“Repair Me if You Can”: Membrane Damage, Response, and Control from the Viral Perspective


Abstract
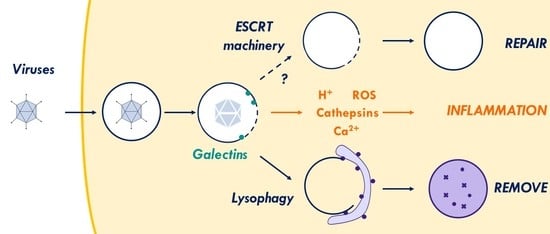
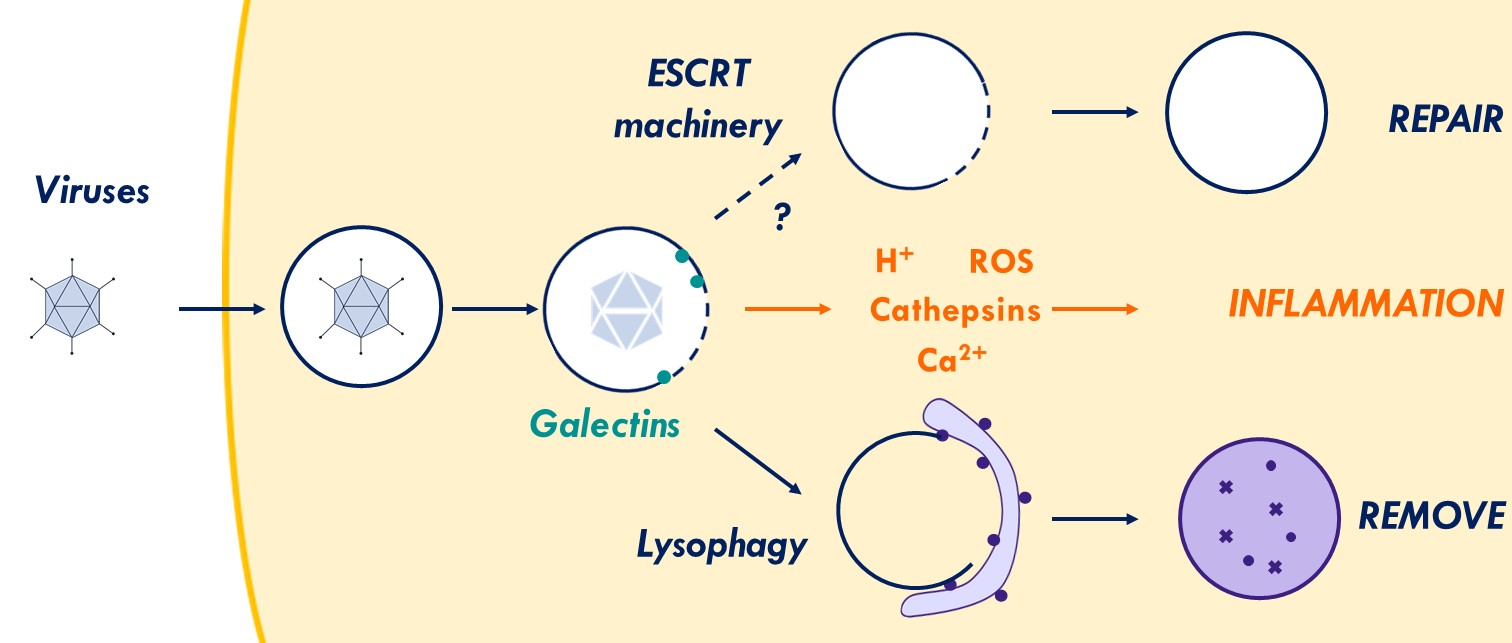{kind=link}
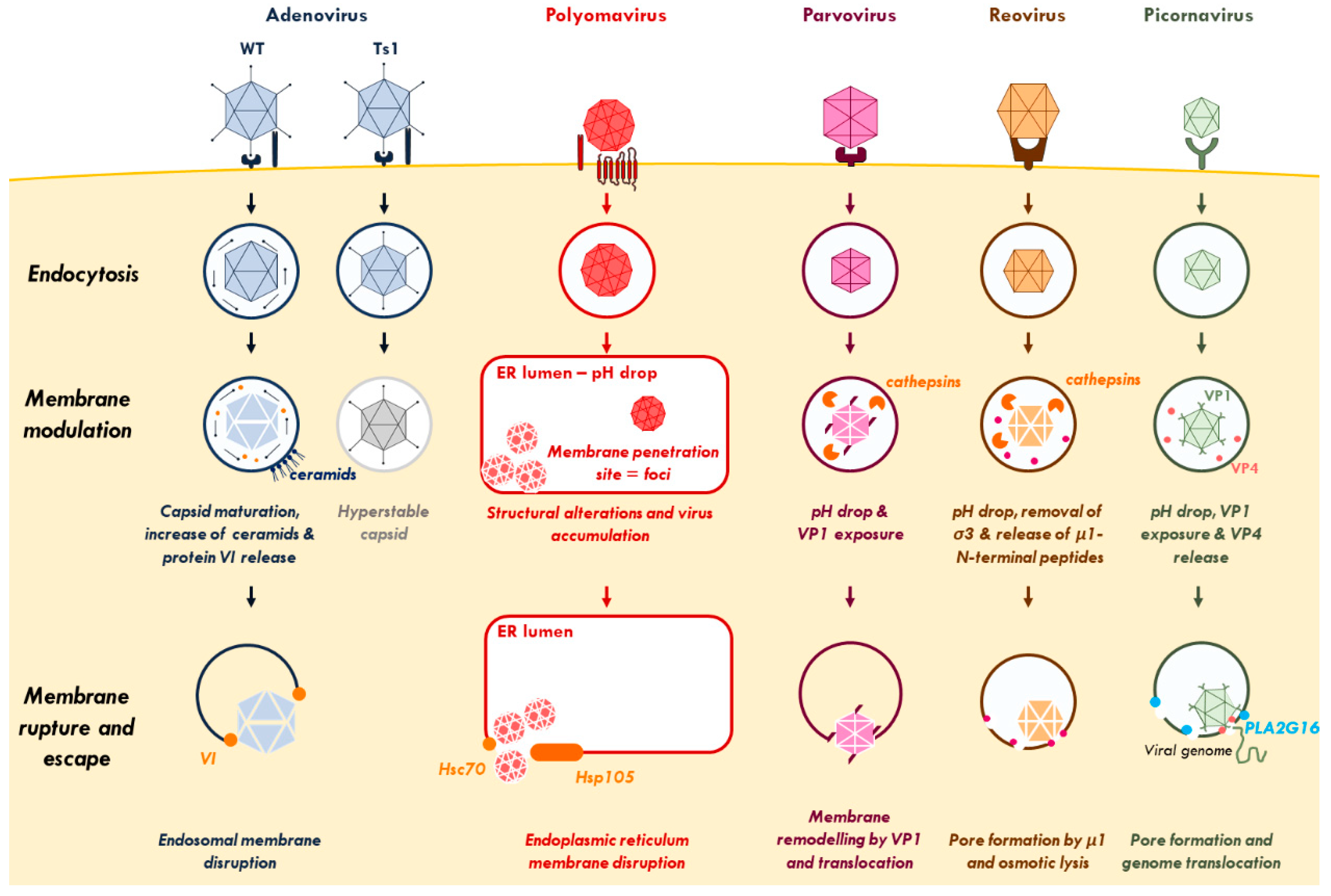{kind=link}
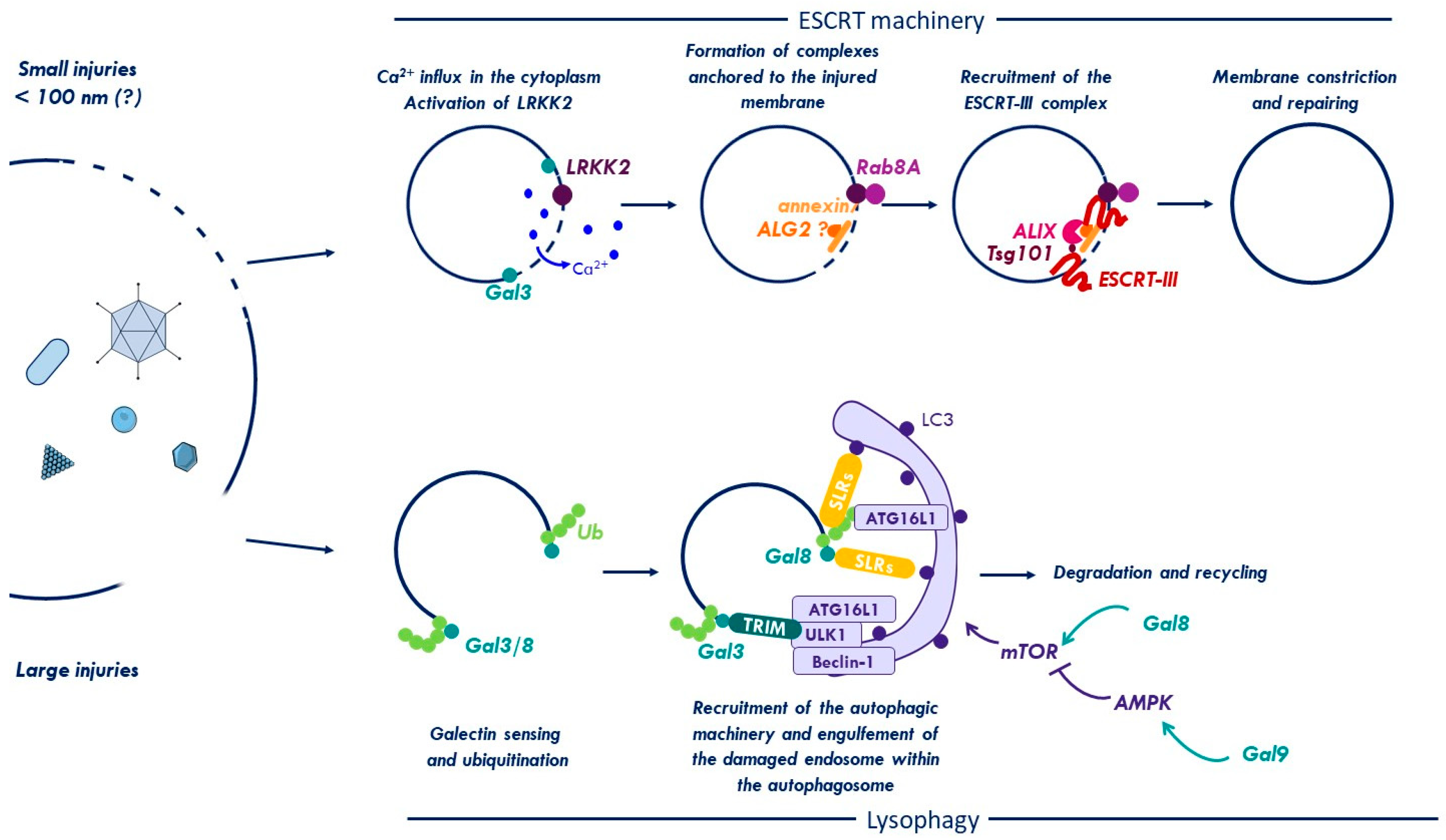{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Virus Inflicted Membrane Damage
