagenpumatucel-L, a TGF-beta2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009, 16, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Song, X.; Su, L.; Zheng, Y.; Li, R.; Sun, J. New therapeutic strategies based on IL-2 to modulate Treg cells for autoimmune diseases. Int. Immunopharmacol. 2019, 72, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Küpcü, Z.; Wallner, G.; Wagner, E. Cytokine gene-modified tumor cells for prophylactic and therapeutic vaccination: IL-2, IFN-gamma, or combination IL-2 + IFN-gamma. Cytokines Cell. Mol. Ther. 1998, 4, 95–103. [Google Scholar]
- Rosenberg, S.A.; Anderson, W.F.; Blaese, M.R.; Ettinghausen, S.E.; Hwu, P.; Karp, S.E.; Kasid, A.; Mule, J.J.; Parkinson, D.R.; Salo, J.C.; et al. Immunization of Cancer Patients Using Autologous Cancer Cells Modified by Insertion of the Gene for Interleukin-2 (National Institutes of Health). Hum. Gene Ther. 1992, 3, 75–90. [Google Scholar] [CrossRef]
- Kircheis, R.; Küpcü, Z.; Wallner, G.; Rössler, V.; Schweighoffer, T.; Wagner, E. Interleukin-2 gene-modified allogeneic melanoma cell vaccines can induce cross-protection against syngeneic tumors in mice. Cancer Gene Ther. 2000, 7, 870–878. [Google Scholar] [CrossRef][Green Version]
- Wagner, E.; Zatloukal, K.; Cotten, M.; Kirlappos, H.; Mechtler, K.; Curiel, D.T.; Birnstiel, M.L. Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes. Proc. Natl. Acad. Sci. USA 1992, 89, 6099–6103. [Google Scholar] [CrossRef]
- Vile, R.; Miller, N.; Chernajovsky, Y.; Hart, I. A comparison of the properties of different retroviral vectors containing the murine tyrosinase promoter to achieve transcriptionally targeted expression of the HSVtk or IL-2 genes. Gene Ther. 1994, 1, 307–316. [Google Scholar]
- He, P.; Tang, Z.Y.; Liu, B.B.; Ye, S.L.; Liu, Y.K. The targeted expression of the human interleukin-2/interferon alpha2b fused gene in alpha-fetoprotein-expressing hepatocellular carcinoma cells. J. Cancer Res. Clin. Oncol. 1999, 125, 77–82. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Hew, P.; Crosley, P.; Sharon, D.; Potts, K.; Agopsowicz, K.; Long, M.; Shi, C.; Hitt, M.M. Breast cancer gene therapy using an adenovirus encoding human IL-2 under control of mammaglobin promoter/enhancer sequences. Cancer Gene Ther. 2016, 23, 178–187. [Google Scholar] [CrossRef]
- Herman, J.M.; Wild, A.T.; Wang, H.; Tran, P.T.; Chang, K.J.; Taylor, G.E.; Donehower, R.C.; Pawlik, T.M.; Ziegler, M.A.; Cai, H.; et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: Final results. J. Clin. Oncol. 2013, 31, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Bottermann, M.; Foss, S.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; Higginson, K.; Hirst, J.C.; et al. TRIM21 mediates antibody inhibition of adenovirus-based gene delivery and vaccination. Proc. Natl. Acad. Sci. USA 2018, 115, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Kircheis, R.; Ostermann, E.; Wolschek, M.F.; Lichtenberger, C.; Magin-Lachmann, C.; Wightman, L.; Kursa, M.; Wagner, E. Tumor-targeted gene delivery of tumor necrosis factor-α induces tumor necrosis and tumor regression without systemic toxicity. Cancer Gene Ther. 2002, 9, 673–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, B.; Cengizeroglu, A.; Farkasova, K.; Viola, J.R.; Anton, M.; Ellwart, J.W.; Haase, R.; Wagner, E.; Ogris, M. Systemic TNFα Gene Therapy Synergizes With Liposomal Doxorubicine in the Treatment of Metastatic Cancer. Mol. Ther. 2013, 21, 300–308. [Google Scholar] [CrossRef]
- Russ, V.; Günther, M.; Halama, A.; Ogris, M.; Wagner, E. Oligoethylenimine-grafted polypropylenimine dendrimers as degradable and biocompatible synthetic vectors for gene delivery. J. Controlled Release 2008, 132, 131–140. [Google Scholar] [CrossRef]
- Schäfer, A.; Pahnke, A.; Schaffert, D.; van Weerden, W.M.; de Ridder, C.M.; Rödl, W.; Vetter, A.; Spitzweg, C.; Kraaij, R.; Wagner, E.; et al. Disconnecting the yin and yang relation of epidermal growth factor receptor (EGFR)-mediated delivery: A fully synthetic, EGFR-targeted gene transfer system avoiding receptor activation. Hum. Gene Ther. 2011, 22, 1463–1473. [Google Scholar] [CrossRef]
- Tandle, A.; Hanna, E.; Lorang, D.; Hajitou, A.; Moya, C.A.; Pasqualini, R.; Arap, W.; Adem, A.; Starker, E.; Hewitt, S.; et al. Tumor vasculature-targeted delivery of tumor necrosis factor-α*. Cancer 2009, 115, 128–139. [Google Scholar] [CrossRef]
- Yuan, Z.; Syrkin, G.; Adem, A.; Geha, R.; Pastoriza, J.; Vrikshajanani, C.; Smith, T.; Quinn, T.J.; Alemu, G.; Cho, H.; et al. Blockade of inhibitors of apoptosis (IAPs) in combination with tumor-targeted delivery of tumor necrosis factor-α leads to synergistic antitumor activity. Cancer Gene Ther. 2013, 20, 46–56. [Google Scholar] [CrossRef]
- Quinn, T.J.; Healy, N.; Sara, A.; Maggi, E.; Claros, C.S.; Kabarriti, R.; Scandiuzzi, L.; Liu, L.; Gorecka, J.; Adem, A.; et al. Preclinical evaluation of radiation and systemic, RGD-targeted, adeno-associated virus phage-TNF gene therapy in a mouse model of spontaneously metastatic melanoma. Cancer Gene Ther. 2017, 24, 13–19. [Google Scholar] [CrossRef]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef]
- Voest, E.E.; Kenyon, B.M.; O’Reilly, M.S.; Truitt, G.; D’Amato, R.J.; Folkman, J. Inhibition of angiogenesis in vivo by interleukin 12. J. Natl. Cancer Inst. 1995, 87, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Boyd, R.; Balkwill, F. IL-12 regulates VEGF and MMPs in a murine breast cancer model. Int. J. Cancer 1998, 78, 361–365. [Google Scholar] [CrossRef]
- Del Vecchio, M.; Bajetta, E.; Canova, S.; Lotze, M.T.; Wesa, A.; Parmiani, G.; Anichini, A. Interleukin-12: Biological properties and clinical application. Clin. Cancer Res. 2007, 13, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. IL-12 Deaths: Explanation and a Puzzle. Science 1995, 270, 908. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Sherman, M.L.; Fisher, G.L.; Buchanan, L.J.; Larsen, G.; Atkins, M.B.; Sosman, J.A.; Dutcher, J.P.; Vogelzang, N.J.; Ryan, J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997, 90, 2541–2548. [Google Scholar] [PubMed]
