Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Classification and Functional Role of Heat Shock Protein 70
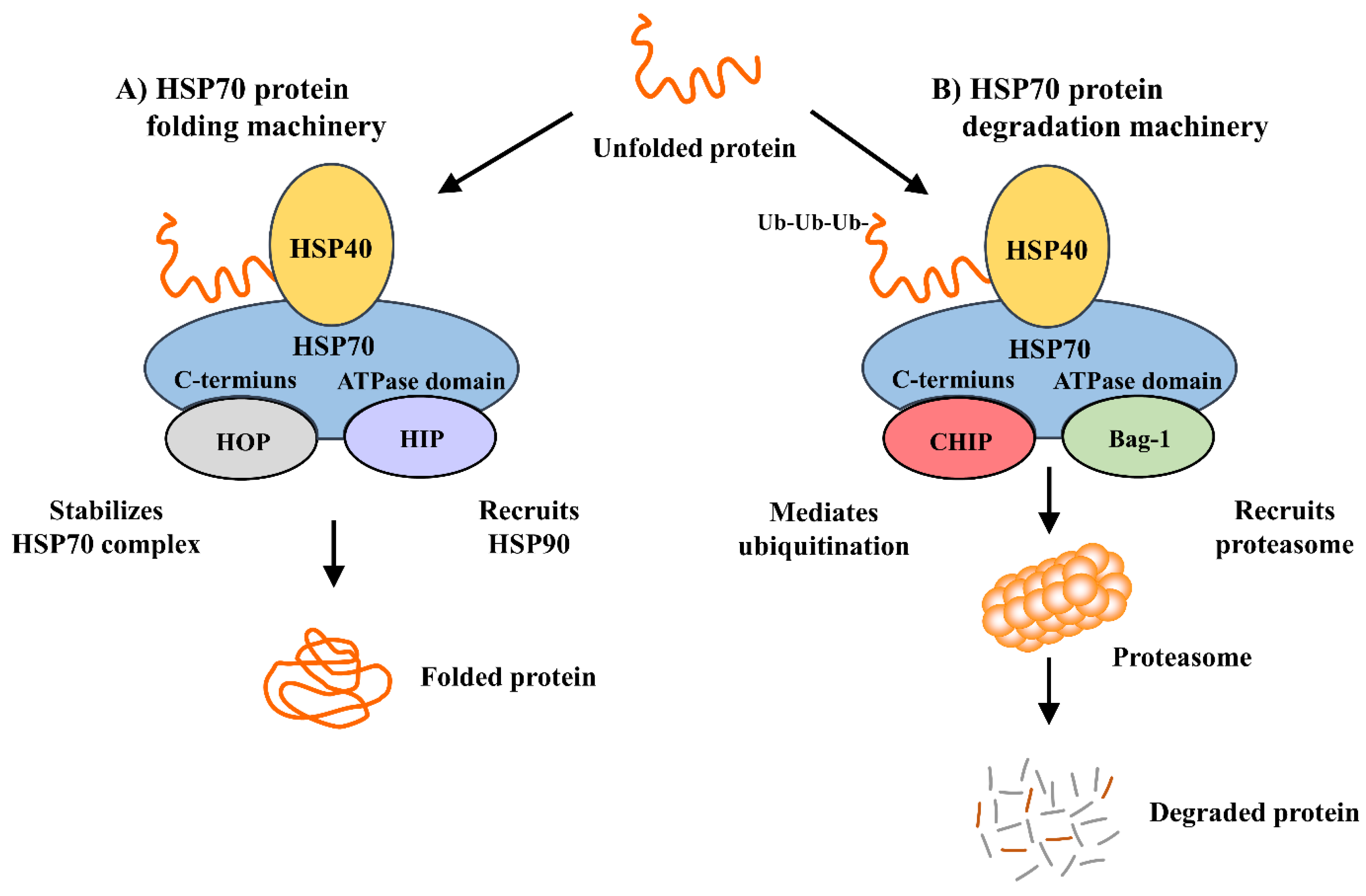
3. Mechanism of Heat Shock Protein 70 Induction and Its