A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope

 ,
,  and
and 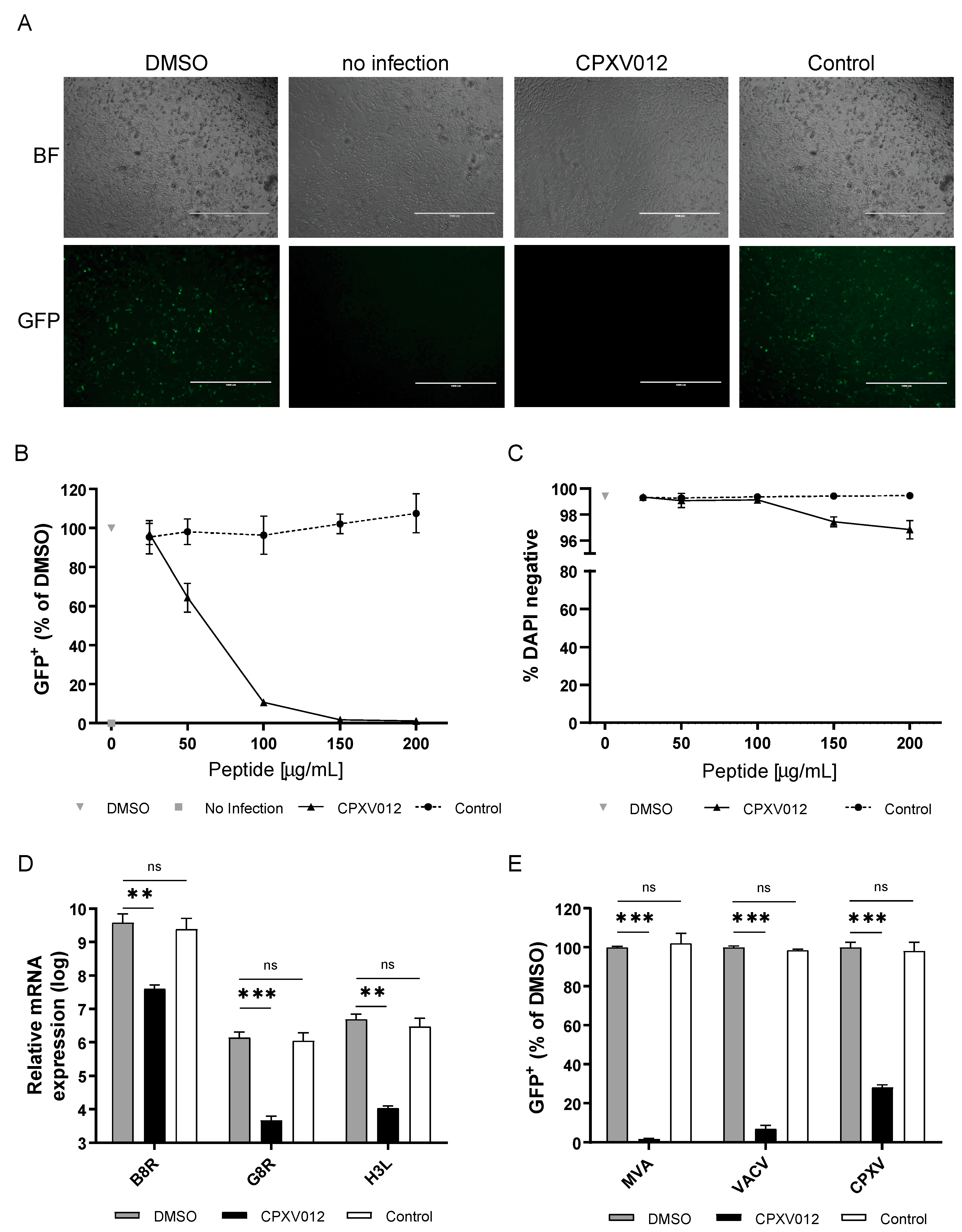{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Peptides
2.3. Cell Viability Assays
2.4. Virus Inhibition Assays
2.4.1. MVA, VACV, and Cowpox
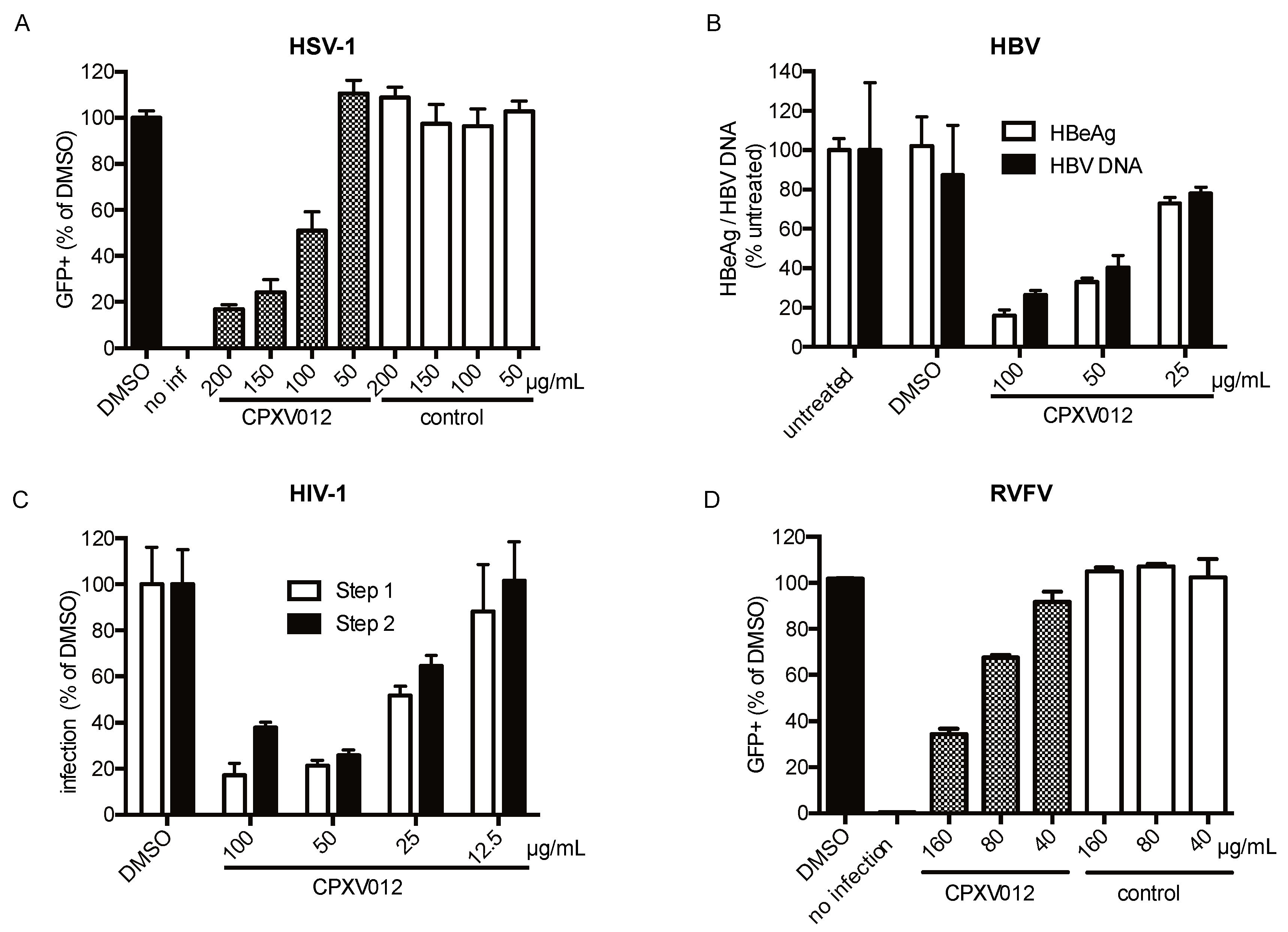
2.4.2. HSV-1
2.4.3. HBV
2.4.4. HIV-1
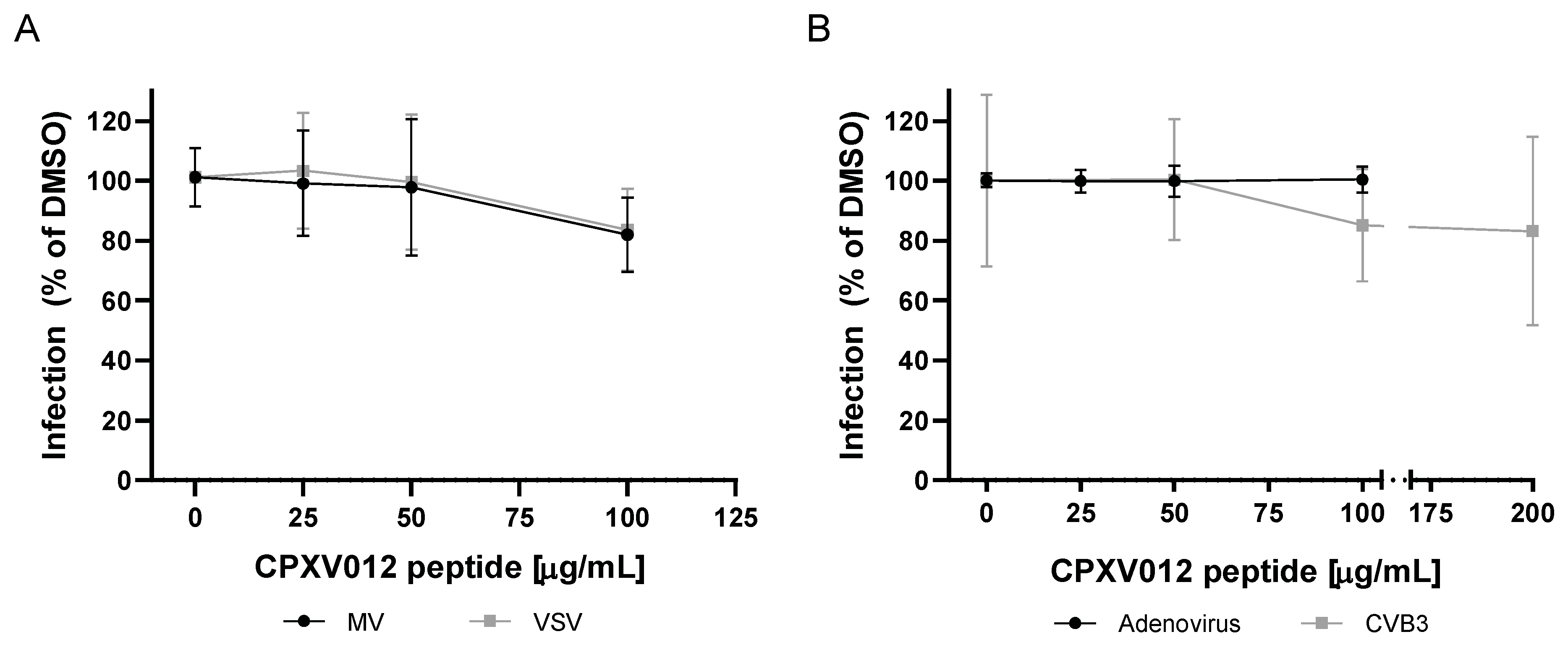
2.4.5. Measles and VSV
2.4.6. Adenovirus
2.4.7. Rift Valley Fever Virus
2.4.8. Coxsackievirus B3
2.5. Preparation of Large Unilamellar Vesicles (LUVs)
2.6. Circular Dichroism
2.7. Langmuir Monolayers
2.8. Membrane Permeability Assay
2.9. Statistical Analysis
3. Results
3.1. CPXV012 Peptide Prevents Infection by Different Viruses in Cell Culture
3.2. CPXV012 May Bind to Viral Particles
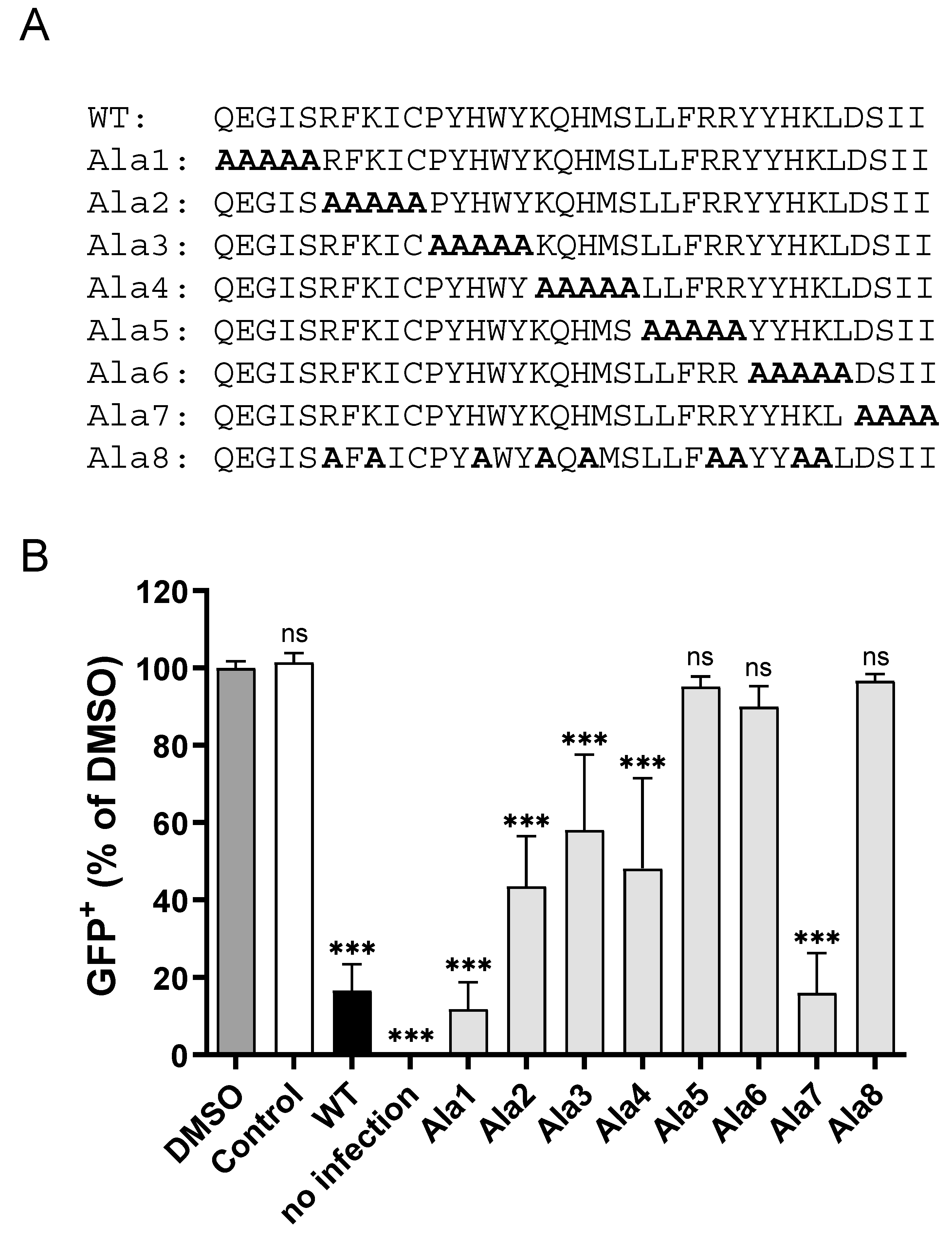
3.3. CPXV012 Peptide Variants Differentially Affect Virus Infection
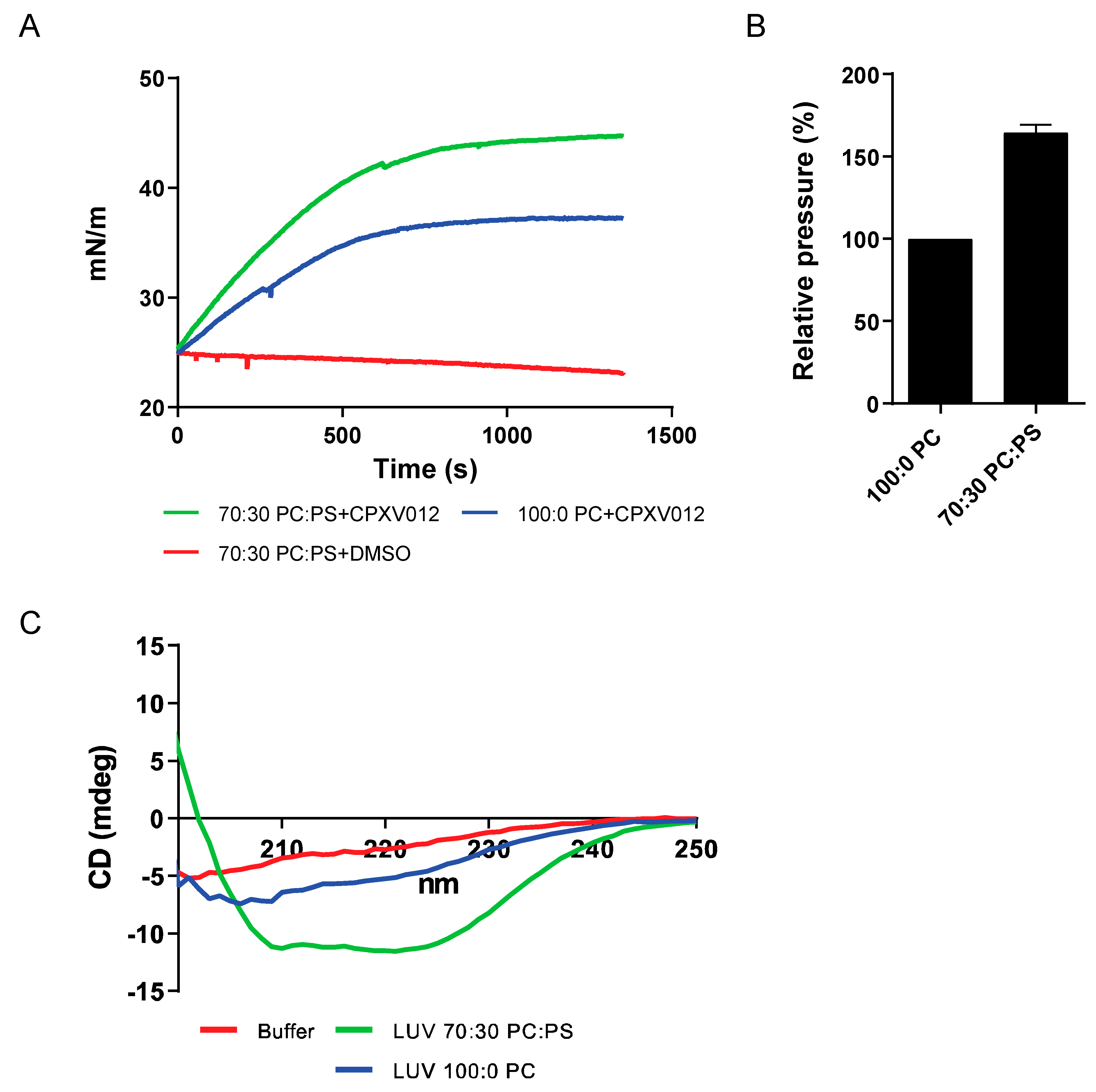
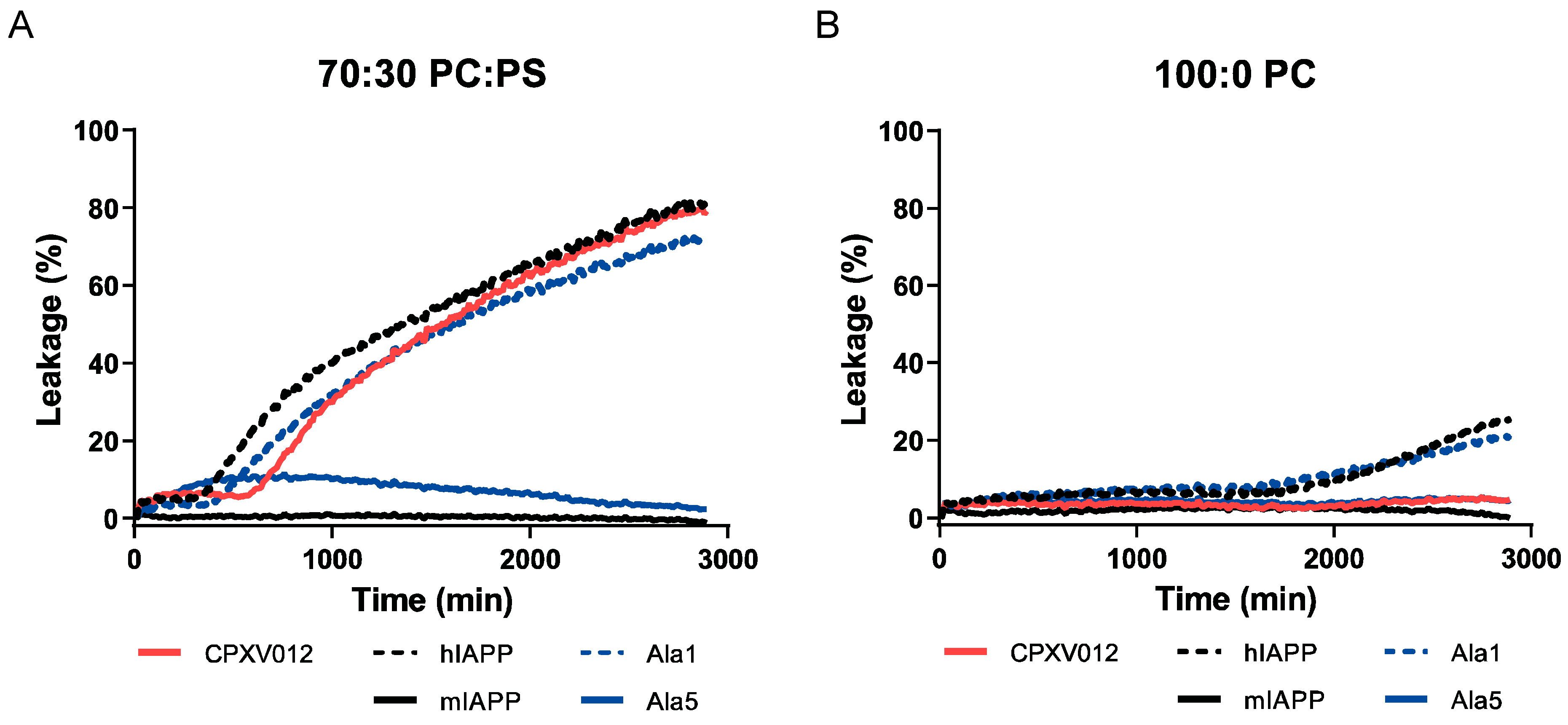
3.4. CPXV012 Peptide Interacts with Charged Phospholipids
4. Discussion
4.1. Poxviruses
4.2. HSV-1
4.3. HBV
4.4. HIV
4.5. RVFV
4.6. Viruses Not Affected by CPXV012 Peptide
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Marston, H.D.; Folkers, G.K.; Morens, D.M.; Fauci, A.S. Emerging viral diseases: Confronting threats with new technologies. Sci. Transl. Med. 2014, 6, 253ps10. [Google Scholar] [CrossRef]
- Zhu, J.D.; Meng, W.; Wang, X.J.; Wang, H.C. Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 517. [Google Scholar] [CrossRef]
- Soares, M.M.; King, S.W.; Thorpe, P.E. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 2008, 14, 1357–1362. [Google Scholar] [CrossRef]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.C.; Freiberg, A.N.; Zhang, T.; Akyol-Ataman, Z.; Grock, A.; Hong, P.W.; Li, J.; Watson, N.F.; Fang, A.Q.; Aguilar, H.C.; et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl Acad. Sci. USA 2010, 107, 3157–3162. [Google Scholar] [CrossRef] [PubMed]