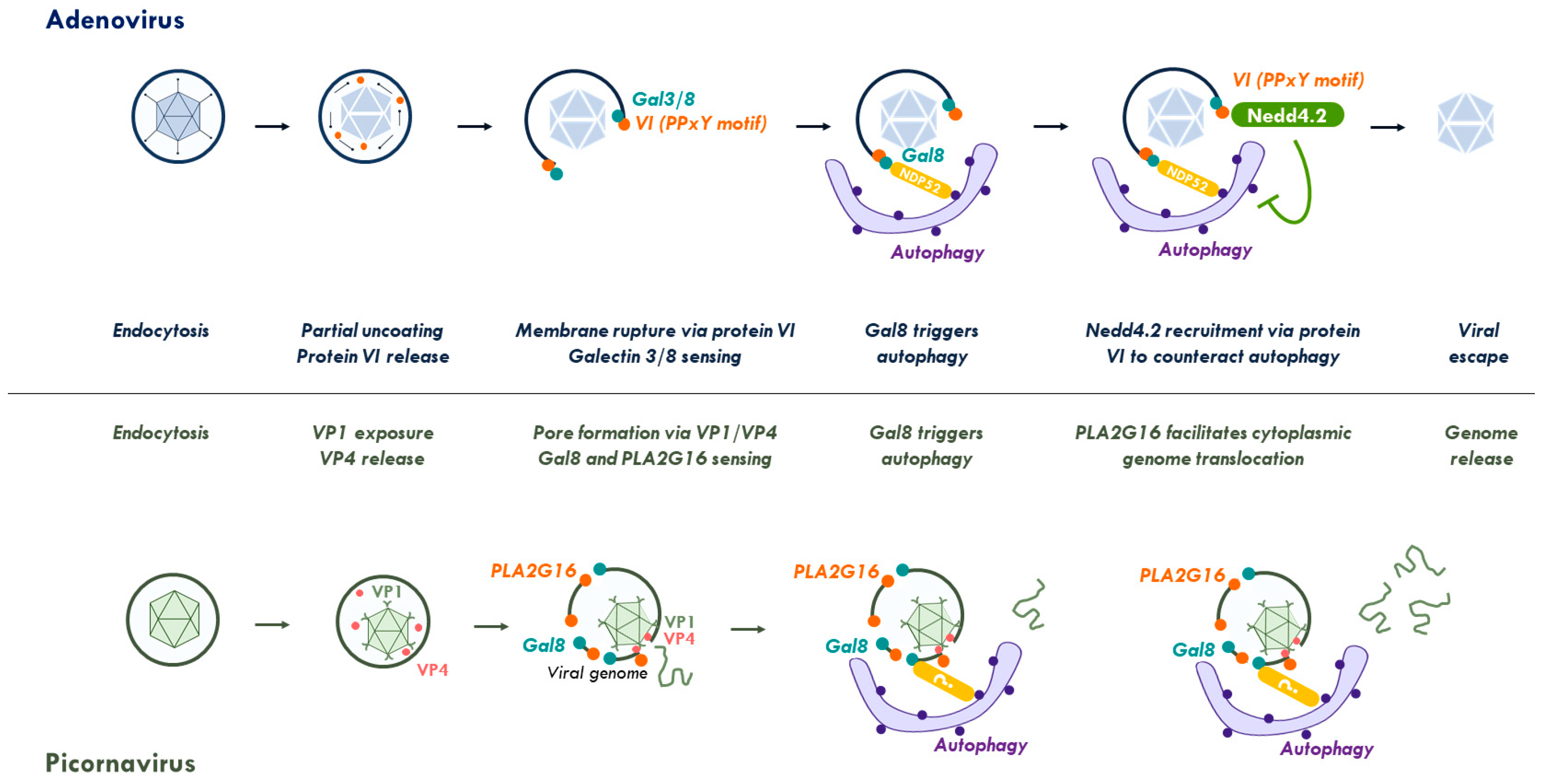
1.1.1. Adenovirus
1.1.2. Polyomavirus
1.1.3. Parvovirus
1.1.4. Reovirus
1.1.5. Picornavirus
1.2. Bacteria Induced Membrane Damage
1.3. Membrane Damage Induced by Other Pathogens
1.4. Non-Pathogenic Membrane Damage
2. Membrane Damage Recognition and Cell Response
2.1. Galectins and Their Role in Membrane Damage Recognition
2.2. Galectins and Ubiquitin Coordinate Autophagy for Membrane Damage Removal
2.3. Viral Control of the Autophagy Response to Membrane Damage Removal
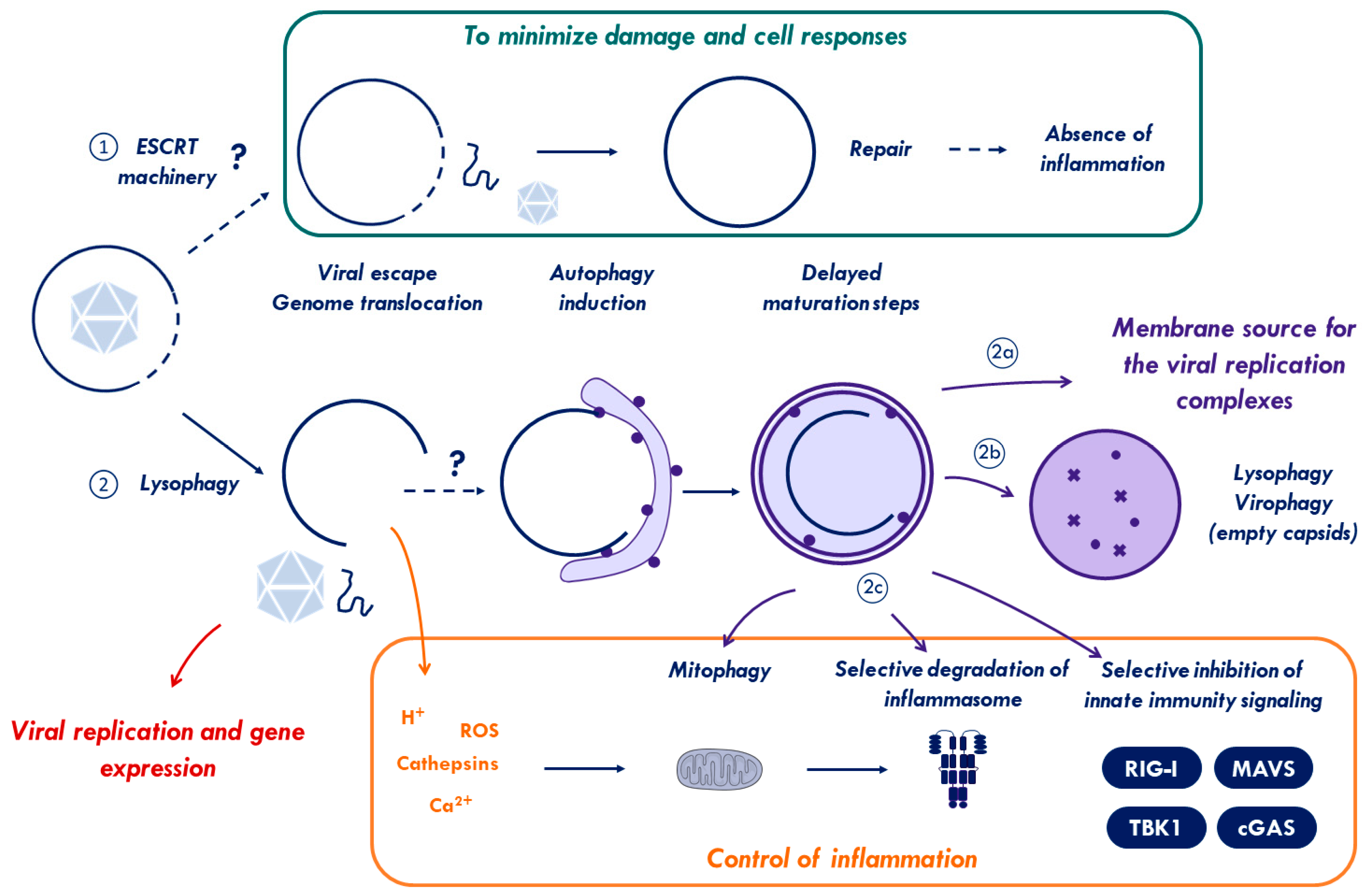
2.4. ESCRT Machinery for Membrane Repair
2.5. Viral Control of the ESCRT-Response to Membrane Repair
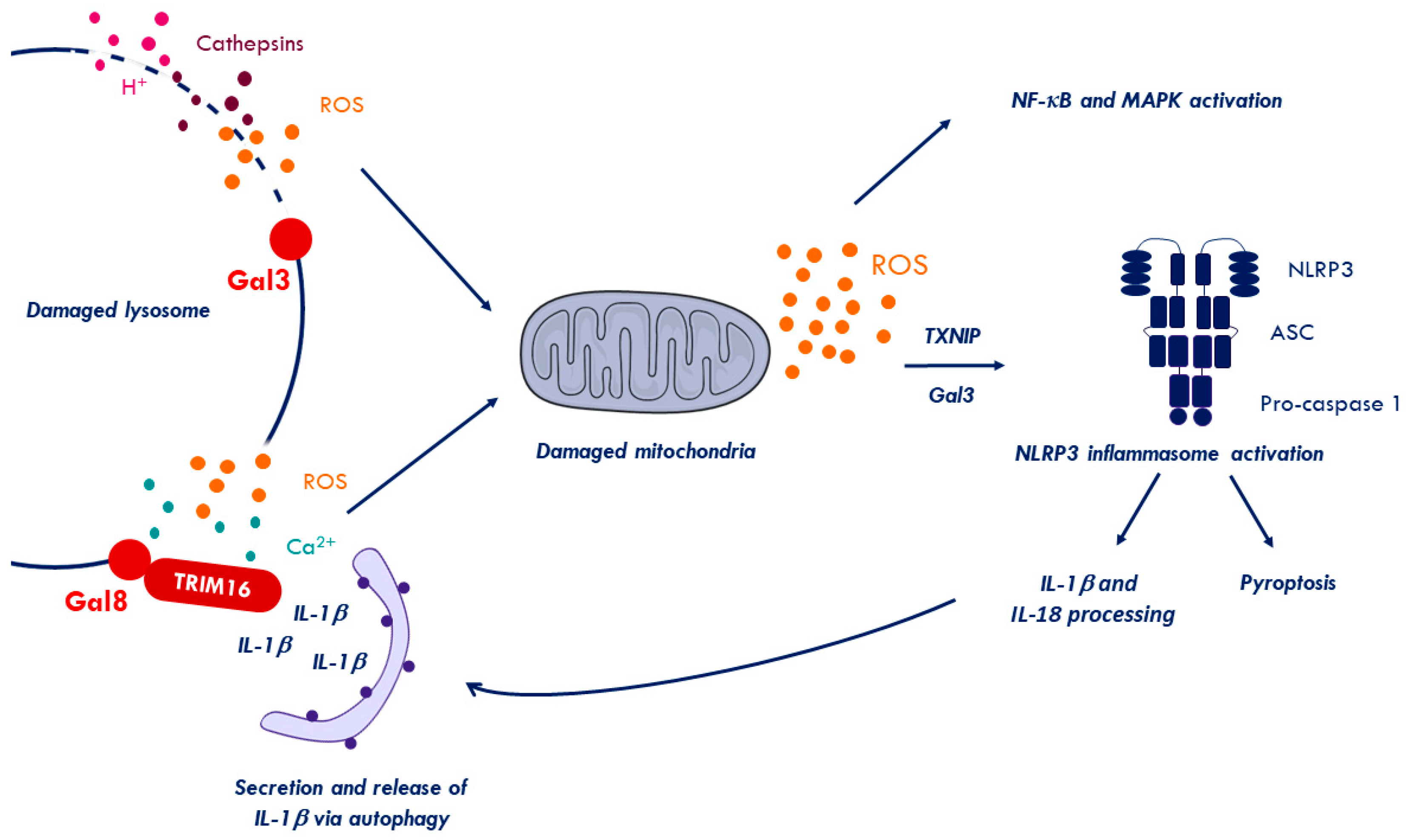
2.6. Inflammation to Signal Membrane Damage
2.7. Inflammation upon Virus Membrane Penetration
3. Crosstalk Between Membrane Damage, Autophagy, and Inflammatory Response
4. Concluding Remarks: What Is in It for the Virus?
Funding
Acknowledgments
Conflicts of Interest
References
- Lombard, J. Once upon a time the cell membranes: 175 years of cell boundary research. Biol. Direct 2014, 9, 1–35. [Google Scholar] [CrossRef]
- Thurston, T.L.M.M.; Wandel, M.P.; Von Muhlinen, N.; Foeglein, Á.; Randow, F.; Foeglein, A.; Randow, F.; Foeglein, Á.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Montespan, C.; Marvin, S.A.; Austin, S.; Burrage, A.M.; Roger, B.; Rayne, F.; Faure, M.; Campell, E.M.; Schneider, C.; Reimer, R.; et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins Control mTOR in Response to Endomembrane Damage. Mol. Cell 2018, 70, 120–135.e8. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Lozach, P.Y.; Huotari, J.; Helenius, A. Late-penetrating viruses. Curr. Opin. Virol. 2011, 1, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Ketter, E.; Randall, G. Virus Impact on Lipids and Membranes. Annu. Rev. Virol. 2019, 6, 319–340. [Google Scholar] [CrossRef]
- Moyer, C.L.; Nemerow, G.R. Viral weapons of membrane destruction: Variable modes of membrane penetration by non-enveloped viruses. Curr. Opin. Virol. 2011, 1, 44–49. [Google Scholar] [CrossRef]
- Stewart, P.L.; Dermody, T.S.; Nemerow, G.R. Structural basis of nonenveloped virus cell entry. Adv. Protein Chem. 2003, 64, 455–491. [Google Scholar] [CrossRef]
- Prchla, E.; Plank, C.; Wagner, E.; Blaas, D.; Fuchs, R. Virus-mediated release of endosomal content in vitro: Different behavior of adenovirus and rhinovirus serotype 2. J. Cell Biol. 1995, 131, 111–123. [Google Scholar] [CrossRef]
- Farr, G.A.; Zhang, L.G.; Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. USA 2005, 102, 17148–17153. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Webster, P.; Weber, J.; Helenius, A. The role of the adenovirus protease in virus entry into cells. EMBO J. 1996, 15, 1766–1777. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Wodrich, H.; Gerace, L.; Nemerow, G.R. Adenovirus Protein VI Mediates Membrane Disruption following Capsid Disassembly. J. Virol. 2005, 79, 1992–2000. [Google Scholar] [CrossRef]
- Martinez, R.; Schellenberger, P.; Vasishtan, D.; Aknin, C.; Austin, S.; Dacheux, D.; Rayne, F.; Siebert, A.; Ruzsics, Z.; Gruenewald, K.; et al. The amphipathic helix of adenovirus capsid protein VI contributes to penton release and postentry sorting. J. Virol. 2015, 89, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Weber, J. Genetic analysis of adenovirus type 2 III. Temperature sensitivity of processing viral proteins. J. Virol. 1976, 17, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Weber, J.M. The adenovirus protease is required for virus entry into host cells. Virology 1995, 213, 494–502. [Google Scholar] [CrossRef]
- Martín, C.S. Latest insights on adenovirus structure and assembly. Viruses 2012, 4, 847–877. [Google Scholar] [CrossRef]
- Wodrich, H.; Henaff, D.; Jammart, B.; Segura-Morales, C.; Seelmeir, S.; Coux, O.; Ruzsics, Z.; Wiethoff, C.M.; Kremer, E.J. A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner, L.B.; Wenk, M.R.; Guan, X.L.; Grzybek, M.; Coskun, U.; Greber, U.F. Co-option of membrane wounding enables virus penetration into cells. Cell Host Microbe 2015, 18, 75–85. [Google Scholar] [CrossRef]
- Wiethoff, C.M.; Nemerow, G.R. Adenovirus membrane penetration: Tickling the tail of a sleeping dragon. Virology 2015, 479–480, 591–599. [Google Scholar] [CrossRef]
- Hernando-Pérez, M.; Martín-González, N.; Pérez-Illana, M.; Suomalainen, M.; Condezo, G.N.; Ostapchuk, P.; Gallardo, J.; Menéndez, M.; Greber, U.F.; Hearing, P.; et al. Dynamic competition for hexon binding between core protein VII and lytic protein VI promotes adenovirus maturation and entry. Proc. Natl. Acad. Sci. USA 2020, 117, 13699–13707. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Galan, D.L.; Wodrich, H.; Wiethoff, C.M. An N-terminal domain of adenovirus protein VI fragments membranes by inducing positive membrane curvature. Virology 2010, 402, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Moyer, C.L.; Wiethoff, C.M.; Maier, O.; Smith, J.G.; Nemerow, G.R. Functional Genetic and Biophysical Analyses of Membrane Disruption by Human Adenovirus. J. Virol. 2011, 85, 2631–2641. [Google Scholar] [CrossRef] [PubMed]
- Pied, N.; Wodrich, H. Imaging the adenovirus infection cycle. FEBS Lett. 2019, 593, 3419–3448. [Google Scholar] [CrossRef]
- Dupzyk, A.; Tsai, B. How polyomaviruses exploit the ERAD machinery to cause infection. Viruses 2016, 8, 242. [Google Scholar] [CrossRef]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef]