- Pasche, N.; Neri, D. Immunocytokines: A novel class of potent armed antibodies. Drug Discov. Today 2012, 17, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Rudman, S.M.; Jameson, M.B.; McKeage, M.J.; Savage, P.; Jodrell, D.I.; Harries, M.; Acton, G.; Erlandsson, F.; Spicer, J.F. A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin. Cancer. Res. 2011, 17, 1998–2005. [Google Scholar] [CrossRef]
- Hernandez-Alcoceba, R.; Poutou, J.; Ballesteros-Briones, M.C.; Smerdou, C. Gene therapy approaches against cancer using in vivo and ex vivo gene transfer of interleukin-12. Immunotherapy 2016, 8, 179–198. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef]
- Sangro, B.; Mazzolini, G.; Ruiz, J.; Herraiz, M.; Quiroga, J.; Herrero, I.; Benito, A.; Larrache, J.; Pueyo, J.; Subtil, J.C.; et al. Phase I Trial of Intratumoral Injection of an Adenovirus Encoding Interleukin-12 for Advanced Digestive Tumors. J. Clin. Oncol. 2004, 22, 1389–1397. [Google Scholar] [CrossRef]
- Triozzi, P.L.; Strong, T.V.; Bucy, R.P.; Allen, K.O.; Carlisle, R.R.; Moore, S.E.; Lobuglio, A.F.; Conry, R.M. Intratumoral administration of a recombinant canarypox virus expressing interleukin 12 in patients with metastatic melanoma. Hum. Gene Ther. 2005, 16, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Allen, K.O.; Carlisle, R.R.; Craig, M.; LoBuglio, A.F.; Conry, R.M. Phase I study of the intratumoral administration of recombinant canarypox viruses expressing B7.1 and interleukin 12 in patients with metastatic melanoma. Clin. Cancer. Res. 2005, 11, 4168–4175. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Hamid, O.; Whitman, E.D.; Nemunaitis, J.J.; Chesney, J.; Agarwala, S.S.; Starodub, A.; Barrett, J.A.; Marsh, A.; Martell, L.A.; et al. A phase I open-label study of Ad-RTS-hIL-12, an adenoviral vector engineered to express hIL-12 under the control of an oral activator ligand, in subjects with unresectable stage III/IV melanoma. J. Clin. Oncol. 2013, 31, 3022. [Google Scholar] [CrossRef]
- Quetglas, J.I.; Labiano, S.; Aznar, M.Á.; Bolaños, E.; Azpilikueta, A.; Rodriguez, I.; Casales, E.; Sánchez-Paulete, A.R.; Segura, V.; Smerdou, C.; et al. Virotherapy with a Semliki Forest Virus–Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol. Res. 2015, 3, 449. [Google Scholar] [CrossRef]
- Yang, X.; Yu, X.; Wei, Y. Lentiviral delivery of novel fusion protein IL12/FasTI for cancer immune/gene therapy. PLoS ONE 2018, 13, e0201100. [Google Scholar] [CrossRef]
- Lucas, M.L.; Heller, L.; Coppola, D.; Heller, R. IL-12 plasmid delivery by in vivo electroporation for the successful treatment of established subcutaneous B16.F10 melanoma. Mol. Ther. 2002, 5, 668–675. [Google Scholar] [CrossRef]
- Heinzerling, L.; Burg, G.; Dummer, R.; Maier, T.; Oberholzer, P.A.; Schultz, J.; Elzaouk, L.; Pavlovic, J.; Moelling, K. Intratumoral injection of DNA encoding human interleukin 12 into patients with metastatic melanoma: Clinical efficacy. Hum. Gene Ther. 2005, 16, 35–48. [Google Scholar] [CrossRef]
- Mahvi, D.M.; Henry, M.B.; Albertini, M.R.; Weber, S.; Meredith, K.; Schalch, H.; Rakhmilevich, A.; Hank, J.; Sondel, P. Intratumoral injection of IL-12 plasmid DNA--results of a phase I/IB clinical trial. Cancer Gene Ther. 2007, 14, 717–723. [Google Scholar] [CrossRef]
- Daud, A.I.; DeConti, R.C.; Andrews, S.; Urbas, P.; Riker, A.I.; Sondak, V.K.; Munster, P.N.; Sullivan, D.M.; Ugen, K.E.; Messina, J.L.; et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J. Clin. Oncol. 2008, 26, 5896–5903. [Google Scholar] [CrossRef]
- Cutrera, J.; King, G.; Jones, P.; Kicenuik, K.; Gumpel, E.; Xia, X.; Li, S. Safety and efficacy of tumor-targeted interleukin 12 gene therapy in treated and non-treated, metastatic lesions. Curr. Gene Ther. 2015, 15, 44–54. [Google Scholar] [CrossRef]
- Cemazar, M.; Ambrozic Avgustin, J.; Pavlin, D.; Sersa, G.; Poli, A.; Krhac Levacic, A.; Tesic, N.; Lampreht Tratar, U.; Rak, M.; Tozon, N. Efficacy and safety of electrochemotherapy combined with peritumoral IL-12 gene electrotransfer of canine mast cell tumours. Vet. Comp. Oncol. 2017, 15, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Cicchelero, L.; Denies, S.; Haers, H.; Vanderperren, K.; Stock, E.; Van Brantegem, L.; de Rooster, H.; Sanders, N.N. Intratumoural interleukin 12 gene therapy stimulates the immune system and decreases angiogenesis in dogs with spontaneous cancer. Vet. Comp. Oncol. 2017, 15, 1187–1205. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo-Garzón, M.; Berraondo, P.; Ochoa, L.; Zulueta, J.J.; González-Aseguinolaza, G. Antitumoral efficacy of DNA nanoparticles in murine models of lung cancer and pulmonary metastasis. Cancer Gene Ther. 2010, 17, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.; Barnes, M.N.; Fewell, J.; Lewis, D.H.; Alvarez, R.D. Phase-I clinical trial of IL-12 plasmid/lipopolymer complexes for the treatment of recurrent ovarian cancer. Gene Ther. 2010, 17, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.; Kelly, F.J.; Chu, C.; Fewell, J.G.; Lewis, D.; Alvarez, R.D. Phase I trial of a formulated IL-12 plasmid in combination with carboplatin and docetaxel chemotherapy in the treatment of platinum-sensitive recurrent ovarian cancer. Gynecol. Oncol. 2013, 131, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Men, K.; Huang, R.; Zhang, X.; Zhang, R.; Zhang, Y.; He, M.; Tong, R.; Yang, L.; Wei, Y.; Duan, X. Local and Systemic Delivery of Interleukin-12 Gene by Cationic Micelles for Cancer Immunogene Therapy. J. Biomed. Nanotechnol. 2018, 14, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lee, S.; McDermott, D.F.; Rao, U.N.; Butterfield, L.H.; Tarhini, A.A.; Leming, P.; Puzanov, I.; Shin, D.; Kirkwood, J.M. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: A randomized clinical trial. JAMA 2014, 312, 1744–1753. [Google Scholar] [CrossRef]
