Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Overview
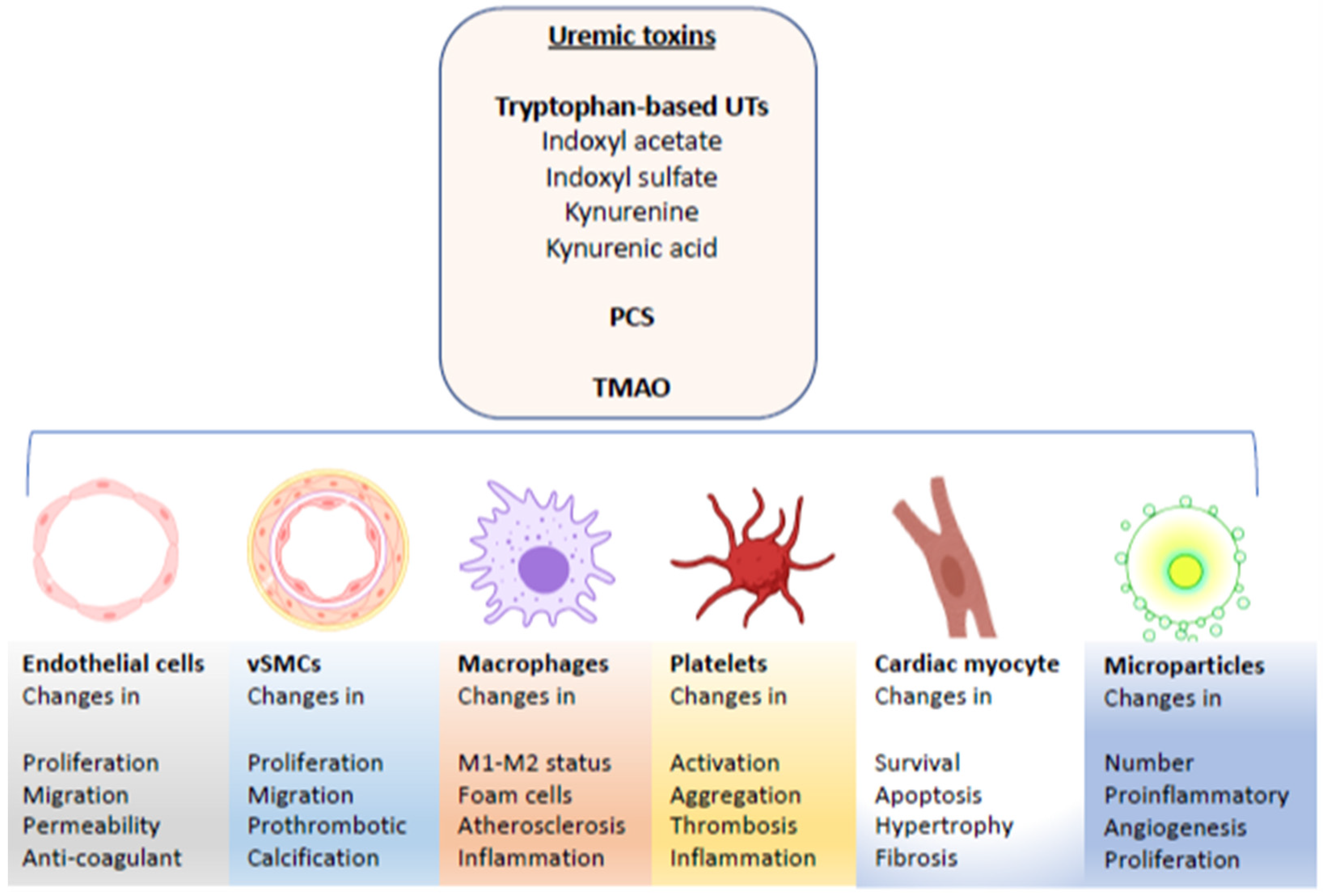
1.2. Uremic Solutes
2. Effects of UTs on Endothelial and Endothelial Progenitor Cells
2.1. Alteration in Mitogenic Signaling in Endothelial Cells and Endothelial Progenitor Cells
2.2. Alteration of the Procoagulant Function of Endothelial Cells
2.3. Inflammation, Oxidative Stress and Atherosclerosis
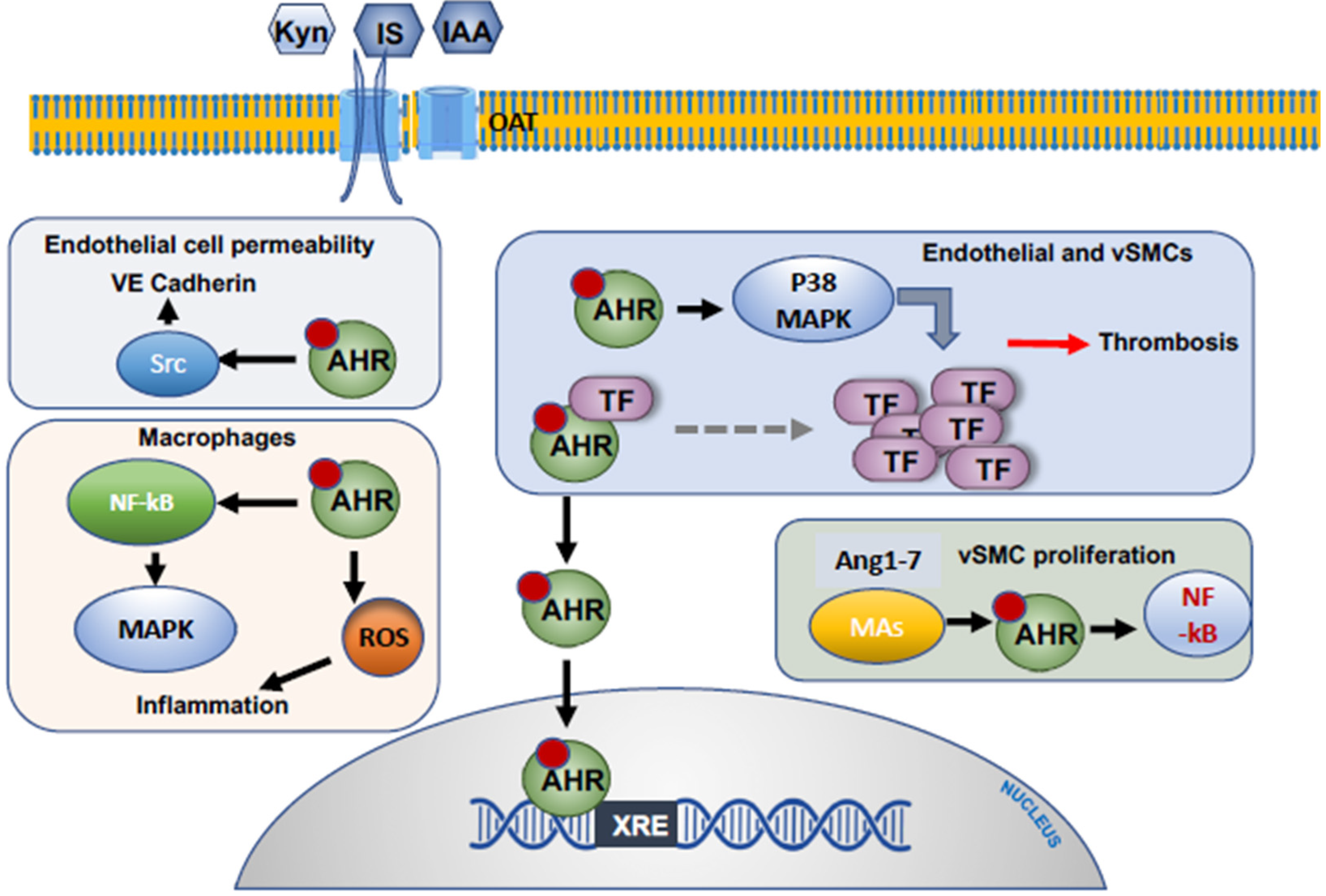
2.4. Endothelial Cells and the Permeability Barrier
3. Effects of UTs on Vascular Smooth Muscle Cells
3.1. Effects of Uremic Toxins on Mitogenic Signaling in Vascular Smooth Muscle Cells
3.2. Effect of Uremic Toxins on Neointimal Hyperplasia and Calcification
3.3. Prothrombotic Effects of Specific Uremic Toxins
4. Effect of Uremic Toxins on Cardiomyocytes
5. The Effects of Uremic Toxins on Monocytes/Macrophages, Polymorphonuclear Cells and Platelets
5.1. The Influence of Uremic Toxins on Macrophages and Monocytes
5.2. Effect of Uremic Toxins on Platelets
6. Uremic Toxin-Induced Microparticles and Cardiovascular Disease
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Temgoua, M.N.; Danwang, C.; Agbor, V.N.; Noubiap, J.J. Prevalence, incidence and associated mortality of cardiovascular disease in patients with chronic kidney disease in low- and middle-income countries: A protocol for a systematic review and meta-analysis. BMJ Open 2017, 7, e016412. [Google Scholar] [CrossRef] [PubMed]
- Weaver, D.J.; Mitsnefes, M. Cardiovascular Disease in Children and Adolescents with Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 559–569. [Google Scholar] [CrossRef]
- Charytan, D.M. Introduction: Cardiovascular Disease in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 541. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Lekawanvijit, S. Cardiotoxicity of Uremic Toxins: A Driver of Cardiorenal Syndrome. Toxins 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.J.; Liu, H.L.; Pan, C.F.; Chuang, C.K.; Jayakumar, T.; Wang, T.J.; Chen, H.H.; Wu, C.J. Indoxyl sulfate predicts cardiovascular disease and renal function deterioration in advanced chronic kidney disease. Arch. Med. Res. 2012, 43, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.C.; Chang, J.C.; Lin, C.N.; Lee, C.C.; Chen, Y.T.; Chu, P.H.; Kou, G.; Lu, Y.A.; Yang, C.W.; Chen, Y.C. Serum indoxyl sulfate predicts adverse cardiovascular events in patients with chronic kidney disease. J. Formos. Med. Assoc. 2019, 118, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, J.R.; House, J.A.; Ocque, A.J.; Zhang, S.; Johnson, C.; Kimber, C.; Schmidt, K.; Gupta, A.; Wetmore, J.B.; Nolin, T.D.; et al. Serum Trimethylamine-N-Oxide is Elevated in CKD and Correlates with Coronary Atherosclerosis Burden. J. Am. Soc. Nephrol. 2016, 27, 305–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, E.R.; Tuseth, N.; Eussen, S.J.; Ueland, P.M.; Strand, E.; Svingen, G.F.; Midttun, O.; Meyer, K.; Mellgren, G.; Ulvik, A.; et al. Associations of plasma kynurenines with risk of acute myocardial infarction in patients with stable angina pectoris. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Sallee, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate-Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205. [Google Scholar] [CrossRef]