- Guerrero, C.A.; Bouyssounade, D.; Zárate, S.; Iša, P.; López, T.; Espinosa, R.; Romero, P.; Méndez, E.; López, S.; Arias, C.F. Heat Shock Cognate Protein 70 Is Involved in Rotavirus Cell Entry. J. Virol. 2002, 76, 4096–4102. [Google Scholar] [CrossRef]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A Cytosolic Chaperone Complexes with Dynamic Membrane J-Proteins and Mobilizes a Nonenveloped Virus out of the Endoplasmic Reticulum. PLoS Pathog. 2014, 10, e1004007. [Google Scholar] [CrossRef]
- Inoue, T.; Tsai, B. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog. 2011, 7, 1002037. [Google Scholar] [CrossRef]
- Spriggs, C.C.; Badieyan, S.; Verhey, K.J.; Cianfrocco, M.A.; Tsai, B. Golgi-associated BICD adaptors couple ER membrane penetration and disassembly of a viral cargo. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Suikkanen, S.; Antila, M.; Jaatinen, A.; Vihinen-Ranta, M.; Vuento, M. Release of canine parvovirus from endocytic vesicles. Virology 2003, 316, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Canaan, S.; Zádori, Z.; Ghomashchi, F.; Bollinger, J.; Sadilek, M.; Moreau, M.E.; Tijssen, P.; Gelb, M.H. Interfacial Enzymology of Parvovirus Phospholipases A2. J. Biol. Chem. 2004, 279, 14502–14508. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, S.; Liebisch, G.; Kaufmann, B.; von Landenberg, P.; Hoffmann, J.H.; Drobnik, W.; Modrow, S. The VP1 Unique Region of Parvovirus B19 and Its Constituent Phospholipase A2-Like Activity. J. Virol. 2002, 76, 2014–2018. [Google Scholar] [CrossRef]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Farsetta, D.L.; Nibert, M.L. Strategy for Nonenveloped Virus Entry: A Hydrophobic Conformer of the Reovirus Membrane Penetration Protein μ1 Mediates Membrane Disruption. J. Virol. 2002, 76, 9920–9933. [Google Scholar] [CrossRef] [PubMed]
- Odegard, A.L.; Chandran, K.; Zhang, X.; Parker, J.S.L.; Baker, T.S.; Nibert, M.L. Putative Autocleavage of Outer Capsid Protein μ1, Allowing Release of Myristoylated Peptide μ1N during Particle Uncoating, Is Critical for Cell Entry by Reovirus. J. Virol. 2004, 78, 8732–8745. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Chandran, K.; Baker, T.S.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell 2002, 108, 283–295. [Google Scholar] [CrossRef]
- Zhang, L.; Agosto, M.A.; Ivanovic, T.; King, D.S.; Nibert, M.L.; Harrison, S.C. Requirements for the Formation of Membrane Pores by the Reovirus Myristoylated μ1N Peptide. J. Virol. 2009, 83, 7004–7014. [Google Scholar] [CrossRef]
- Agosto, M.A.; Ivanovic, T.; Nibert, M.L. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc. Natl. Acad. Sci. USA 2006, 103, 16496–16501. [Google Scholar] [CrossRef]
- Ivanovic, T.; Agosto, M.A.; Zhang, L.; Chandran, K.; Harrison, S.C.; Nibert, M.L. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008, 27, 1289–1298. [Google Scholar] [CrossRef]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2010; Volume 343, pp. 43–89. [Google Scholar]
- Schober, D.; Kronenberger, P.; Prchla, E.; Blaas, D.; Fuchs, R. Major and Minor Receptor Group Human Rhinoviruses Penetrate from Endosomes by Different Mechanisms. J. Virol. 1998, 72, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.J.; Rowlands, D.J.; Stonehouse, N.J.; Hogle, J.M.; Tuthill, T.J. Capsid Protein VP4 of Human Rhinovirus Induces Membrane Permeability by the Formation of a Size-Selective Multimeric Pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Staring, J.; Raaben, M.; Brummelkamp, T.R. Viral escape from endosomes and host detection at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Staring, J.; Von Castelmur, E.; Blomen, V.A.; Van Den Hengel, L.G.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; Van Kuppeveld, F.J.M.M.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017, 541, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.J.; Stanway, G. The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. J. Gen. Virol. 2000, 81, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Knodler, L.A.; Vallance, B.A.; Celli, J.; Winfree, S.; Hansen, B.; Montero, M.; Steele-Mortimer, O. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc. Natl. Acad. Sci. USA 2010, 107, 17733–17738. [Google Scholar] [CrossRef]
- Mellouk, N.; Enninga, J. Cytosolic access of intracellular bacterial pathogens: The Shigella paradigm. Front. Cell. Infect. Microbiol. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Mellouk, N.; Weiner, A.; Aulner, N.; Schmitt, C.; Elbaum, M.; Shorte, S.L.; Danckaert, A.; Enninga, J. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 2014, 16, 517–530. [Google Scholar] [CrossRef]
- Weiner, A.; Mellouk, N.; Lopez-Montero, N.; Chang, Y.-Y.Y.; Souque, C.; Schmitt, C.; Enninga, J. Macropinosomes are Key Players in Early Shigella Invasion and Vacuolar Escape in Epithelial Cells. PLoS Pathog. 2016, 12, e1005602. [Google Scholar] [CrossRef]
- Birmingham, C.L.; Smith, A.C.; Bakowski, M.A.; Yoshimori, T.; Brumell, J.H. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 2006, 281, 11374–11383. [Google Scholar] [CrossRef]
- Radtke, A.L.; Delbridge, L.M.; Balachandran, S.; Barber, G.N.; O’Riordan, M.X.D. TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 2007, 3. [Google Scholar] [CrossRef]