- Findlay, E.G.; Currie, S.M.; Davidson, D.J. Cationic host defence peptides: Potential as antiviral therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.C.L.; Lima, L.A.; Miranda, V.J.; Dias, S.C.; Franco, O.L. Current scenario of peptide-based drugs: The key roles of cationic antitumor and antiviral peptides. Front. Microbiol. 2013, 4, 321. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef]
- Alzhanova, D.; Edwards, D.M.; Hammarlund, E.; Scholz, I.G.; Horst, D.; Wagner, M.J.; Upton, C.; Wiertz, E.J.; Slifka, M.K.; Früh, K. Cowpox Virus Inhibits the Transporter Associated with Antigen Processing to Evade T Cell Recognition. Cell Host Microbe 2009, 6, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.; Verweij, M.C.; Pickup, D.J.; Wiertz, E.J.H.J.; Hansen, T.H.; Yokoyama, W.M. Two Mechanistically Distinct Immune Evasion Proteins of Cowpox Virus Combine to Avoid Antiviral CD8 T Cells. Cell Host Microbe 2009, 6, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Eggensperger, S.; Hank, S.; Wycisk, A.I.; Wieneke, R.; Mayerhofer, P.U.; Tampé, R. A negative feedback modulator of antigen processing evolved from a frameshift in the cowpox virus genome. PLoS Pathog. 2014, 10, e1004554. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.D.; Hoelen, H.; Kruse, E.; van Leeuwen, W.F.; Grootens, J.; Horst, D.; Koorengevel, M.; Drijfhout, J.W.; Kremmer, E.; Früh, K.; et al. Cowpox Virus Protein CPXV012 Eludes CTLs by Blocking ATP Binding to TAP. J. Immunol. 2014, 193, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Praest, P.; Liaci, A.M.; Förster, F.; Wiertz, E.J.H.J. New insights into the structure of the MHC class I peptide-loading complex and mechanisms of TAP inhibition by viral immune evasion proteins. Mol. Immunol. 2019, 113, 103–114. [Google Scholar] [CrossRef]