- Abbasi, J. TMAO and Heart Disease: The New Red Meat Risk? JAMA 2019, 321, 2149–2151. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Cheung, W.; Keski-Rahkonen, P.; Assi, N.; Ferrari, P.; Freisling, H.; Rinaldi, S.; Slimani, N.; Zamora-Ros, R.; Rundle, M.; Frost, G.; et al. A metabolomic study of biomarkers of meat and fish intake. Am. J. Clin. Nutr. 2017, 105, 600–608. [Google Scholar] [CrossRef]
- Wattanakit, K.; Cushman, M. Chronic kidney disease and venous thromboembolism: Epidemiology and mechanisms. Curr. Opin. Pulm. Med. 2009, 15, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Sarnak, M.J. Cardiovascular complications in chronic kidney disease. Am. J. Kidney Dis. 2003, 41, 11–17. [Google Scholar] [CrossRef]
- Casserly, L.F.; Dember, L.M. Thrombosis in end-stage renal disease. Semin. Dial. 2003, 16, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Morimoto, T.; Kozuma, K.; Honda, Y.; Kume, T.; Aizawa, T.; Mitsudo, K.; Miyazaki, S.; Yamaguchi, T.; Hiyoshi, E.; et al. Comparisons of baseline demographics, clinical presentation, and long-term outcome among patients with early, late, and very late stent thrombosis of sirolimus-eluting stents: Observations from the Registry of Stent Thrombosis for Review and Reevaluation (RESTART). Circulation 2010, 122, 52–61. [Google Scholar] [PubMed] [Green Version]
- Iakovou, I.; Schmidt, T.; Bonizzoni, E.; Ge, L.; Sangiorgi, G.M.; Stankovic, G.; Airoldi, F.; Chieffo, A.; Montorfano, M.; Carlino, M.; et al. Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents. JAMA 2005, 293, 2126–2130. [Google Scholar] [CrossRef]
- Belardi, J.A.; Albertal, M. Coronary stent thrombosis in patients with chronic kidney disease: Balancing anti-ischemic efficacy and hemorrhagic risk. Catheter. Cardiovasc. Interv. 2012, 80, 368–369. [Google Scholar] [CrossRef]
- Beinart, R.; Abu Sham’a, R.; Segev, A.; Hod, H.; Guetta, V.; Shechter, M.; Boyko, V.; Behar, S.; Matetzky, S. The incidence and clinical predictors of early stent thrombosis in patients with acute coronary syndrome. Am. Heart J. 2010, 159, 118–124. [Google Scholar] [CrossRef]
- Chua, S.K.; Hung, H.F.; Cheng, J.J.; Wang, J.H.; Lo, H.M.; Kuan, P.; Lee, S.H.; Lin, S.C.; Liou, J.Y.; Chang, C.M.; et al. Incidence, predictors and outcomes of subacute stent thrombosis following primary stenting for ST-elevation myocardial infarction. J. Formos. Med. Assoc. 2010, 109, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Dangas, G.; Aymong, E.D.; Mehran, R.; Tcheng, J.E.; Grines, C.L.; Cox, D.A.; Garcia, E.; Griffin, J.J.; Guagliumi, G.; Stuckey, T.; et al. Predictors of and outcomes of early thrombosis following balloon angioplasty versus primary stenting in acute myocardial infarction and usefulness of abciximab (the CADILLAC trial). Am. J. Cardiol. 2004, 94, 983–988. [Google Scholar] [CrossRef]