- Boyle, K.B.; Thurston, T.L.M.; Randow, F. TBK1 directs WIPI2 against Salmonella. Autophagy 2016, 12, 2508–2509. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.; Boyle, K.B.; Allen, M.; Ravenhill, B.J.; Karpiyevich, M.; Bloor, S.; Kaul, A.; Noad, J.; Foeglein, A.; Matthews, S.A.; et al. Recruitment of TBK 1 to cytosol-invading Salmonella induces WIPI 2-dependent antibacterial autophagy. EMBO J. 2016, 35, 1779–1792. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Duchateau, M.; Fredlund, J.; Weiner, A.; Mallet, A.; Schmitt, C.; Matondo, M.; Hourdel, V.; Chamot-Rooke, J.; Enninga, J. The COPII complex and lysosomal VAMP7 determine intracellular Salmonella localization and growth. Cell. Microbiol. 2015, 17, 1699–1720. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Jamieson, A.; Cresswell, P. GILT is a critical host factor for Listeria monocytogenes infection. Nature 2008, 455, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Radtke, A.L.; Anderson, K.L.; Davis, M.J.; DiMagno, M.J.; Swanson, J.A.; O’Riordan, M.X. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc. Natl. Acad. Sci. USA 2011, 108, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Gedde, M.M.; Higgins, D.E.; Tilney, L.G.; Portnoy, D.A. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect. Immun. 2000, 68, 999–1003. [Google Scholar] [CrossRef]
- Alberti-Segui, C.; Goeden, K.R.; Higgins, D.E. Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell-to-cell spread. Cell. Microbiol. 2007, 9, 179–195. [Google Scholar] [CrossRef]
- Malet, J.K.; Cossart, P.; Ribet, D. Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol-dependent cytolysins. Cell. Microbiol. 2017, 19, 1–11. [Google Scholar] [CrossRef]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef]
- Li, F.Y.; Weng, I.C.; Lin, C.H.; Kao, M.C.; Wu, M.S.; Chen, H.Y.; Liu, F.T. Helicobacter pylori induces intracellular galectin-8 aggregation around damaged lysosomes within gastric epithelial cells in a host O-glycan-dependent manner. Glycobiology 2018, 29, 151–162. [Google Scholar] [CrossRef] [PubMed]
- van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; Van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J.M. tuberculosis and M. leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, M.I.; Pehau-Arnaudet, G.; Fretz, M.M.; Romain, F.; Bottai, D.; Brodin, P.; Honoré, N.; Marchal, G.; Jiskoot, W.; England, P.; et al. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J. Bacteriol. 2007, 189, 6028–6034. [Google Scholar] [CrossRef] [PubMed]
- Akimana, C.; Al-Khodor, S.; Kwaik, Y.A. Host factors required for modulation of phagosome biogenesis and proliferation of francisella tularensis within the cytosol. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- De Leon, J.; Jiang, G.; Ma, Y.; Rubin, E.; Fortune, S.; Sun, J. Mycobacterium tuberculosis ESAT-6 exhibits a unique membrane-interacting activity that is not found in its ortholog from non-pathogenic Mycobacterium smegmatis. J. Biol. Chem. 2012, 287, 44184–44191. [Google Scholar] [CrossRef]
- Peng, X.; Sun, J. Mechanism of ESAT-6 membrane interaction and its roles in pathogenesis of Mycobacterium tuberculosis. Toxicon 2016, 116, 29–34. [Google Scholar] [CrossRef]
- Pilatz, S.; Breitbach, K.; Hein, N.; Fehlhaber, B.; Schulze, J.; Brenneke, B.; Eberl, L.; Steinmetz, I. Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect. Immun. 2006, 74, 3576–3586. [Google Scholar] [CrossRef]
- Whitworth, T.; Popov, V.L.; Yu, X.J.; Walker, D.H.; Bouyer, D.H. Expression of the Rickettsia prowazekii pld or tlyC gene in Salmonella enterica serovar typhimurium mediates phagosomal escape. Infect. Immun. 2005, 73, 6668–6673. [Google Scholar] [CrossRef]
- Renesto, P.; Dehoux, P.; Gouin, E.; Touqui, L.; Cossart, P.; Raoult, D. Identification and Characterization of a Phospholipase D–Superfamily Gene in Rickettsiae. J. Infect. Dis. 2003, 188, 1276–1283. [Google Scholar] [CrossRef]
- Clemens, D.L.; Lee, B.Y.; Horwitz, M.A. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 2004, 72, 3204–3217. [Google Scholar] [CrossRef]
- Clemens, D.L.; Ge, P.; Lee, B.Y.; Horwitz, M.A.; Zhou, Z.H. Atomic structure of T6SS reveals interlaced array essential to function. Cell 2015, 160, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Chong, A.; Wehrly, T.D.; Nair, V.; Fischer, E.R.; Barker, J.R.; Klose, K.E.; Celli, J. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect. Immun. 2008, 76, 5488–5499. [Google Scholar] [CrossRef] [PubMed]
- Berman, J. Morphogenesis and cell cycle progression in Candida albicans. Curr. Opin. Microbiol. 2006, 9, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Westman, J.; Moran, G.; Mogavero, S.; Hube, B.; Grinsteina, S. crossm Candida albicans Hyphal Expansion Causes Phagosomal. MBio 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Vylkova, S.; Carman, A.J.; Danhof, H.A.; Collette, J.R.; Zhou, H.; Lorenz, M.C. The fungal pathogen candida albicans autoinduces hyphal morphogenesis by raising extracellular pH. MBio 2011, 2. [Google Scholar] [CrossRef]
- De Souza, W.; De Carvalho, T.M.U.; Barrias, E.S. Review on Trypanosoma cruzi: Host cell interaction. Int. J. Cell Biol. 2010, 2010. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD 1, UBXD 1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-Synuclein Induces Lysosomal Rupture and Cathepsin Dependent Reactive Oxygen Species Following Endocytosis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Villamil Giraldo, A.M.; Appelqvist, H.; Ederth, T.; Öllinger, K. Lysosomotropic agents: Impact on lysosomal membrane permeabilization and cell death. Biochem. Soc. Trans. 2014, 42, 1460–1464. [Google Scholar] [CrossRef]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.-M.M.; Hämälistö, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Høgh, S.; Farkas, T.; Jonassen, A.H.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef] [PubMed]