- Kremb, S.; Helfer, M.; Heller, W.; Hoffmann, D.; Wolff, H.; Kleinschmidt, A.; Cepok, S.; Hemmer, B.; Durner, J.; Brack-Werner, R. EASY-HIT: HIV full-replication technology for broad discovery of multiple classes of HIV inhibitors. Antimicrob. Agents Chemother. 2010, 54, 5257–5268. [Google Scholar] [CrossRef]
- Gasteiger, G.; Kastenmuller, W.; Ljapoci, R.; Sutter, G.; Drexler, I. Cross-Priming of Cytotoxic T Cells Dictates Antigen Requisites for Modified Vaccinia Virus Ankara Vector Vaccines. J. Virol. 2007, 81, 11925–11936. [Google Scholar] [CrossRef]
- Staib, C.; Drexler, I.; Sutter, G. Construction and isolation of recombinant MVA. Methods Mol. Biol. 2004, 269, 77–100. [Google Scholar] [CrossRef]
- Lucifora, J.; Arzberger, S.; Durantel, D.; Belloni, L.; Strubin, M.; Levrero, M.; Zoulim, F.; Hantz, O.; Protzer, U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 2011, 55, 996–1003. [Google Scholar] [CrossRef]
- Duprex, W.P.; McQuaid, S.; Roscic-Mrkic, B.; Cattaneo, R.; McCallister, C.; Rima, B.K. In vitro and in vivo infection of neural cells by a recombinant measles virus expressing enhanced green fluorescent protein. J. Virol. 2000, 74, 7972–7979. [Google Scholar] [CrossRef] [PubMed]
- Wohlleber, D.; Kashkar, H.; Gartner, K.; Frings, M.K.; Odenthal, M.; Hegenbarth, S.; Borner, C.; Arnold, B.; Hammerling, G.; Nieswandt, B.; et al. TNF-induced target cell killing by CTL activated through cross-presentation. Cell Rep. 2012, 2, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, J.; Oreshkova, N.; Cobos-Jimenez, V.; Vloet, R.P.; Potgieter, C.A.; Moormann, R.J. Creation of a nonspreading Rift Valley fever virus. J. Virol. 2011, 85, 12622–12630. [Google Scholar] [CrossRef] [PubMed]
- Lanke, K.H.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, H.S.; Duinkerken, G.; Benckhuijsen, W.E.; Amons, R.; de Vries, R.R.; Roep, B.O.; Drijfhout, J.W. The identification of CD4+ T cell epitopes with dedicated synthetic peptide libraries. Proc. Natl. Acad. Sci. USA 1997, 94, 10313–10318. [Google