- Sterner, R.M.; Cox, M.J.; Sakemura, R.; Kenderian, S.S. Using CRISPR/Cas9 to Knock Out GM-CSF in CAR-T Cells. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Meng, W.; Xue, S.; Chen, Y. The role of CXCL12 in tumor microenvironment. Gene 2018, 641, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wang, J.; Li, B.; Xiang, H.; Ultsch, M.; Coons, M.; Wong, T.; Chiang, N.Y.; Clark, S.; Clark, R.; et al. Development and preclinical characterization of a humanized antibody targeting CXCL12. Clin. Cancer Res. 2013, 19, 4433–4445. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, T.J.; Zhou, Y.; Musetti, S.N.; Liu, R.; Huang, L. Local and transient gene expression primes the liver to resist cancer metastasis. Sci. Transl. Med. 2016, 8, 364ra153. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Li, J.; Liu, Q.; Feng, R.; Das, M.; Lin, C.M.; Goodwin, T.J.; Dorosheva, O.; Liu, R.; Huang, L. Transient and Local Expression of Chemokine and Immune Checkpoint Traps To Treat Pancreatic Cancer. ACS Nano 2017, 11, 8690–8706. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Haynes, M.T.; Wang, Y.; Liu, F.; Huang, L. A highly efficient synthetic vector: Nonhydrodynamic delivery of DNA to hepatocyte nuclei in vivo. ACS Nano 2013, 7, 5376–5384. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, A.C.; Rothenberg, M.L.; Dupont, J.; Cooper, W.; Chevalier, P.; Sternas, L.; Buzenet, G.; Koehler, E.; Sosman, J.A.; Schwartz, L.H.; et al. Phase I Study of Intravenous Vascular Endothelial Growth Factor Trap, Aflibercept, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2009, 28, 207–214. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Frankel, P.; Margolin, K.A.; Christensen, S.; Ruel, C.; Shipe-Spotloe, J.; Gandara, D.R.; Chen, A.; Kirkwood, J.M. Aflibercept (VEGF Trap) in Inoperable Stage III or Stage IV Melanoma of Cutaneous or Uveal Origin. Clin. Cancer Res. 2011, 17, 6574. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Melosky, B.; Reardon, D.A.; Nixon, A.B.; Subramanian, J.; Bair, A.H.; Jacobs, I. Bevacizumab biosimilars: Scientific justification for extrapolation of indications. Future Oncol. 2018, 14, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Vennepureddy, A.; Singh, P.; Rastogi, R.; Atallah, J.P.; Terjanian, T. Evolution of ramucirumab in the treatment of cancer - A review of literature. J. Oncol. Pharm. Pract. 2017, 23, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Tekade, R.K.; Tekade, M.; Kesharwani, P.; D’Emanuele, A. RNAi-combined nano-chemotherapeutics to tackle resistant tumors. Drug Discov. Today 2016, 21, 1761–1774. [Google Scholar] [CrossRef]
- Kanazawa, T.; Sugawara, K.; Tanaka, K.; Horiuchi, S.; Takashima, Y.; Okada, H. Suppression of tumor growth by systemic delivery of anti-VEGF siRNA with cell-penetrating peptide-modified MPEG-PCL nanomicelles. Eur. J. Pharm. Biopharm. 2012, 81, 470–477. [Google Scholar] [CrossRef]
- Egorova, A.; Shubina, A.; Sokolov, D.; Selkov, S.; Baranov, V.; Kiselev, A. CXCR4-targeted modular peptide carriers for efficient anti-VEGF siRNA delivery. Int. J. Pharm. 2016, 515, 431–440. [Google Scholar] [CrossRef]
- Chung, J.Y.; Ul Ain, Q.; Lee, H.L.; Kim, S.M.; Kim, Y.H. Enhanced Systemic Anti-Angiogenic siVEGF Delivery Using PEGylated Oligo-d-arginine. Mol. Pharm. 2017, 14, 3059–3068. [Google Scholar] [CrossRef]
- Lee, Y.W.; Hwang, Y.E.; Lee, J.Y.; Sohn, J.H.; Sung, B.H.; Kim, S.C. VEGF siRNA Delivery by a Cancer-Specific Cell-Penetrating Peptide. J. Microbiol. Biotechnol. 2018, 28, 367–374. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. Local and systemic delivery of VEGF siRNA using polyelectrolyte complex micelles for effective treatment of cancer. J. Controlled Release 2008, 129, 107–116. [Google Scholar] [CrossRef]
- Jiang, G.; Park, K.; Kim, J.; Kim, K.S.; Hahn, S.K. Target specific intracellular delivery of siRNA/PEI-HA complex by receptor mediated endocytosis. Mol. Pharm. 2009, 6, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Shukla, R.; Liu, H.; Jain, A.; Barve, A.; Cheng, K. Development of a Biocompatible Copolymer Nanocomplex to Deliver VEGF siRNA for Triple Negative Breast Cancer. Theranostics 2019, 9, 4508–4524. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; Jo, S.D.; Yhee, J.Y.; Lee, B.S.; Lee, S.J.; Park, S.G.; Kang, S.W.; Kim, S.H.; Jeong, J.H. Synergistic anti-tumor effects of bevacizumab and tumor targeted polymerized VEGF siRNA nanoparticles. Biochem. Biophys. Res. Commun. 2017, 489, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Li, J.Q.; Wang, Z.Z.; Dong, D.W.; Qi, X.R. Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials 2014, 35, 5226–5239. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Chen, W.L.; Wang, D.D.; Zhou, Y.J.; You, B.G.; Liu, Y.; Qu, C.X.; Yang, S.D.; Chen, M.T.; et al. Co-delivery of VEGF siRNA and Etoposide for Enhanced Anti-angiogenesis and Anti-proliferation Effect via Multi-functional Nanoparticles for Orthotopic Non-Small Cell Lung Cancer Treatment. Theranostics 2019, 9, 5886–5898. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Bao, C.; Tan, Y.; Cui, D.; Edelman, E.R.; Azevedo, H.S.; Byrne, H.J.; Artzi, N.; Tian, F. Dual targeted immunotherapy via in vivo delivery of biohybrid RNAi-peptide nanoparticles to tumour-associated macrophages and cancer cells. Adv. Funct. Mater. 2015, 25, 4183–4194. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, X.; Cui, C.; Li, J.; Wang, Y. Doxorubicin and anti-VEGF siRNA co-delivery via nano-graphene oxide for enhanced cancer therapy in vitro and in vivo. Int. J. Nanomed. 2018, 13, 3713–3728. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccin. Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-β signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Liu, F.; Ji, K.; Liu, N.; He, Y.; Zhang, W.; Wang, L. MicroRNA-381 inhibits the metastasis of gastric cancer by targeting TMEM16A expression. J. Exp. Clin. Cancer Res. 2017, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.J.; Bao, R.F.; Jiang, L.; Wang, Z.; Wang, X.A.; Zhang, F.; Liang, H.B.; Li, H.F.; Ye, Y.Y.; Xiang, S.S.; et al. MicroRNA-29c-5p suppresses gallbladder carcinoma progression by directly targeting CPEB4 and inhibiting the MAPK pathway. Cell Death Differ. 2017, 24, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Huang, B.; Sun, S.; Xiao, M.; Guo, J.; Yi, X.; Cai, J.; Wang, Z. miR-27a inhibits cervical adenocarcinoma progression by downregulating the TGF-βRI signaling pathway. Cell Death Dis. 2018, 9, 395. [Google Scholar] [CrossRef]