- Malyszko, J. Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta 2010, 411, 1412–1420. [Google Scholar] [CrossRef]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-kappaB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Kuo, K.L.; Huang, H.L.; Lin, C.C.; Tsai, T.H.; Wang, C.H.; Chen, J.W.; Lin, S.J.; Huang, P.H.; Tarng, D.C. Indoxyl sulfate suppresses endothelial progenitor cell-mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef] [Green Version]
- de Groot, K.; Bahlmann, F.H.; Sowa, J.; Koenig, J.; Menne, J.; Haller, H.; Fliser, D. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004, 66, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Hung, S.C.; Kuo, K.L.; Tarng, D.C. Impact of Indoxyl Sulfate on Progenitor Cell-Related Neovascularization of Peripheral Arterial Disease and Post-Angioplasty Thrombosis of Dialysis Vascular Access. Toxins 2017, 9, 25. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Sabatier, F.; Calaf, R.; Cerini, C.; Robert, S.; Camoin-Jau, L.; Charpiot, P.; Argiles, A.; Dignat-George, F.; et al. Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J. Thromb. Haemost. 2009, 7, 1576–1584. [Google Scholar] [CrossRef]
- Roy-Chaudhury, P.; Sukhatme, V.P.; Cheung, A.K. Hemodialysis vascular access dysfunction: A cellular and molecular viewpoint. J. Am. Soc. Nephrol. 2006, 17, 1112–1127. [Google Scholar] [CrossRef]
- Chen, T.Y.; Lin, T.T.; Hsieh, M.Y.; Lin, L.; Yang, C.W.; Chuang, S.Y.; Huang, P.H.; Wu, C.C. Circulating Progenitor Cells Affect Thrombosis of Dialysis Arteriovenous Fistulas. Am. J. Nephrol. 2016, 44, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallee, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, D.H.; Monti, S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Semin. Immunopathol. 2013, 35, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Petrulis, J.R.; Perdew, G.H. The role of chaperone proteins in the aryl hydrocarbon receptor core complex. Chem. Biol. Interact. 2002, 141, 25–40. [Google Scholar] [CrossRef]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Claro, L.M.; Moreno-Amaral, A.N.; Gadotti, A.C.; Dolenga, C.J.; Nakao, L.S.; Azevedo, M.L.V.; de Noronha, L.; Olandoski, M.; de Moraes, T.P.; Stinghen, A.E.M.; et al. The Impact of Uremic Toxicity Induced Inflammatory Response on the Cardiovascular Burden in Chronic Kidney Disease. Toxins 2018, 10, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amdur, R.L.; Feldman, H.I.; Dominic, E.A.; Anderson, A.H.; Beddhu, S.; Rahman, M.; Wolf, M.; Reilly, M.; Ojo, A.; Townsend, R.R.; et al. Use of Measures of Inflammation and Kidney Function for Prediction of Atherosclerotic Vascular Disease Events and Death in Patients With CKD: Findings From the CRIC Study. Am. J. Kidney Dis. 2019, 73, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taki, K.; Tsuruta, Y.; Niwa, T. Indoxyl sulfate and atherosclerotic risk factors in hemodialysis patients. Am. J. Nephrol. 2007, 27, 30–35. [Google Scholar] [CrossRef]