- Uchimoto, T.; Nohara, H.; Kamehara, R.; Iwamura, M.; Watanabe, N.; Kobayashi, Y. Mechanism of apoptosis induced by a lysosomotropic agent, L-leucyl-L- leucine methyl ester. Apoptosis 1999, 4, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Du Rietz, H.; Hedlund, H.; Wilhelmson, S.; Nordenfelt, P.; Wittrup, A. Imaging small molecule-induced endosomal escape of siRNA. Nat. Commun. 2020, 11, 1809. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, A.; Ai, A.; Liu, X.; Hamar, P.; Trifonova, R.; Charisse, K.; Manoharan, M.; Kirchhausen, T.; Lieberman, J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015, 33, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Fraire, J.C.; Houthaeve, G.; Liu, J.; Raes, L.; Vermeulen, L.; Stremersch, S.; Brans, T.; García-Díaz Barriga, G.; De Keulenaer, S.; Van Nieuwerburgh, F.; et al. Vapor nanobubble is the more reliable photothermal mechanism for inducing endosomal escape of siRNA without disturbing cell homeostasis. J. Control. Release 2020, 319, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.; Miu, K.; Lung, P.; Zhang, S.; Zhao, S.; Chang, H.C.; Lin, G.; Li, Q. Rapid endosomal escape of prickly nanodiamonds: Implications for gene delivery. Sci. Rep. 2015, 5, 11661. [Google Scholar] [CrossRef] [PubMed]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.W.; Cummings, R.D.; Drickamer, K.; Felzi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.I.; et al. Galectins: A family of animal β-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Hirabayashi, J.; Kasai, K.I. The family of metazoan metal-independent β-galactoside-binding lectins: Structure, function and molecular evolution. Glycobiology 1993, 3, 297–304. [Google Scholar] [CrossRef]
- Wang, W.H.; Lin, C.Y.; Chang, M.R.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Chen, Y.H.; Liu, F.T.; Wang, S.F. The role of galectins in virus infection—A systemic literature review. J. Microbiol. Immunol. Infect. 2019. [Google Scholar] [CrossRef]
- Baum, L.G.; Garner, O.B.; Schaefer, K.; Lee, B. Microbe-host interactions are positively and negatively regulated by galectin-glycan interactions. Front. Immunol. 2014, 5, 284. [Google Scholar] [CrossRef]
- Paz, I.; Sachse, M.; Dupont, N.; Mounier, J.; Cederfur, C.; Enninga, J.; Leffler, H.; Poirier, F.; Prevost, M.C.; Lafont, F.; et al. Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 2010, 12, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Maier, O.; Marvin, S.A.; Wodrich, H.; Campbell, E.M.; Wiethoff, C.M. Spatiotemporal Dynamics of Adenovirus Membrane Rupture and Endosomal Escape. J. Virol. 2012, 86, 10821–10828. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Burrage, A.M.; Wiethoff, C.M.; Wodrich, H. High temporal resolution imaging reveals endosomal membrane penetration and escape of adenoviruses in real time. Methods Mol. Biol. 2013, 1064, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P. The mechanism and physiological function of macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Suzuki, K.; Noda, T.; Ohsumi, Y. Interrelationships among Atg proteins during autophagy inSaccharomyces cerevisiae. Yeast 2004, 21, 1057–1065. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Ravenhill, B.J.; Boyle, K.B.; von Muhlinen, N.; Ellison, C.J.; Masson, G.R.; Otten, E.G.; Foeglein, A.; Williams, R.; Randow, F. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol. Cell 2019, 74, 320–329.e6. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef]
- Roberts, R.; Ktistakis, N.T. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013, 55, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNARE s and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Weng, I.C.; Chen, H.L.; Lo, T.H.; Lin, W.H.; Chen, H.Y.; Hsu, D.K.; Liu, F.T. Cytosolic galectin-3 and -8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology 2018, 28, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Feeley, E.M.; Pilla-Moffett, D.M.; Zwack, E.E.; Piro, A.S.; Finethy, R.; Kolb, J.P.; Martinez, J.; Brodsky, I.E.; Coers, J. Galectin-3 directs antimicrobial guanylate binding proteins to vacuoles furnished with bacterial secretion systems. Proc. Natl. Acad. Sci. USA 2017, 114, E1698–E1706. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Dikic, I. Regulation of Salmonella-host cell interactions via the ubiquitin system. Int. J. Med. Microbiol. 2018, 308, 176–184. [Google Scholar] [CrossRef]
- Dupont, N.; Lacas-Gervais, S.; Bertout, J.; Paz, I.; Freche, B.; Van Nhieu, G.T.; van der Goot, F.G.; Sansonetti, P.J.; Lafont, F. Shigella Phagocytic Vacuolar Membrane Remnants Participate in the Cellular Response to Pathogen Invasion and Are Regulated by Autophagy. Cell Host Microbe 2009, 6, 137–149. [Google Scholar] [CrossRef]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef]
- Luisoni, S.; Bauer, M.; Prasad, V.; Boucke, K.; Papadopoulos, C.; Meyer, H.; Hemmi, S.; Suomalainen, M.; Greber, U. Endosomophagy clears disrupted early endosomes but not virus particles during virus entry into cells. Matters 2016, 1–9. [Google Scholar] [CrossRef]