- Schlingensiepen, R.; Goldbrunner, M.; Szyrach, M.N.; Stauder, G.; Jachimczak, P.; Bogdahn, U.; Schulmeyer, F.; Hau, P.; Schlingensiepen, K.H. Intracerebral and intrathecal infusion of the TGF-beta 2-specific antisense phosphorothioate oligonucleotide AP 12009 in rabbits and primates: Toxicology and safety. Oligonucleotides 2005, 15, 94–104. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Schlingensiepen, R.; Steinbrecher, A.; Hau, P.; Bogdahn, U.; Fischer-Blass, B.; Jachimczak, P. Targeted tumor therapy with the TGF-beta 2 antisense compound AP 12009. Cytokine Growth Factor Rev. 2006, 17, 129–139. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Hau, P.; Jachimczak, P.; Schlingensiepen, R.; Schulmeyer, F.; Jauch, T.; Steinbrecher, A.; Brawanski, A.; Proescholdt, M.; Schlaier, J.; Buchroithner, J.; et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: From preclinical to phase I/II studies. Oligonucleotides 2007, 17, 201–212. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF-β2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef]
- Zhu, J.; Liu, J.Q.; Shi, M.; Cheng, X.; Ding, M.; Zhang, J.C.; Davis, J.P.; Varikuti, S.; Satoskar, A.R.; Lu, L.; et al. IL-27 gene therapy induces depletion of Tregs and enhances the efficacy of cancer immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Ueda, R.; Narumi, K.; Heike, Y.; Yoshida, T.; Aoki, K. Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther. 2014, 21, 532–541. [Google Scholar] [CrossRef]
- Hirata, A.; Hashimoto, H.; Shibasaki, C.; Narumi, K.; Aoki, K. Intratumoral IFN-α gene delivery reduces tumor-infiltrating regulatory T cells through the downregulation of tumor CCL17 expression. Cancer Gene Ther. 2019, 26, 334–343. [Google Scholar] [CrossRef]
- Byrne, W.L.; Mills, K.H.G.; Lederer, J.A.; Sullivan, G.C. Targeting Regulatory T Cells in Cancer. Cancer Res. 2011, 71, 6915. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.F.M.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.M.; Adema, G.J. Regulatory T cells in melanoma: The final hurdle towards effective immunotherapy? Lancet Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Pfeffer, L.M.; Dinarello, C.A.; Herberman, R.B.; Williams, B.R.G.; Borden, E.C.; Bordens, R.; Walter, M.R.; Nagabhushan, T.L.; Trotta, P.P.; Pestka, S. Biological Properties of Recombinant α-Interferons: 40th Anniversary of the Discovery of Interferons. Cancer Res. 1998, 58, 2489. [Google Scholar] [PubMed]
- Iqbal Ahmed, C.M.; Johnson, D.E.; Demers, G.W.; Engler, H.; Howe, J.A.; Wills, K.N.; Wen, S.F.; Shinoda, J.; Beltran, J.; Nodelman, M.; et al. Interferon alpha2b gene delivery using adenoviral vector causes inhibition of tumor growth in xenograft models from a variety of cancers. Cancer Gene Ther. 2001, 8, 788–795. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Sadreddini, S.; Baradaran, B.; Aghebati-Maleki, A.; Sadreddini, S.; Shanehbandi, D.; Fotouhi, A.; Aghebati-Maleki, L. Immune checkpoint blockade opens a new way to cancer immunotherapy. J. Cell. Physiol. 2019, 234, 8541–8549. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Taams, L.S.; de Gruijl, T.D. Immune checkpoint inhibition: From molecules to clinical application. Clin. Exp. Immunol. 2020, 200, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016, 6, 827. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future Challenges in Cancer Resistance to Immunotherapy. Cancers 2020, 12, 935. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef]
- Lamichhane, P.; Amin, N.P.; Agarwal, M.; Lamichhane, N. Checkpoint Inhibition: Will Combination with Radiotherapy and Nanoparticle-Mediated Delivery Improve Efficacy? Medicines 2018, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Salem, J.-E.; Cohen, J.V.; Chandra, S.; Menzer, C.; Ye, F.; Zhao, S.; Das, S.; Beckermann, K.E.; Ha, L.; et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Urwyler, P.; Earnshaw, I.; Bermudez, M.; Perucha, E.; Wu, W.; Ryan, S.; McDonald, L.; Karagiannis, S.N.; Taams, L.S.; Powell, N.; et al. Mechanisms of checkpoint inhibition-induced adverse events. Clin. Exp. Immunol. 2020, 200, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, S.K.; Boczkowski, D.; de Rosa, N.; Haley, N.R.; Morse, M.A.; Tyler, D.S.; Dannull, J.; Nair, S. Enhancement of anti-tumor immunity through local modulation of CTLA-4 and GITR by dendritic cells. Eur. J. Immunol. 2011, 41, 3553–3563. [Google Scholar] [CrossRef]
- Goodwin, T.J.; Shen, L.; Hu, M.; Li, J.; Feng, R.; Dorosheva, O.; Liu, R.; Huang, L. Liver specific gene immunotherapies resolve immune suppressive ectopic lymphoid structures of liver metastases and prolong survival. Biomaterials 2017, 141, 260–271. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef] [PubMed]
- Teo, P.Y.; Yang, C.; Whilding, L.M.; Parente-Pereira, A.C.; Maher, J.; George, A.J.; Hedrick, J.L.; Yang, Y.Y.; Ghaem-Maghami, S. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: Strategies to enhance T cell killing. Adv. Healthc. Mater. 2015, 4, 1180–1189. [Google Scholar] [CrossRef]
- Wang, D.; Wang, T.; Liu, J.; Yu, H.; Jiao, S.; Feng, B.; Zhou, F.; Fu, Y.; Yin, Q.; Zhang, P.; et al. Acid-Activatable Versatile Micelleplexes for PD-L1 Blockade-Enhanced Cancer Photodynamic Immunotherapy. Nano Lett. 2016, 16, 5503–5513. [Google Scholar] [CrossRef]
- Kwak, G.; Kim, D.; Nam, G.H.; Wang, S.Y.; Kim, I.S.; Kim, S.H.; Kwon, I.C.; Yeo, Y. Programmed Cell Death Protein Ligand-1 Silencing with Polyethylenimine-Dermatan Sulfate Complex for Dual Inhibition of Melanoma Growth. ACS Nano 2017, 11, 10135–10146. [Google Scholar] [CrossRef]
- Li, G.; Gao, Y.; Gong, C.; Han, Z.; Qiang, L.; Tai, Z.; Tian, J.; Gao, S. Dual-Blockade Immune Checkpoint for Breast Cancer Treatment Based on a Tumor-Penetrating Peptide Assembling Nanoparticle. ACS Appl. Mater. Interfaces 2019, 11, 39513–39524. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Wan, W.J.; Tong, Y.; Chen, M.T.; Wang, D.D.; Wang, Y.; You, B.G.; Liu, Y.; Zhang, X.N. Stimuli-responsive nanoparticles for the codelivery of chemotherapeutic agents doxorubicin and siPD-L1 to enhance the antitumor effect. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 1710–1724. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene. Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef]