- Eloueyk, A.; Osta, B.; Alameldinne, R.; Awad, D. Uremic Serum Induces Inflammation in Cultured Human Endothelial Cells and Triggers Vascular Repair Mechanisms. Inflammation 2019, 42, 2003–2010. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E-/- mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.D.; Santos, S.S.; Meireles, T.; Romero, N.; Glorieux, G.; Pecoits-Filho, R.; Zhang, D.D.; Nakao, L.S. Uremic toxins promote accumulation of oxidized protein and increased sensitivity to hydrogen peroxide in endothelial cells by impairing the autophagic flux. Biochem. Biophys. Res. Commun. 2019, 523, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-acetylcysteine. Sci. World J. 2015, 2015, 620826. [Google Scholar] [CrossRef] [Green Version]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Katome, T.; Taguchi, K.; Kobayashi, T. Direct Impairment of the Endothelial Function by Acute Indoxyl Sulfate through Declined Nitric Oxide and Not Endothelium-Derived Hyperpolarizing Factor or Vasodilator Prostaglandins in the Rat Superior Mesenteric Artery. Biol. Pharm. Bull. 2019, 42, 1236–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assefa, E.G.; Yan, Q.; Gezahegn, S.B.; Salissou, M.T.M.; He, S.; Wu, N.; Zuo, X.; Ying, C. Role of Resveratrol on Indoxyl Sulfate-Induced Endothelial Hyperpermeability via Aryl Hydrocarbon Receptor (AHR)/Src-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 5847040. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of cell-cell junctions by active vitamin D ameliorates uraemia-induced loss of human endothelial barrier function. Nephrol. Dial. Transplant. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Maciel, R.A.P.; Cunha, R.S.; Busato, V.; Franco, C.R.C.; Gregorio, P.C.; Dolenga, C.J.R.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Henaut, L.; Mary, A.; Chillon, J.M.; Kamel, S.; Massy, Z.A. The Impact of Uremic Toxins on Vascular Smooth Muscle Cell Function. Toxins 2018, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Tsuruoka, S.; Ioka, T.; Ando, H.; Ito, C.; Akimoto, T.; Fujimura, A.; Asano, Y.; Kusano, E. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006, 69, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Yisireyili, M.; Saito, S.; Abudureyimu, S.; Adelibieke, Y.; Ng, H.Y.; Nishijima, F.; Takeshita, K.; Murohara, T.; Niwa, T. Indoxyl sulfate-induced activation of (pro)renin receptor promotes cell proliferation and tissue factor expression in vascular smooth muscle cells. PLoS ONE 2014, 9, e109268. [Google Scholar] [CrossRef] [Green Version]
- Muteliefu, G.; Enomoto, A.; Niwa, T. Indoxyl sulfate promotes proliferation of human aortic smooth muscle cells by inducing oxidative stress. J. Ren. Nutr. 2009, 19, 29–32. [Google Scholar] [CrossRef]