- Yoshida, Y.; Yasuda, S.; Fujita, T.; Hamasaki, M.; Murakami, A.; Kawawaki, J.; Iwai, K.; Saeki, Y.; Yoshimori, T.; Matsuda, N.; et al. Ubiquitination of exposed glycoproteins by SCFFBXO27 directs damaged lysosomes for autophagy. Proc. Natl. Acad. Sci. USA 2017, 114, 8574–8579. [Google Scholar] [CrossRef]
- Fujita, N.; Morita, E.; Itoh, T.; Tanaka, A.; Nakaoka, M.; Osada, Y.; Umemoto, T.; Saitoh, T.; Nakatogawa, H.; Kobayashi, S.; et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J. Cell Biol. 2013, 203, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.P.; Ho, M.W.L.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Huett, A.; Heath, R.J.; Begun, J.; Sassi, S.O.; Baxt, L.A.; Vyas, J.M.; Goldberg, M.B.; Xavier, R.J. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular salmonella typhimurium. Cell Host Microbe 2012, 12, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Noad, J.; Von Der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2017, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Koerver, L.; Papadopoulos, C.; Liu, B.; Kravic, B.; Rota, G.; Brecht, L.; Veenendaal, T.; Polajnar, M.; Bluemke, A.; Ehrmann, M.; et al. The ubiquitin-conjugating enzyme UBE 2 QL 1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Seczynska, M.; Dikic, I. Removing the waste bags: How p97 drives autophagy of lysosomes. EMBO J. 2017, 36, 129–131. [Google Scholar] [CrossRef]
- Falcon, B.; Noad, J.; McMahon, H.; Randow, F.; Goedert, M. Galectin-8-mediated selective autophagy protects against seeded tau aggregation. J. Biol. Chem. 2018, 293, 2438–2451. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Thurston, T.L.M.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Pilli, M.; Arko-Mensah, J.; Ponpuak, M.; Roberts, E.; Master, S.; Mandell, M.A.; Dupont, N.; Ornatowski, W.; Jiang, S.; Bradfute, S.B.; et al. TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 2012, 37, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A.; Children, T. Signals from the lysosome. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Brady, O.A.; Martina, J.A.; Puertollano, R. Emerging roles for TFEB in the immune response and inflammation. Autophagy 2018, 14, 181–189. [Google Scholar] [CrossRef]
- Nazio, F.; Carinci, M.; Valacca, C.; Bielli, P.; Strappazzon, F.; Antonioli, M.; Ciccosanti, F.; Rodolfo, C.; Campello, S.; Fimia, G.M.; et al. Fine-tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J. Cell Biol. 2016, 215, 841–856. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343. [Google Scholar] [CrossRef]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef]
- Radulovic, M.; Schink, K.O.; Wenzel, E.M.; Nähse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018, 37, 1–15. [Google Scholar] [CrossRef]
- Hurley, J.H. ESCRTs are everywhere. Embo J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef]
- Skowyra, M.L.; Schlesinger, P.H.; Naismith, T.V.; Hanson, P.I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 2018, 360. [Google Scholar] [CrossRef]
- Repnik, U.; Distefano, M.B.; Speth, M.T.; Wui Ng, M.Y.; Progida, C.; Hoflack, B.; Gruenberg, J.; Griffiths, G. L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J. Cell Sci. 2017, 130, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins Control MTOR and AMPK in Response to Lysosomal Damage to Induce Autophagy; Taylor and Francis Inc.: Abingdon, UK, 2019; Volume 15, pp. 169–171. [Google Scholar]
- Herbst, S.; Campbell, P.; Harvey, J.; Bernard, E.M.; Papayannopoulos, V.; Wood, N.W.; Morris, H.R.; Gutierrez, M.G. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020, 44, 1–14. [Google Scholar] [CrossRef]
- Mittal, E.; Skowyra, M.L.; Uwase, G.; Tinaztepe, E.; Mehra, A.; Köster, S.; Hanson, P.I.; Philips, J.A. Mycobacterium tuberculosis type VII secretion system effectors differentially impact the ESCRT endomembrane damage response. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kreibich, S.; Emmenlauer, M.; Fredlund, J.; Dehio, C. Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1 Graphical Abstract Highlights d RNAi screen implicates autophagy in Salmonella-containing vacuole (SCV) maintenance d Autophagy promotes repair of TTSS-1-damaged SCV membranes and TTSS-2 expression d In parallel, autophagy eliminates cytosolic Salmonella d Autophagy could play a general role in maintaining endosomal membrane integrity. Cell Host Microbe 2015, 18, 527–537. [Google Scholar] [CrossRef] [PubMed]
- López-Jiménez, A.T.; Cardenal-Muñoz, E.; Leuba, F.; Gerstenmaier, L.; Barisch, C.; Hagedorn, M.; King, J.S.; Soldati, T. The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PLOS Pathog. 2018, 14, e1007501. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Emr, S.D. The ESCRT complexes: Structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 277–298. [Google Scholar] [CrossRef]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef]
- Ellison, C.J.; Kukulski, W.; Boyle, K.B.; Munro, S.; Randow, F. Transbilayer Movement of Sphingomyelin Precedes Catastrophic Breakage of Enterobacteria-Containing Vacuoles. Curr. Biol. 2020, 30, 2974–2983.e6. [Google Scholar] [CrossRef]
- Gout, E.; Gutkowska, M.; Takayama, S.; Reed, J.C.; Chroboczek, J. Co-chaperone BAG3 and adenovirus penton base protein partnership. J. Cell. Biochem. 2010, 111, 699–708. [Google Scholar] [CrossRef]