- Wing, J.B.; Tay, C.; Sakaguchi, S. Control of Regulatory T Cells by Co-signal Molecules. Adv. Exp. Med. Biol. 2019, 1189, 179–210. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Lan, Q.; Xia, S.; Wang, Q.; Xu, W.; Huang, H.; Jiang, S.; Lu, L. Development of oncolytic virotherapy: From genetic modification to combination therapy. Front. Med. 2020, 14, 160–184. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Rabkin, S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016, 21, 26–34. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; Kirn, D.; Eckhardt, S.G.; Rodriguez, G.I.; Soutar, D.S.; Otto, R.; Robertson, A.G.; Park, O.; Gulley, M.L.; Heise, C.; et al. A Phase I Study of Onyx-015, an E1B Attenuated Adenovirus, Administered Intratumorally to Patients with Recurrent Head and Neck Cancer. Clin. Cancer Res. 2000, 6, 798. [Google Scholar] [PubMed]
- Aghi, M.; Martuza, R.L. Oncolytic viral therapies - the clinical experience. Oncogene 2005, 24, 7802–7816. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Eggermont, A.; Sautès-Fridman, C.; Galon, J.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch. OncoImmunology 2013, 2, e24612. [Google Scholar] [CrossRef] [PubMed]
- Miest, T.S.; Cattaneo, R. New viruses for cancer therapy: Meeting clinical needs. Nat. Rev. Microbiol. 2014, 12, 23–34. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Peters, C.; Rabkin, S.D. Designing Herpes Viruses as Oncolytics. Mol. Ther. Oncolytics 2015, 2, 15010. [Google Scholar] [CrossRef]
- Achard, C.; Surendran, A.; Wedge, M.-E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef]
- Lemay, C.G.; Keller, B.A.; Edge, R.E.; Abei, M.; Bell, J.C. Oncolytic Viruses: The Best is Yet to Come. Curr. Cancer Drug Targets 2018, 18, 109–123. [Google Scholar] [CrossRef]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Fountzilas, C.; Moseley, J.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Coffey, M.; Thompson, B.; Sarantopoulos, J. A phase II study of REOLYSIN(®) (pelareorep) in combination with carboplatin and paclitaxel for patients with advanced malignant melanoma. Cancer Chemother. Pharmacol. 2017, 79, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.J.; Chang, J.H.; Zhang, L.; Jiang, W.Q.; Guan, Z.Z.; Liu, J.W.; Zhang, Y.; Hu, X.H.; Wu, G.H.; Wang, H.Q.; et al. Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng 2004, 23, 1666–1670. [Google Scholar] [PubMed]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Packiam Vignesh, T.; Campanile Alexa, N.; Barocas Daniel, A.; Chamie, K.; Davis, I.I.I.R.L.; Kader, A.K.; Lamm Donald, L.; Yeung Alex, W.; Steinberg Gary, D. MP13–19 a phase II/III trial of CG0070, an oncolytic adenovirus, for BCG-refractory non-muscle-invasive bladder cancer (NMIBC). J. Urol. 2016, 195, e142. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Harrington, K.J.; Puzanov, I.; Hecht, J.R.; Hodi, F.S.; Szabo, Z.; Murugappan, S.; Kaufman, H.L. Clinical development of talimogene laherparepvec (T-VEC): A modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev. Anticancer Ther. 2015, 15, 1389–1403. [Google Scholar] [CrossRef]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef]
- Ino, Y.; Todo, T. CLINICAL DEVELOPMENT OF A THIRD-GENERATION ONCOLYTIC HSV-1 (G47Δ) FOR MALIGNANT GLIOMA. Gene Ther. Regul. 2010, 05, 101–111. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Brown, K.T.; Heo, J.; Borad, M.J.; Luca, A.; Pelusio, A.; Agathon, D.; Lusky, M.; et al. PHOCUS: A phase 3 randomized, open-label study comparing the oncolytic immunotherapy Pexa-Vec followed by sorafenib (SOR) vs SOR in patients with advanced hepatocellular carcinoma (HCC) without prior systemic therapy. J. Clin. Oncol. 2016, 34, TPS4146. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Kühnel, F. Oncolytic viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Huang, B.; Deng, L.; Hu, Z. Progress in gene therapy using oncolytic vaccinia virus as vectors. J. Cancer Res. Clin. Oncol. 2018, 144, 2433–2440. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.-H.; Moon, A.; Burke, J.; Ribas, A.; Stephenson, J.; Breitbach, C.J.; Daneshmand, M.; De Silva, N.; Parato, K.; Diallo, J.-S.; et al. A Mechanistic Proof-of-concept Clinical Trial With JX-594, a Targeted Multi-mechanistic Oncolytic Poxvirus, in Patients With Metastatic Melanoma. Mol. Ther. 2011, 19, 1913–1922. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Bell, J.C.; Hwang, T.H.; Kirn, D.H.; Burke, J. The emerging therapeutic potential of the oncolytic immunotherapeutic Pexa-Vec (JX-594). Oncolytic Virother. 2015, 4, 25–31. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Parato, K.; Burke, J.; Hwang, T.H.; Bell, J.C.; Kirn, D.H. Pexa-Vec double agent engineered vaccinia: Oncolytic and active immunotherapeutic. Curr. Opin. Virol. 2015, 13, 49–54. [Google Scholar] [CrossRef]
- Bourhill, T.; Mori, Y.; Rancourt, D.E.; Shmulevitz, M.; Johnston, R.N. Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection. Viruses 2018, 10, 421. [Google Scholar] [CrossRef]
- Abdullahi, S.; Jäkel, M.; Behrend, S.J.; Steiger, K.; Topping, G.; Krabbe, T.; Colombo, A.; Sandig, V.; Schiergens, T.S.; Thasler, W.E.; et al. A Novel Chimeric Oncolytic Virus Vector for Improved Safety and Efficacy as a Platform for the Treatment of Hepatocellular Carcinoma. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Ammi, R.; De Waele, J.; Willemen, Y.; Van Brussel, I.; Schrijvers, D.M.; Lion, E.; Smits, E.L. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther. 2015, 146, 120–131. [Google Scholar] [CrossRef]
- Smith, M.; García-Martínez, E.; Pitter, M.R.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Toll-like receptor agonists in cancer immunotherapy. Oncoimmunology 2018, 7, e1526250. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.F. Clinical update of the AS04-adjuvanted human papillomavirus-16/18 cervical cancer vaccine, Cervarix. Adv. Ther. 2009, 26, 983–998. [Google Scholar] [CrossRef] [PubMed]
- ‘Mac’ Cheever, M.A. Twelve immunotherapy drugs that could cure cancers. Immunol. Rev. 2008, 222, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Pretto, S.; Tagliabue, E.; Balsari, A.; Sfondrini, L. Exploiting poly(I:C) to induce cancer cell apoptosis. Cancer Biol. Ther. 2017, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef]