- Shimizu, H.; Hirose, Y.; Nishijima, F.; Tsubakihara, Y.; Miyazaki, H. ROS and PDGF-beta [corrected] receptors are critically involved in indoxyl sulfate actions that promote vascular smooth muscle cell proliferation and migration. Am. J. Physiol. Cell Physiol. 2009, 297, C389–C396. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.Y.; Bolati, W.; Lee, C.T.; Chien, Y.S.; Yisireyili, M.; Saito, S.; Pei, S.N.; Nishijima, F.; Niwa, T. Indoxyl Sulfate Downregulates Mas Receptor via Aryl Hydrocarbon Receptor/Nuclear Factor-kappa B, and Induces Cell Proliferation and Tissue Factor Expression in Vascular Smooth Muscle Cells. Nephron 2016, 133, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Simoes e Silva, A.C.; Silveira, K.D.; Ferreira, A.J.; Teixeira, M.M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.Y.; Hsu, S.C.; Lee, H.S.; Lin, S.H.; Tsai, C.S.; Huang, S.M.; Shih, C.C.; Hsu, Y.J. Enhanced expression of glucose transporter-1 in vascular smooth muscle cells via the Akt/tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR)/ribosomal S6 protein kinase (S6K) pathway in experimental renal failure. J. Vasc. Surg. 2013, 57, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef]
- Han, H.; Chen, Y.; Zhu, Z.; Su, X.; Ni, J.; Du, R.; Zhang, R.; Jin, W. p-Cresyl sulfate promotes the formation of atherosclerotic lesions and induces plaque instability by targeting vascular smooth muscle cells. Front. Med. 2016, 10, 320–329. [Google Scholar] [CrossRef]
- Kokubo, T.; Ishikawa, N.; Uchida, H.; Chasnoff, S.E.; Xie, X.; Mathew, S.; Hruska, K.A.; Choi, E.T. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J. Am. Soc. Nephrol. 2009, 20, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Rotmans, J.I.; Pasterkamp, G.; Verhagen, H.J.; Pattynama, P.M.; Blankestijn, P.J.; Stroes, E.S. Hemodialysis access graft failure: Time to revisit an unmet clinical need? J. Nephrol. 2005, 18, 9–20. [Google Scholar]
- Wu, Y.; Han, X.; Wang, L.; Diao, Z.; Liu, W. Indoxyl sulfate promotes vascular smooth muscle cell calcification via the JNK/Pit-1 pathway. Ren. Fail. 2016, 38, 1702–1710. [Google Scholar] [CrossRef] [Green Version]
- Muteliefu, G.; Enomoto, A.; Jiang, P.; Takahashi, M.; Niwa, T. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [Green Version]
- Torremade, N.; Bozic, M.; Panizo, S.; Barrio-Vazquez, S.; Fernandez-Martin, J.L.; Encinas, M.; Goltzman, D.; Arcidiacono, M.V.; Fernandez, E.; Valdivielso, J.M. Vascular Calcification Induced by Chronic Kidney Disease Is Mediated by an Increase of 1alpha-Hydroxylase Expression in Vascular Smooth Muscle Cells. J. Bone Miner. Res. 2016, 31, 1865–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Jiang, H.; Gao, F.; Liang, S.; Wei, M.; Chen, L. Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-kappaB signaling pathway. Microsc. Res. Tech. 2019, 82, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef]