Scholar] [CrossRef] [PubMed]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Rouser, G.; Kritchevsky, G.; Simon, G.; Nelson, G.J. Quantitative analysis of brain and spinach leaf lipids employing silicic acid column chromatography and acetone for elution of glycolipids. Lipids 1967, 2, 37–40. [Google Scholar] [CrossRef]
- Van de Weijer, M.L.; Luteijn, R.D.; Wiertz, E.J.H.J. Viral immune evasion: Lessons in MHC class I antigen presentation. Semin. Immunol. 2015, 27, 125–137. [Google Scholar] [CrossRef]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, L.E.; Dunkelberger, E.B.; Tran, H.Q.; Cheng, P.N.; Chiu, C.C.; Cao, P.; Raleigh, D.P.; de Pablo, J.J.; Nowick, J.S.; Zanni, M.T. Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient beta-sheet. Proc. Natl. Acad. Sci. USA 2013, 110, 19285–19290. [Google Scholar] [CrossRef]
- Eiriksdottir, E.; Konate, K.; Langel, U.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta 2010, 1798, 1119–1128. [Google Scholar] [CrossRef]
- Shepherd, C.M.; Vogel, H.J.; Tieleman, D.P. Interactions of the designed antimicrobial peptide MB21 and truncated dermaseptin S3 with lipid bilayers: Molecular-dynamics simulations. Biochem. J. 2003, 370, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Vogel, H.J. Structure-function relationships of antimicrobial peptides. Biochem. Cell Biol. 1998, 76, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Baltzer, S.A.; Brown, M.H. Antimicrobial peptides: Promising alternatives to conventional antibiotics. J. Mol. Microbiol. Biotechnol. 2011, 20, 228–235. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Currie, S.M.; Gwyer Findlay, E.; McFarlane, A.J.; Fitch, P.M.; Bottcher, B.; Colegrave, N.; Paras, A.; Jozwik, A.; Chiu, C.; Schwarze, J.; et al. Cathelicidins Have Direct Antiviral Activity against Respiratory Syncytial Virus In Vitro and Protective Function In Vivo in Mice and Humans. J. Immunol. 2016, 196, 2699–2710. [Google Scholar] [CrossRef]