- Zhao, M.; Antunes, F.; Eaton, J.W.; Brunk, U.T. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 2003, 270, 3778–3786. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–226. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Claude-Taupin, A.; Kumar, S.; Won Choi, S.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; et al. Cellular and molecular mechanism for secretory autophagy. Autophagy 2017, 13, 1084–1085. [Google Scholar] [CrossRef]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Gabandé-Rodríguez, E.; Pérez-Cañamás, A.; Soto-Huelin, B.; Mitroi, D.N.; Sánchez-Redondo, S.; Martínez-Sáez, E.; Venero, C.; Peinado, H.; Ledesma, M.D. Lipid-induced lysosomal damage after demyelination corrupts microglia protective function in lysosomal storage disorders. EMBO J. 2019, 38, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Leyns, C.E.G.; Holtzman, D.M. Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Schoonbroodt, S.; Ferreira, V.; Best-Belpomme, M.; Boelaert, J.R.; Legrand-Poels, S.; Korner, M.; Piette, J. Crucial Role of the Amino-Terminal Tyrosine Residue 42 and the Carboxyl-Terminal PEST Domain of IκBα in NF-κB Activation by an Oxidative Stress. J. Immunol. 2000, 164, 4292–4300. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65. Evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef]
- Anderson, F.L.; Coffey, M.M.; Berwin, B.L.; Havrda, M.C. Inflammasomes: An Emerging Mechanism Translating Environmental Toxicant Exposure Into Neuroinflammation in Parkinson’s Disease. Toxicol. Sci. 2018, 166, 3–15. [Google Scholar] [CrossRef]
- Siew, J.J.; Chen, H.M.; Chen, H.Y.; Chen, H.L.; Chen, C.M.; Soong, B.W.; Wu, Y.R.; Chang, C.P.; Chan, Y.C.; Lin, C.H.; et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Streetly, M.J.; Maharaj, L.; Joel, S.; Schey, S.A.; Gribben, J.G.; Cotter, F.E. GCS-100, a novel galectin-3 antagonist, modulates MCL-1, NOXA, and cell cycle to induce myeloma cell death. Blood 2010, 115, 3939–3948. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis E Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Takahasi, K.; Yoneyama, M.; Nishihori, T.; Hirai, R.; Kumeta, H.; Narita, R.; Gale, M.; Inagaki, F.; Fujita, T. Nonself RNA-Sensing Mechanism of RIG-I Helicase and Activation of Antiviral Immune Responses. Mol. Cell 2008, 29, 428–440. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Pichlmair, A.; Reis e Sousa, C. Innate Recognition of Viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 59-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic: Polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J.M. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS Mediate Type I Interferon Responses to Coxsackie B Virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yanagi, Y.; Ichinohe, T. Encephalomyocarditis Virus Viroporin 2B Activates NLRP3 Inflammasome. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Rajan, J.V.; Rodriguez, D.; Miao, E.A.; Aderem, A. The NLRP3 Inflammasome Detects Encephalomyocarditis Virus and Vesicular Stomatitis Virus Infection. J. Virol. 2011, 85, 4167–4172. [Google Scholar] [CrossRef]
- da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Xu, Z.; Tian, J.; Palmer, D.J.; Ng, P.; Byrnes, A.P. The role of endosomal escape and mitogen-activated protein kinases in adenoviral activation of the innate immune response. PLoS ONE 2011, 6, e26755. [Google Scholar] [CrossRef] [PubMed]
- Nociari, M.; Ocheretina, O.; Murphy, M.; Falck-Pedersen, E. Adenovirus Induction of IRF3 Occurs through a Binary Trigger Targeting Jun N-Terminal Kinase and TBK1 Kinase Cascades and Type I Interferon Autocrine Signaling. J. Virol. 2009, 83, 4081–4091. [Google Scholar] [CrossRef][Green Version]
- Stein, S.C.; Falck-Pedersen, E. Sensing Adenovirus Infection: Activation of Interferon Regulatory Factor 3 in RAW 264.7 Cells. J. Virol. 2012, 86, 4527–4537. [Google Scholar] [CrossRef]
- Eichholz, K.; Bru, T.; Tran, T.T.P.; Fernandes, P.; Welles, H.; Mennechet, F.J.D.; Manel, N.; Alves, P.; Perreau, M.; Kremer, E.J. Immune-Complexed Adenovirus Induce AIM2-Mediated Pyroptosis in Human Dendritic Cells. PLoS Pathog. 2016, 12, e1005871. [Google Scholar] [CrossRef]
- Barlan, A.U.; Griffin, T.M.; Mcguire, K.A.; Wiethoff, C.M. Adenovirus Membrane Penetration Activates the NLRP3 Inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef]
- McGuire, K.A.; Barlan, A.U.; Griffin, T.M.; Wiethoff, C.M. Adenovirus Type 5 Rupture of Lysosomes Leads to Cathepsin B-Dependent Mitochondrial Stress and Production of Reactive Oxygen Species. J. Virol. 2011, 85, 10806–10813. [Google Scholar] [CrossRef]
- Tibbles, L.A.; Spurrell, J.C.L.; Bowen, G.P.; Liu, Q.; Lam, M.; Zaiss, A.K.; Robbins, S.M.; Hollenberg, M.D.; Wickham, T.J.; Muruve, D.A. Activation of p38 and ERK Signaling during Adenovirus Vector Cell Entry Lead to Expression of the C-X-C Chemokine IP-10. J. Virol. 2002, 76, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Anghelina, D.; Lam, E.; Falck-Pedersen, E. Diminished Innate Antiviral Response to Adenovirus Vectors in cGAS/STING-Deficient Mice Minimally Impacts Adaptive Immunity. J. Virol. 2016, 90, 5915–5927. [Google Scholar] [CrossRef] [PubMed]
- Maler, M.D.; Nielsen, P.J.; Stichling, N.; Cohen, I.; Ruzsics, Z.; Wood, C.; Engelhard, P.; Suomalainen, M.; Gyory, I.; Huber, M.; et al. Key role of the scavenger receptor MARCO in mediating adenovirus infection and subsequent innate responses of macrophages. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, R.E.; McEwan, W.A.; Tam, J.C.H.; Vaysburd, M.; James, L.C. TRIM21 Promotes cGAS and RIG-I Sensing of Viral Genomes during Infection by Antibody-Opsonized Virus. PLoS Pathog. 2015, 11, e1005253. [Google Scholar] [CrossRef]