- Pawlak, K.; Tankiewicz, J.; Mysliwiec, M.; Pawlak, D. Tissue factor/its pathway inhibitor system and kynurenines in chronic kidney disease patients on conservative treatment. Blood Coagul. Fibrinolysis 2009, 20, 590–594. [Google Scholar] [CrossRef]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Shashar, M.; Belghasem, M.E.; Matsuura, S.; Walker, J.; Richards, S.; Alousi, F.; Rijal, K.; Kolachalama, V.B.; Balcells, M.; Odagi, M.; et al. Targeting STUB1-tissue factor axis normalizes hyperthrombotic uremic phenotype without increasing bleeding risk. Sci. Transl. Med. 2017, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef]
- Shashar, M.; Francis, J.; Chitalia, C.V. Thrombosis in the uremic milieu--emerging role of “thrombolome”. Semin. Dial. 2015, 28, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, R.N.; Parfrey, P.S.; Harnett, J.D.; Kent, G.M.; Martin, C.J.; Murray, D.C.; Barre, P.E. Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int. 1995, 47, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Xu, X.; Nie, L.; Xiao, T.; Guan, X.; He, T.; Yu, Y.; Liu, L.; Huang, Y.; Zhang, J.; et al. Indoxyl sulfate induces oxidative stress and hypertrophy in cardiomyocytes by inhibiting the AMPK/UCP2 signaling pathway. Toxicol. Lett. 2015, 234, 110–119. [Google Scholar] [CrossRef]
- Yisireyili, M.; Shimizu, H.; Saito, S.; Enomoto, A.; Nishijima, F.; Niwa, T. Indoxyl sulfate promotes cardiac fibrosis with enhanced oxidative stress in hypertensive rats. Life Sci. 2013, 92, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Cao, X.S.; Zhang, P.; Xiang, F.F.; Teng, J.; Zou, J.Z.; Ding, X.Q. Endoplasmic reticulum stress associated apoptosis as a novel mechanism in indoxyl sulfateinduced cardiomyocyte toxicity. Mol. Med. Rep. 2018, 18, 5117–5122. [Google Scholar] [PubMed] [Green Version]
- Han, H.; Zhu, J.; Zhu, Z.; Ni, J.; Du, R.; Dai, Y.; Chen, Y.; Wu, Z.; Lu, L.; Zhang, R. p-Cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J. Am. Heart Assoc. 2015, 4, e001852. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.S.; Ding, H.C.; Lin, Y.T.; Syu, J.P.; Chen, Y.; Wang, S.M. Uremic toxin p-cresol induces disassembly of gap junctions of cardiomyocytes. Toxicology 2012, 302, 11–17. [Google Scholar] [CrossRef]
- Nahrendorf, M. Myeloid cell contributions to cardiovascular health and disease. Nat. Med. 2018, 24, 711–720. [Google Scholar] [CrossRef]
- Cochain, C.; Zernecke, A. Macrophages in vascular inflammation and atherosclerosis. Pflugers Arch. 2017, 469, 485–499. [Google Scholar] [CrossRef]
- Decano, J.L.; Mattson, P.C.; Aikawa, M. Macrophages in Vascular Inflammation: Origins and Functions. Curr. Atheroscler. Rep. 2016, 18, 34. [Google Scholar] [CrossRef]
- Blankenberg, S.; Barbaux, S.; Tiret, L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003, 170, 191–203. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm. Des. 2012, 18, 4266–4288. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yamamoto, S.; Wakamatsu, T.; Takahashi, Y.; Kawamura, K.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Narita, I. Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins 2015, 7, 3155–3166. [Google Scholar] [CrossRef] [Green Version]