- Howell, M.D.; Jones, J.F.; Kisich, K.O.; Streib, J.E.; Gallo, R.L.; Leung, D.Y. Selective killing of vaccinia virus by LL-37: Implications for eczema vaccinatum. J. Immunol. 2004, 172, 1763–1767. [Google Scholar] [CrossRef]
- Tripathi, S.; Tecle, T.; Verma, A.; Crouch, E.; White, M.; Hartshorn, K.L. The human cathelicidin LL-37 inhibits influenza A viruses through a mechanism distinct from that of surfactant protein D or defensins. J. Gen. Virol. 2013, 94, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, M.E.; Amatore, D.; Villa, S.; Casciaro, B.; Aimola, P.; Franci, G.; Grieco, P.; Galdiero, M.; Palamara, A.T.; Mangoni, M.L.; et al. The Amphibian Antimicrobial Peptide Temporin B Inhibits In Vitro Herpes Simplex Virus 1 Infection. Antimicrob. Agents Chemother. 2018, 62, e02367-17. [Google Scholar] [CrossRef] [PubMed]
- Holthausen, D.J.; Lee, S.H.; Kumar, V.T.; Bouvier, N.M.; Krammer, F.; Ellebedy, A.H.; Wrammert, J.; Lowen, A.C.; George, S.; Pillai, M.R.; et al. An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses. Immunity 2017, 46, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Migallon, M.P.; Aranda, F.J.; Gomez-Fernandez, J.C. Role of phosphatidylserine and diacylglycerol in the fusion of chromaffin granules with target membranes. Arch. Biochem. Biophys. 1994, 314, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Teissier, E.; Pecheur, E.I. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur. Biophys. J. 2007, 36, 887–899. [Google Scholar] [CrossRef]
- Dowall, S.D.; Graham, V.A.; Corbin-Lickfett, K.; Empig, C.; Schlunegger, K.; Bruce, C.B.; Easterbrook, L.; Hewson, R. Effective binding of a phosphatidylserine-targeting antibody to Ebola virus infected cells and purified virions. J. Immunol. Res. 2015, 2015, 347903. [Google Scholar] [CrossRef]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Kondratowicz, A.S.; Davey, R.A.; Rennert, P.D.; Maury, W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 2013, 87, 8327–8341. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Chung, K.N.; Gross, R.W. Lipid rafts are enriched in arachidonic acid and plasmenylethanolamine and their composition is independent of caveolin-1 expression: A quantitative electrospray ionization/mass spectrometric analysis. Biochemistry 2002, 41, 2075–2088. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wilk, T.; Fuller, S.D. Do lipid rafts mediate virus assembly and pseudotyping? J. Gen. Virol. 2003, 84, 757–768. [Google Scholar] [CrossRef]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Smith, G.L. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008, 16, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, Y.; Oie, M. The activation of vaccinia virus infectivity by the transfer of phosphatidylserine from the plasma membrane. Virology 1983, 130, 306–317. [Google Scholar] [CrossRef]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Huang, L.C.; Romanowski, E.G.; Yates, K.A.; Proske, R.J.; McDermott, A.M. Human cathelicidin (LL-37), a multifunctional peptide, is expressed by ocular surface epithelia and has potent antibacterial and antiviral activity. Curr. Eye Res. 2005, 30, 385–394. [Google Scholar] [CrossRef]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar] [CrossRef]
- Crump, C. Virus assembly and egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar]
- Jiang, B.; Himmelsbach, K.; Ren, H.; Boller, K.; Hildt, E. Subviral Hepatitis B Virus Filaments, like Infectious Viral Particles, Are Released via Multivesicular Bodies. J. Virol. 2016, 90, 3330–3341. [Google Scholar] [CrossRef]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- De Meyer, S.; Gong, Z.; Depla, E.; Maertens, G.; Yap, S.H. Involvement of phosphatidylserine and non-phospholipid components of the hepatitis B virus envelope in human Annexin V binding and in HBV infection in vitro. J. Hepatol. 1999, 31, 783–790. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Vanlandschoot, P.; Leroux-Roels, G. Viral apoptotic mimicry: An immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 2003, 24, 144–147. [Google Scholar] [CrossRef]