- Estornes, Y.; Toscano, F.; Virard, F.; Jacquemin, G.; Pierrot, A.; Vanbervliet, B.; Bonnin, M.; Lalaoui, N.; Mercier-Gouy, P.; Pachéco, Y.; et al. dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 2012, 19, 1482–1494. [Google Scholar] [CrossRef]
- Feldman, S.; Hughes, W.T.; Darlington, R.W.; Kim, H.K. Evaluation of Topical Polyinosinic Acid-Polycytidylic Acid in Treatment of Localized Herpes Zoster in Children with Cancer: A Randomized, Double-Blind Controlled Study. Antimicrob. Agents Chemother. 1975, 8, 289. [Google Scholar] [CrossRef][Green Version]
- Robinson, R.A.; DeVita, V.T.; Levy, H.B.; Baron, S.; Hubbard, S.P.; Levine, A.S. A Phase I–II Trial of Multiple-Dose Polyriboinosinic-Polyribocytidylic Acid in Patients With Leukemia or Solid Tumors. J. Natl. Cancer Inst. 1976, 57, 599–602. [Google Scholar] [CrossRef]
- Herr, H.W.; Kemeny, N.; Yagoda, A.; Whitmore, W.F., Jr. Poly I:C immunotherapy in patients with papillomas or superficial carcinomas of the bladder. Natl. Cancer Inst. Monogr. 1978, 325. [Google Scholar]
- Nordlund, J.J.; Wolff, S.M.; Levy, H.B. Inhibition of biologic activity of poly I: Poly C by human plasma. Proc. Soc. Exp. Biol. Med. 1970, 133, 439–444. [Google Scholar] [CrossRef]
- Levy, H.B.; Baer, G.; Baron, S.; Buckler, C.E.; Gibbs, C.J.; Iadarola, M.J.; London, W.T.; Rice, J. A Modified Polyriboinosinic-Polyribocytidylic Acid Complex That Induces Interferon in Primates. J. Infect. Dis. 1975, 132, 434–439. [Google Scholar] [CrossRef]
- Levine, A.S.; Sivulich, M.; Wiernik, P.H.; Levy, H.B. Initial clinical trials in cancer patients of polyriboinosinic-polyribocytidylic acid stabilized with poly-L-lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective interferon inducer. Cancer Res. 1979, 39, 1645–1650. [Google Scholar]
- Patchett, A.L.; Tovar, C.; Corcoran, L.M.; Lyons, A.B.; Woods, G.M. The toll-like receptor ligands Hiltonol® (polyICLC) and imiquimod effectively activate antigen-specific immune responses in Tasmanian devils (Sarcophilus harrisii). Dev. Comp. Immunol. 2017, 76, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ruiz, M.E.; Perez-Gracia, J.L.; Rodríguez, I.; Alfaro, C.; Oñate, C.; Pérez, G.; Gil-Bazo, I.; Benito, A.; Inogés, S.; López-Diaz de Cerio, A.; et al. Combined immunotherapy encompassing intratumoral poly-ICLC, dendritic-cell vaccination and radiotherapy in advanced cancer patients. Ann. Oncol. 2018, 29, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.M.; Corthésy, B.; Merkle, H.P. Particulate formulations for the delivery of poly(I:C) as vaccine adjuvant. Adv. Drug Deliv. Rev. 2013, 65, 1386–1399. [Google Scholar] [CrossRef] [PubMed]
- Shir, A.; Ogris, M.; Wagner, E.; Levitzki, A. EGF receptor-targeted synthetic double-stranded RNA eliminates glioblastoma, breast cancer, and adenocarcinoma tumors in mice. PLoS Med. 2006, 3, e6. [Google Scholar] [CrossRef]
- Shir, A.; Ogris, M.; Roedl, W.; Wagner, E.; Levitzki, A. EGFR-Homing dsRNA Activates Cancer-Targeted Immune Response and Eliminates Disseminated EGFR-Overexpressing Tumors in Mice. Clin. Cancer Res. 2011, 17, 1033. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, D.; Kiss, M.; Rödl, W.; Shir, A.; Levitzki, A.; Ogris, M.; Wagner, E. Poly(I:C)-mediated tumor growth suppression in EGF-receptor overexpressing tumors using EGF-polyethylene glycol-linear polyethylenimine as carrier. Pharm. Res. 2011, 28, 731–741. [Google Scholar] [CrossRef]
- Abourbeh, G.; Shir, A.; Mishani, E.; Ogris, M.; Rödl, W.; Wagner, E.; Levitzki, A. PolyIC GE11 polyplex inhibits EGFR-overexpressing tumors. IUBMB Life 2012, 64, 324–330. [Google Scholar] [CrossRef]
- Lächelt, U.; Wittmann, V.; Müller, K.; Edinger, D.; Kos, P.; Höhn, M.; Wagner, E. Synthetic polyglutamylation of dual-functional MTX ligands for enhanced combined cytotoxicity of poly(I:C) nanoplexes. Mol. Pharm. 2014, 11, 2631–2639. [Google Scholar] [CrossRef]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Hanagata, N. CpG oligodeoxynucleotide nanomedicines for the prophylaxis or treatment of cancers, infectious diseases, and allergies. Int. J. Nanomedicine 2017, 12, 515–531. [Google Scholar] [CrossRef]
- Adamus, T.; Kortylewski, M. The revival of CpG oligonucleotide-based cancer immunotherapies. Contemp. Oncol. (Pozn) 2018, 22, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. Development of TLR9 agonists for cancer therapy. J. Clin. Invest. 2007, 117, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hossain, D.M.; Duttagupta, P.; Moreira, D.; Zhao, X.; Won, H.; Buettner, R.; Nechaev, S.; Majka, M.; Zhang, B.; et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood 2016, 127, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Nikitczuk, K.P.; Schloss, R.S.; Yarmush, M.L.; Lattime, E.C. PLGA-polymer encapsulating tumor antigen and CpG DNA administered into the tumor microenvironment elicits a systemic antigen-specific IFN-γ response and enhances survival. J. Cancer Ther. 2013, 4, 280–290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, T.; Miao, J.; Kai, D.; Zhang, H. Polyethylenimine-Mediated CpG Oligodeoxynucleotide Delivery Stimulates Bifurcated Cytokine Induction. ACS Biomater. Sci. Eng. 2018, 4, 1013–1018. [Google Scholar] [CrossRef]
- Kwong, B.; Liu, H.; Irvine, D.J. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials 2011, 32, 5134–5147. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Kim, D.; Park, B.K.; Wu, G.; Park, M.C.; Ha, Y.W.; Kwon, H.J.; Lee, Y. Induction of immunological memory response by vaccination with TM4SF5 epitope-CpG-DNA-liposome complex in a mouse hepatocellular carcinoma model. Oncol. Rep. 2013, 29, 735–740. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kwon, S.; Kim, Y.E.; Park, J.A.; Kim, D.S.; Kwon, H.J.; Lee, Y. Therapeutic effect of a TM4SF5-specific peptide vaccine against colon cancer in a mouse model. BMB Rep. 2014, 47, 215–220. [Google Scholar] [CrossRef]