- Trojanowicz, B.; Ulrich, C.; Seibert, E.; Fiedler, R.; Girndt, M. Uremic conditions drive human monocytes to pro-atherogenic differentiation via an angiotensin-dependent mechanism. PLoS ONE 2014, 9, e102137. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl Sulfate Promotes Macrophage IL-1beta Production by Activating Aryl Hydrocarbon Receptor/NF-kappa/MAPK Cascades, but the NLRP3 inflammasome Was Not Activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lu, X.; Xiong, R.; Wang, S. High Neutrophil-to-Lymphocyte Ratio Predicts Cardiovascular Mortality in Chronic Hemodialysis Patients. Mediators Inflamm. 2017, 2017, 9327136. [Google Scholar] [CrossRef]
- Schmidt, S.; Westhoff, T.H.; Krauser, P.; Ignatius, R.; Jankowski, J.; Jankowski, V.; Zidek, W.; van der Giet, M. The uraemic toxin phenylacetic acid impairs macrophage function. Nephrol. Dial. Transplant. 2008, 23, 3485–3493. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G.; Raupachova, J.; Horl, W.H. The uraemic toxin phenylacetic acid contributes to inflammation by priming polymorphonuclear leucocytes. Nephrol. Dial. Transplant. 2013, 28, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Thijs, A.; Nanayakkara, P.W.; Ter Wee, P.M.; Huijgens, P.C.; van Guldener, C.; Stehouwer, C.D. Mild-to-moderate renal impairment is associated with platelet activation: A cross-sectional study. Clin. Nephrol. 2008, 70, 325–331. [Google Scholar]
- Boccardo, P.; Remuzzi, G.; Galbusera, M. Platelet dysfunction in renal failure. Semin. Thromb. Hemost. 2004, 30, 579–589. [Google Scholar] [CrossRef]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood 2017, 129, 2667–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesse, A.; Martinez, M.C.; Hugel, B.; Chalupsky, K.; Muller, C.D.; Meziani, F.; Mitolo-Chieppa, D.; Freyssinet, J.M.; Andriantsitohaina, R. Upregulation of proinflammatory proteins through NF-kappaB pathway by shed membrane microparticles results in vascular hyporeactivity. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2522–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hugel, B.; Martinez, M.C.; Kunzelmann, C.; Freyssinet, J.M. Membrane microparticles: Two sides of the coin. Physiology (Bethesda) 2005, 20, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Chironi, G.N.; Boulanger, C.M.; Simon, A.; Dignat-George, F.; Freyssinet, J.M.; Tedgui, A. Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335, 143–151. [Google Scholar] [CrossRef]
- Burger, D.; Schock, S.; Thompson, C.S.; Montezano, A.C.; Hakim, A.M.; Touyz, R.M. Microparticles: Biomarkers and beyond. Clin. Sci. (Lond.) 2013, 124, 423–441. [Google Scholar] [CrossRef] [Green Version]
- Lemoinne, S.; Thabut, D.; Housset, C.; Moreau, R.; Valla, D.; Boulanger, C.M.; Rautou, P.E. The emerging roles of microvesicles in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 350–361. [Google Scholar] [CrossRef]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic Role of Blood and Endothelial Cells in Uremia through Phosphatidylserine Exposure and Microparticle Release. PLoS ONE 2015, 10, e0142835. [Google Scholar] [CrossRef]