- Gomez-Gutierrez, J.; Rodriguez-Crespo, I.; Peterson, D.L.; Gavilanes, F. Reconstitution of hepatitis B surface antigen proteins into phospholipid vesicles. Biochim. Biophys. Acta 1994, 1192, 45–52. [Google Scholar] [CrossRef]
- Callahan, M.K.; Popernack, P.M.; Tsutsui, S.; Truong, L.; Schlegel, R.A.; Henderson, A.J. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J. Immunol. 2003, 170, 4840–4845. [Google Scholar] [CrossRef]
- Lorizate, M.; Sachsenheimer, T.; Glass, B.; Habermann, A.; Gerl, M.J.; Krausslich, H.G.; Brugger, B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell Microbiol. 2013, 15, 292–304. [Google Scholar] [CrossRef]
- Renkonen, O.; Kaariainen, L.; Pettersson, R.; Oker-Blom, N. The phospholipid composition of Uukuniemi virus, a non-cubical tick-borne arbovirus. Virology 1972, 50, 899–901. [Google Scholar] [CrossRef]
- Ellis, D.S.; Shirodaria, P.V.; Fleming, E.; Simpson, D.I. Morphology and development of Rift Valley fever virus in Vero cell cultures. J. Med. Virol. 1988, 24, 161–174. [Google Scholar] [CrossRef]
- Kuismanen, E.; Hedman, K.; Saraste, J.; Pettersson, R.F. Uukuniemi virus maturation: Accumulation of virus particles and viral antigens in the Golgi complex. Mol. Cell Biol. 1982, 2, 1444–1458. [Google Scholar] [CrossRef]
- Hall, W.W.; Martin, S.J. Structure and function relationships of the envelope of measles virus. Med. Microbiol. Immunol. 1974, 160, 143–154. [Google Scholar] [CrossRef]
- Schlegel, R.; Tralka, T.S.; Willingham, M.C.; Pastan, I. Inhibition of VSV binding and infectivity by phosphatidylserine: Is phosphatidylserine a VSV-binding site? Cell 1983, 32, 639–646. [Google Scholar] [CrossRef]