- Zhao, D.; Alizadeh, D.; Zhang, L.; Liu, W.; Farrukh, O.; Manuel, E.; Diamond, D.J.; Badie, B. Carbon nanotubes enhance CpG uptake and potentiate antiglioma immunity. Clin. Cancer Res. 2011, 17, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Hashida, Y.; Kawakami, S.; Mihara, J.; Umeyama, T.; Imahori, H.; Murakami, T.; Yamashita, F.; Hashida, M. Preparation of immunostimulatory single-walled carbon nanotube/CpG DNA complexes and evaluation of their potential in cancer immunotherapy. Int. J. Pharm. 2014, 471, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kwon, H.K.; An, S.; Kim, D.; Kim, S.; Yu, M.K.; Lee, J.H.; Lee, T.S.; Im, S.H.; Jon, S. Imageable antigen-presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.Y.; Almeida, J.P.; Bear, A.; Liu, N.; Luo, L.; Foster, A.E.; Drezek, R.A. Gold nanoparticle delivery of modified CpG stimulates macrophages and inhibits tumor growth for enhanced immunotherapy. PLoS ONE 2013, 8, e63550. [Google Scholar] [CrossRef]
- Cha, B.G.; Jeong, J.H.; Kim, J. Extra-Large Pore Mesoporous Silica Nanoparticles Enabling Co-Delivery of High Amounts of Protein Antigen and Toll-like Receptor 9 Agonist for Enhanced Cancer Vaccine Efficacy. ACS Cent. Sci. 2018, 4, 484–492. [Google Scholar] [CrossRef]
- Schüller, V.J.; Heidegger, S.; Sandholzer, N.; Nickels, P.C.; Suhartha, N.A.; Endres, S.; Bourquin, C.; Liedl, T. Cellular immunostimulation by CpG-sequence-coated DNA origami structures. ACS Nano 2011, 5, 9696–9702. [Google Scholar] [CrossRef]
- Wang, C.; Sun, W.; Wright, G.; Wang, A.Z.; Gu, Z. Inflammation-Triggered Cancer Immunotherapy by Programmed Delivery of CpG and Anti-PD1 Antibody. Adv. Mater. 2016, 28, 8912–8920. [Google Scholar] [CrossRef]
- Guo, L.; Yan, D.D.; Yang, D.; Li, Y.; Wang, X.; Zalewski, O.; Yan, B.; Lu, W. Combinatorial Photothermal and Immuno Cancer Therapy Using Chitosan-Coated Hollow Copper Sulfide Nanoparticles. ACS Nano 2014, 8, 5670–5681. [Google Scholar] [CrossRef]
- Tao, Y.; Ju, E.; Ren, J.; Qu, X. Immunostimulatory oligonucleotides-loaded cationic graphene oxide with photothermally enhanced immunogenicity for photothermal/immune cancer therapy. Biomaterials 2014, 35, 9963–9971. [Google Scholar] [CrossRef]
- Tao, Y.; Ju, E.; Liu, Z.; Dong, K.; Ren, J.; Qu, X. Engineered, self-assembled near-infrared photothermal agents for combined tumor immunotherapy and chemo-photothermal therapy. Biomaterials 2014, 35, 6646–6656. [Google Scholar] [CrossRef]
- Speiser, D.E.; Schwarz, K.; Baumgaertner, P.; Manolova, V.; Devevre, E.; Sterry, W.; Walden, P.; Zippelius, A.; Conzett, K.B.; Senti, G.; et al. Memory and effector CD8 T-cell responses after nanoparticle vaccination of melanoma patients. J. Immunother. 2010, 33, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Takabayashi, K.; Cheng, P.M.; Nguyen, M.D.; Corr, M.; Tuck, S.; Raz, E. Immunostimulatory DNA-based vaccines induce cytotoxic lymphocyte activity by a T-helper cell-independent mechanism. Nat. Biotechnol. 2000, 18, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Gungor, B.; Yagci, F.C.; Tincer, G.; Bayyurt, B.; Alpdundar, E.; Yildiz, S.; Ozcan, M.; Gursel, I.; Gursel, M. CpG ODN nanorings induce IFNα from plasmacytoid dendritic cells and demonstrate potent vaccine adjuvant activity. Sci Transl. Med. 2014, 6, 235ra261. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, H.J.; Wittig, B.; Arnold, D.; Riera-Knorrenschild, J.; Nitsche, D.; Kroening, H.; Mayer, F.; Andel, J.; Ziebermayr, R.; Scheithauer, W. Maintenance treatment with the immunomodulator MGN1703, a Toll-like receptor 9 (TLR9) agonist, in patients with metastatic colorectal carcinoma and disease control after chemotherapy: A randomised, double-blind, placebo-controlled trial. J. Cancer Res. Clin. Oncol. 2014, 140, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Weihrauch, M.R.; Richly, H.; von Bergwelt-Baildon, M.S.; Becker, H.J.; Schmidt, M.; Hacker, U.T.; Shimabukuro-Vornhagen, A.; Holtick, U.; Nokay, B.; Schroff, M.; et al. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur. J. Cancer 2015, 51, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Schmidt, M.; Scheithauer, W.; Schmoll, H.J. MGN1703, an immunomodulator and toll-like receptor 9 (TLR-9) agonist: From bench to bedside. Crit. Rev. Oncol. Hematol. 2015, 94, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Müller, K.; Wagner, E. RNAi-Based Nano-Oncologicals: Delivery and Clinical Applications. In Nano-Oncologicals: New Targeting and Delivery Approaches; Alonso, M.J., Garcia-Fuentes, M., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 245–268. [Google Scholar]
- Allen, K.E.; Weiss, G.J. Resistance may not be futile: microRNA biomarkers for chemoresistance and potential therapeutics. Mol. Cancer Ther. 2010, 9, 3126–3136. [Google Scholar] [CrossRef]
- Kong, Y.W.; Ferland-McCollough, D.; Jackson, T.J.; Bushell, M. microRNAs in cancer management. Lancet Oncol. 2012, 13, e249–e258. [Google Scholar] [CrossRef]
- Kobayashi, E.; Hornicek, F.J.; Duan, Z. MicroRNA Involvement in Osteosarcoma. Sarcoma 2012, 2012, 359739. [Google Scholar] [CrossRef]
- Iyer, A.K.; Duan, Z.; Amiji, M.M. Nanodelivery Systems for Nucleic Acid Therapeutics in Drug Resistant Tumors. Mol. Pharm. 2014, 11, 2511–2526. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, R.; Rathbone, M.J.; Hansbro, P.M.; Bebawy, M.; Dua, K. Therapeutic prospects of microRNAs in cancer treatment through nanotechnology. Drug Deliv. Transl. Res. 2018, 8, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Winkler, M. The promise of microRNA replacement therapy. Cancer Res. 2010, 70, 7027–7030. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E. Biomaterials in RNAi therapeutics: Quo vadis? Biomater. Sci. 2013, 1, 804–809. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Bio. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13, R45. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Oak, P.S.; Wagner, E.; Roidl, A. miR-200c sensitizes breast cancer cells to doxorubicin treatment by decreasing TrkB and Bmi1 expression. PLoS ONE 2012, 7, e50469. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Wagner, E.; Roidl, A. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 2014, 5, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, M.; Raza, U.; Saatci, Ö.; Eyüpoğlu, E.; Yurdusev, E.; Şahin, Ö. miR-200c: A versatile watchdog in cancer progression, EMT, and drug resistance. J. Mol. Med. 2016, 94, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Klein, P.M.; Heissig, P.; Roidl, A.; Wagner, E. EGF receptor targeted lipo-oligocation polyplexes for antitumoral siRNA and miRNA delivery. Nanotechnology 2016, 27, 464001. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, e321–e327. [Google Scholar] [CrossRef] [PubMed]