- Hare, D.; Mossman, K.L. Novel paradigms of innate immune sensing of viral infections. Cytokine 2013, 63, 219–224. [Google Scholar] [CrossRef]
- Collins, S.E.; Mossman, K.L. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev. 2014, 25, 525–531. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy as an innate immunity paradigm: Expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 2012, 24, 21–31. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Santeford, A.; Wiley, L.A.; Park, S.; Bamba, S.; Nakamura, R.; Gdoura, A.; Ferguson, T.A.; Rao, P.K.; Guan, J.L.; Saitoh, T.; et al. Impaired autophagy in macrophages promotes inflammatory eye disease. Autophagy 2016, 12, 1876–1885. [Google Scholar] [CrossRef]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef]
- Bechelli, J.; Vergara, L.; Smalley, C.; Buzhdygan, T.P.; Bender, S.; Zhang, W.; Liu, Y.; Popov, V.L.; Wang, J.; Garg, N.; et al. Atg5 supports Rickettsia australis infection in macrophages in vitro and in vivo. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 1–15. [Google Scholar] [CrossRef]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O’Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1β secretion by targeting Pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef]
- Shi, C.-S.S.; Shenderov, K.; Huang, N.-N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Liu, T.; Tang, Q.; Liu, K.; Xie, W.; Liu, X.; Wang, H.; Wang, R.F.; Cui, J. TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via p62-Dependent Selective Autophagy. Cell Rep. 2016, 16, 1988–2002. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef]
- Van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM proteins and their roles in antiviral host defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 Negatively Regulates Autophagic Processes through an Association with Beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.J.; Huang, H.Y.; Huang, M.P.; Liou, W.; Chang, Y.T.; Wu, C.C.; Ojcius, D.M.; Chang, Y.S. The microtubule-associated protein EB1 links AIM2 inflammasomes with autophagy-dependent secretion. J. Biol. Chem. 2014, 289, 29322–29333. [Google Scholar] [CrossRef]
- Deretic, V. Multiple regulatory and effector roles of autophagy in immunity. Curr. Opin. Immunol. 2009, 21, 53–62. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, X.-D.; Gong, X.; Eissa, N.T. Signaling pathway of autophagy associated with innate immunity. Autophagy 2008, 4, 110–112. [Google Scholar] [CrossRef]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef]
- Shi, C.-S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5-Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef]
- Jin, S.; Tian, S.; Chen, Y.; Zhang, C.; Xie, W.; Xia, X.; Cui, J.; Wang, R. USP 19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin-1. EMBO J. 2016, 35, 866–880. [Google Scholar] [CrossRef]
- Jin, S.; Tian, S.; Luo, M.; Xie, W.; Liu, T.; Duan, T.; Wu, Y.; Cui, J. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol. Cell 2017, 68, 308–322.e4. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Duan, T.; Feng, Y.; Liu, Q.; Lin, M.; Cui, J.; Wang, R. LRRC25 inhibits type I IFN signaling by targeting ISG15-associated RIG-I for autophagic degradation. EMBO J. 2018, 37, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.P.; Ha, T.; et al. Crosstalk between the cGAS DNA sensor and beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef]
- Prabakaran, T.; Bodda, C.; Krapp, C.; Zhang, B.; Christensen, M.H.; Sun, C.; Reinert, L.; Cai, Y.; Jensen, S.B.; Skouboe, M.K.; et al. Attenuation of c GAS-STING signaling is mediated by a p62/ SQSTM 1-dependent autophagy pathway activated by TBK1. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Niida, M.; Tanaka, M.; Kamitani, T. Downregulation of active IKKβ by Ro52-mediated autophagy. Mol. Immunol. 2010, 47, 2378–2387. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef]
- Sparrer, K.M.J.; Gableske, S.; Zurenski, M.A.; Parker, Z.M.; Full, F.; Baumgart, G.J.; Kato, J.; Pacheco-Rodriguez, G.; Liang, C.; Pornillos, O.; et al. TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2017, 2, 1543–1557. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Münch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Labzin, L.I.; Bottermann, M.; Rodriguez-Silvestre, P.; Foss, S.; Andersen, J.T.; Vaysburd, M.; Clift, D.; James, L.C. Antibody and DNA sensing pathways converge to activate the inflammasome during primary human macrophage infection. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.A.; Jackson, W.T. Picornavirus subversion of the autophagy pathway. Viruses 2011, 3, 1549–1561. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daussy, C.F.; Wodrich, H. “Repair Me if You Can”: Membrane Damage, Response, and Control from the Viral Perspective. Cells 2020, 9, 2042. https://doi.org/10.3390/cells9092042
Daussy CF, Wodrich H. “Repair Me if You Can”: Membrane Damage, Response, and Control from the Viral Perspective. Cells. 2020; 9(9):2042. https://doi.org/10.3390/cells9092042
Chicago/Turabian StyleDaussy, Coralie F., and Harald Wodrich. 2020. "“Repair Me if You Can”: Membrane Damage, Response, and Control from the Viral Perspective" Cells 9, no. 9: 2042. https://doi.org/10.3390/cells9092042
APA StyleDaussy, C. F., & Wodrich, H. (2020). “Repair Me if You Can”: Membrane Damage, Response, and Control from the Viral Perspective. Cells, 9(9), 2042. https://doi.org/10.3390/cells9092042