- Coil, D.A.; Miller, A.D. Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus. J. Virol. 2004, 78, 10920–10926. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Coyne, C.B. Picornavirus entry. Adv. Exp. Med. Biol. 2013, 790, 24–41. [Google Scholar] [CrossRef]
- Marjomaki, V.; Turkki, P.; Huttunen, M. Infectious Entry Pathway of Enterovirus B Species. Viruses 2015, 7, 6387–6399. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Siman-Tov, G.; Hall, G.; Bhalla, N.; Narayanan, A. Human Antimicrobial Peptides as Therapeutics for Viral Infections. Viruses 2019, 11, 704. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luteijn, R.D.; Praest, P.; Thiele, F.; Sadasivam, S.M.; Singethan, K.; Drijfhout, J.W.; Bach, C.; de Boer, S.M.; Lebbink, R.J.; Tao, S.; et al. A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope. Cells 2020, 9, 1989. https://doi.org/10.3390/cells9091989
Luteijn RD, Praest P, Thiele F, Sadasivam SM, Singethan K, Drijfhout JW, Bach C, de Boer SM, Lebbink RJ, Tao S, et al. A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope. Cells. 2020; 9(9):1989. https://doi.org/10.3390/cells9091989
Chicago/Turabian StyleLuteijn, Rutger D., Patrique Praest, Frank Thiele, Saravanan Manikam Sadasivam, Katrin Singethan, Jan W. Drijfhout, Christian Bach, Steffen Matthijn de Boer, Robert J. Lebbink, Sha Tao, and et al. 2020. "A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope" Cells 9, no. 9: 1989. https://doi.org/10.3390/cells9091989
APA StyleLuteijn, R. D., Praest, P., Thiele, F., Sadasivam, S. M., Singethan, K., Drijfhout, J. W., Bach, C., de Boer, S. M., Lebbink, R. J., Tao, S., Helfer, M., Bach, N. C., Protzer, U., Costa, A. I., Killian, J. A., Drexler, I., & Wiertz, E. J. H. J. (2020). A Broad-Spectrum Antiviral Peptide Blocks Infection of Viruses by Binding to Phosphatidylserine in the Viral Envelope. Cells, 9(9), 1989. https://doi.org/10.3390/cells9091989