- Papadouli, I.; Mueller-Berghaus, J.; Beuneu, C.; Ali, S.; Hofner, B.; Petavy, F.; Tzogani, K.; Miermont, A.; Norga, K.; Kholmanskikh, O.; et al. EMA Review of Axicabtagene Ciloleucel (Yescarta) for the Treatment of Diffuse Large B-Cell Lymphoma. Oncologist 2020. [Google Scholar] [CrossRef] [PubMed]
- Tejeda-Mansir, A.; García-Rendón, A.; Guerrero-Germán, P. Plasmid-DNA lipid and polymeric nanovaccines: A new strategic in vaccines development. Biotechnol. Genet. Eng. Rev. 2019, 35, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Caruso, H.G.; Tanaka, R.; Liang, J.; Ling, X.; Sabbagh, A.; Henry, V.K.; Collier, T.L.; Heimberger, A.B. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J. Neurooncol. 2019, 145, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Iurescia, S.; Fioretti, D.; Rinaldi, M. A blueprint for DNA vaccine design. Methods Mol. Biol. (Clifton, N.J.) 2014, 1143, 3–10. [Google Scholar] [CrossRef]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef]
- Di, S.; Zhou, M.; Pan, Z.; Sun, R.; Chen, M.; Jiang, H.; Shi, B.; Luo, H.; Li, Z. Combined Adjuvant of Poly I:C Improves Antitumor Effects of CAR-T Cells. Front. Oncol. 2019, 9, 241. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, F.; Zhang, A.; Zhang, D.; Nie, W.; Xu, T.; Han, B.; Seth, P.; Wang, H.; Yang, Y.; et al. Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 2020, 348, 104041. [Google Scholar] [CrossRef]
- Grunwitz, C.; Kranz, L.M. mRNA Cancer Vaccines-Messages that Prevail. Curr. Top. Microbiol. Immunol. 2017, 405, 145–164. [Google Scholar] [CrossRef]





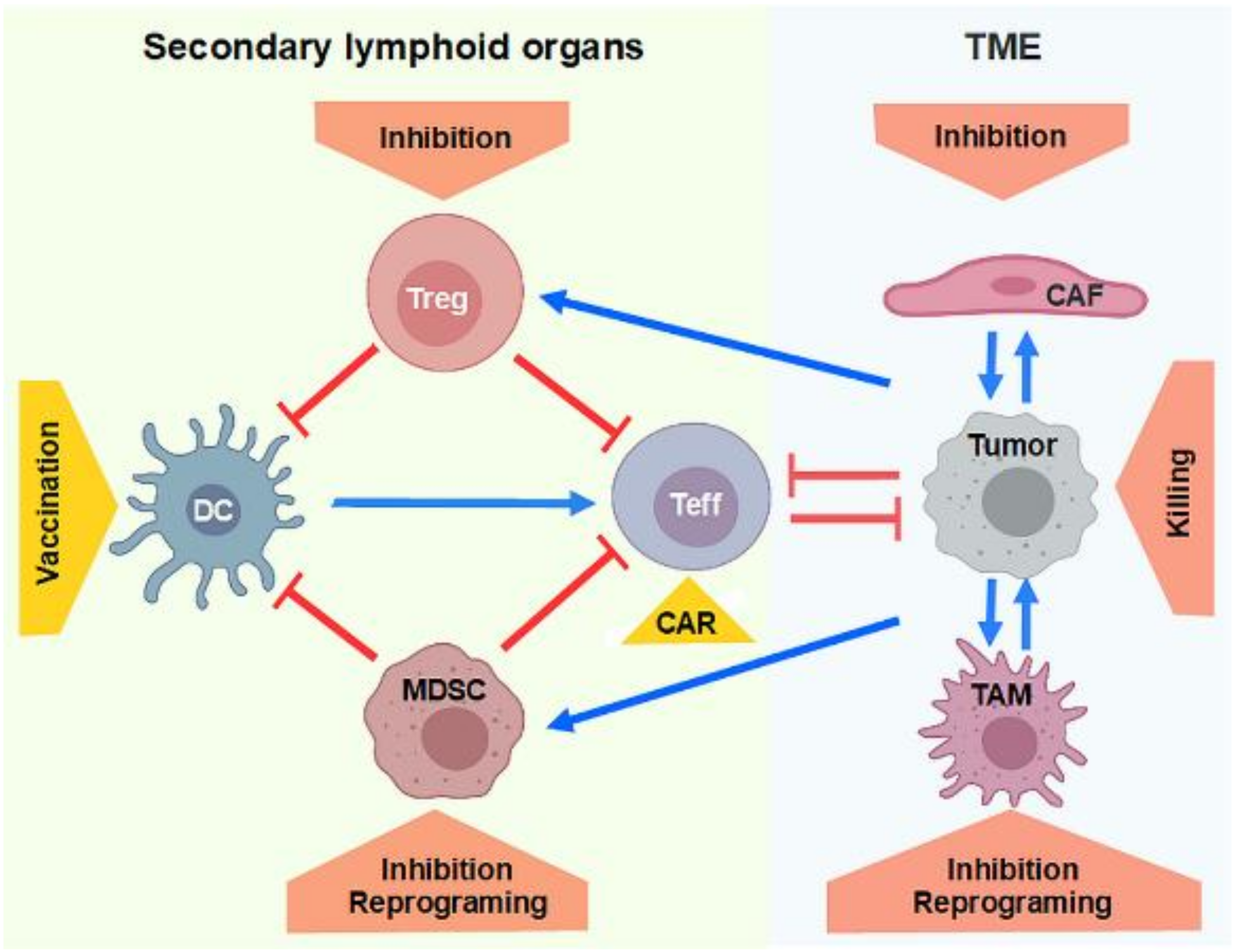{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Molecule | Therapy Strategy | Application Route | Treated Cancer | Clinical State | References |
|---|---|---|---|---|---|
| IL-2 | Syngeneic tumor cell vaccine modified with IL-2 gene ex vivo | Intradermal or subcutaneous injection | Metastatic melanoma | Phase I | [319] |
| Allogeneic tumor cell vaccine modified with IL-2 gene ex vivo | Subcutaneous injection | Metastatic melanoma | Pilot study | [320] | |
| Phase I–II | [321] | ||||
| Allogeneic tumor cell vaccine modified with IL-2 gene ex vivo | Subcutaneous injection | Relapsed neuroblastoma | Phase I | [322] | |
| TNF-α | TNFerade, a replication-deficient adenoviral vector encoding for TNF-α under the control of a radiation inducible promotor (erg-1 gene promotor) | Intratumoral injection | Various cancer types, e.g., advanced pancreatic cancer | Phase III | [323,324] |
| IL-12 | Ad–RTS–hIL-12, an adenoviral vector encoding for IL-12 transgene designed with a ligand-inducible expression switch | Injection in the resection cavity | Recurrent high-grade glioma | Phase I | [325] |
| GM-CSF | GVAX, an allogeneic tumor cell vaccine modified with GM-CSF gene ex vivo,(in combination with immune checkpoint inhibitors and/or cyclophosphamide and Listeria monocytogenes-expressing mesothelin (CRS-207)) | Intradermal injection | Advanced pancreatic cancer | Phase Ib | [326] |
| Phase II | [327] | ||||
| Phase IIb | [328] | ||||
| Phase II | [329] | ||||
| IFN-α | Instiladrin® (rAdIFNα2b/Syn3), an IFN-α encoding adenoviral vector | Intravesical injection | BCG unresponsive bladder cancer | Phase III—results pending (NCT02773849) | [330] |
| TGF-β (inhibition) | Belagenpneumatucel-L, an allogeneic tumor cell vaccine altered to express ASO directed against TGF-β | Intradermal injection | Advanced non-small cell lung cancer | Phase II | [331,332] |
| Phase III | [333] |
| Oncolytic Virus | Genetic Modification | Treated Cancer | Clinical State | Reference |
|---|---|---|---|---|
| Wild-Type Virus | ||||
| RIGVIR® (wild-type ECHO-7; (+)ssRNA virus) | – | Melanoma | Approved in Lativa in 2004 | [471] |
| Reolysin® (pelareorep, type 3 Dearing (T3D) strain reovirus; dsRNA virus) | – | Many advanced malignancies (e.g., melanoma, sarcomas, non-small cell lung cancer, pancreatic adenocarcinoma) | Phase I and II | [457,472,473] |
| Advanced, metastatic head and neck cancer | Phase III | [472] | ||
| Oncolytic Adenovirus (dsDNA virus) | ||||
| Oncorine® (rAdV H101) | Deletion in E1B-55K and E3 genes | Nasopharyngeal carcinoma | Approved in China in 2005 | [474,475] |
| CG0070 (AdV-5) | Deletion in E3 gene; insertion of GM-CSF gene | Non-muscle-invasive bladder cancer | Phase II/III (BOND, NCT01438112); phase II (BOND2, NCT02365818) | [456,476] |
| Oncolytic Herpes Simplex Virus, HSV-1 (dsDNA virus) | ||||
| T-Vec (talminogene laherparepvec) | Deletion in ICP34.5 and ICP47 genes; insertion of GM-CSF gene | Advanced melanoma | Approved by FDA and EMA in 2015 | [477,478] |
| M032 | Deletion in ICP34.5 gene; insertion of IL-12 gene | Glioblastoma multiforme | Phase I | [479] |
| G47Δ | Deletion in ICP34.5, ICP47 and ICP6 genes; insertion of GM-CSF gene | Recurrent glioblastoma, castration resistant prostate cancer, recurrent olfactory neuroblastoma | Clinical trials in Japan | [456,480,481] |
| Oncolytic Vaccinia Virus (dsDNA virus) | ||||
| Pexa-Vec (JX-594, pexastimogene devacirepvec) | Mutation in TK gene; insertion of GM-CSF gene | Advanced hepatocellular carcinoma | Phase III (in combination with sorafenib) | [482] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hager, S.; Fittler, F.J.; Wagner, E.; Bros, M. Nucleic Acid-Based Approaches for Tumor Therapy. Cells 2020, 9, 2061. https://doi.org/10.3390/cells9092061
Hager S, Fittler FJ, Wagner E, Bros M. Nucleic Acid-Based Approaches for Tumor Therapy. Cells. 2020; 9(9):2061. https://doi.org/10.3390/cells9092061
Chicago/Turabian StyleHager, Simone, Frederic Julien Fittler, Ernst Wagner, and Matthias Bros. 2020. "Nucleic Acid-Based Approaches for Tumor Therapy" Cells 9, no. 9: 2061. https://doi.org/10.3390/cells9092061
APA StyleHager, S., Fittler, F. J., Wagner, E., & Bros, M. (2020). Nucleic Acid-Based Approaches for Tumor Therapy. Cells, 9(9), 2061. https://doi.org/10.3390/cells9092061