- Ryu, J.H.; Park, H.; Kim, S.J. The effects of indoxyl sulfate-induced endothelial microparticles on neointimal hyperplasia formation in an ex vivo model. Ann. Surg. Treat. Res. 2017, 93, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sekula, M.; Janawa, G.; Stankiewicz, E.; Stepien, E. Endothelial microparticle formation in moderate concentrations of homocysteine and methionine in vitro. Cell. Mol. Biol. Lett. 2011, 16, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijers, B.K.; De Loor, H.; Bammens, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate and indoxyl sulfate in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1932–1938. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.H.; Kim, S.J. Clopidogrel effectively suppresses endothelial microparticle generation induced by indoxyl sulfate via inhibition of the p38 mitogen-activated protein kinase pathway. Blood Purif. 2011, 32, 186–194. [Google Scholar] [CrossRef]
- Gao, C.; Ji, S.; Dong, W.; Qi, Y.; Song, W.; Cui, D.; Shi, J. Indolic uremic solutes enhance procoagulant activity of red blood cells through phosphatidylserine exposure and microparticle release. Toxins 2015, 7, 4390–4403. [Google Scholar] [CrossRef] [Green Version]
- Amabile, N.; Guerin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef] [Green Version]
- Soriano, S.; Carmona, A.; Trivino, F.; Rodriguez, M.; Alvarez-Benito, M.; Martin-Malo, A.; Alvarez-Lara, M.A.; Ramirez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Renal Physiol. 2014, 307, F1302–F1311. [Google Scholar] [CrossRef] [Green Version]
- Morel, O.; Morel, N.; Jesel, L.; Freyssinet, J.M.; Toti, F. Microparticles: A critical component in the nexus between inflammation, immunity, and thrombosis. Semin. Immunopathol. 2011, 33, 469–486. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Sabatier, F.; Roux, V.; Anfosso, F.; Camoin, L.; Sampol, J.; Dignat-George, F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002, 99, 3962–3970. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Jeon, E.Y.; Kim, S.J. Indoxyl Sulfate-Induced Extracellular Vesicles Released from Endothelial Cells Stimulate Vascular Smooth Muscle Cell Proliferation by Inducing Transforming Growth Factor-Beta Production. J. Vasc. Res. 2019, 56, 129–138. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravid, J.D.; Chitalia, V.C. Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes. Cells 2020, 9, 2024. https://doi.org/10.3390/cells9092024
Ravid JD, Chitalia VC. Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes. Cells. 2020; 9(9):2024. https://doi.org/10.3390/cells9092024
Chicago/Turabian StyleRavid, Jonathan D., and Vipul C. Chitalia. 2020. "Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes" Cells 9, no. 9: 2024. https://doi.org/10.3390/cells9092024
APA StyleRavid, J. D., & Chitalia, V. C. (2020). Molecular Mechanisms Underlying the Cardiovascular Toxicity of Specific Uremic Solutes. Cells, 9(9), 2024. https://doi.org/10.3390/cells9092024