Interactions with Cochaperones and Client Proteins
4. Role of Heat Shock Protein 70 in Brain Injury
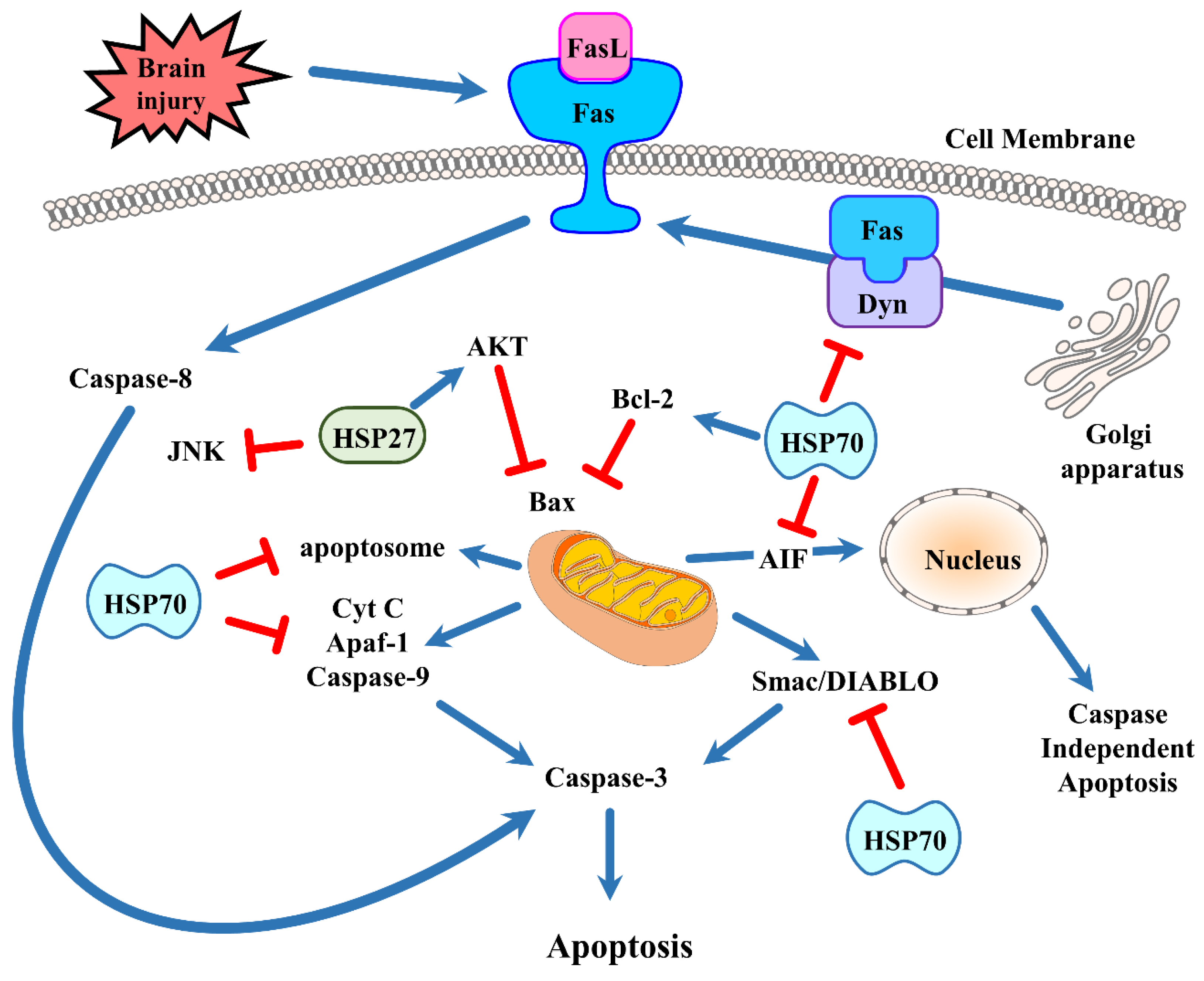
5. The Neuroprotective Effect of Heat Shock Protein 70 via Cell Death Pathways
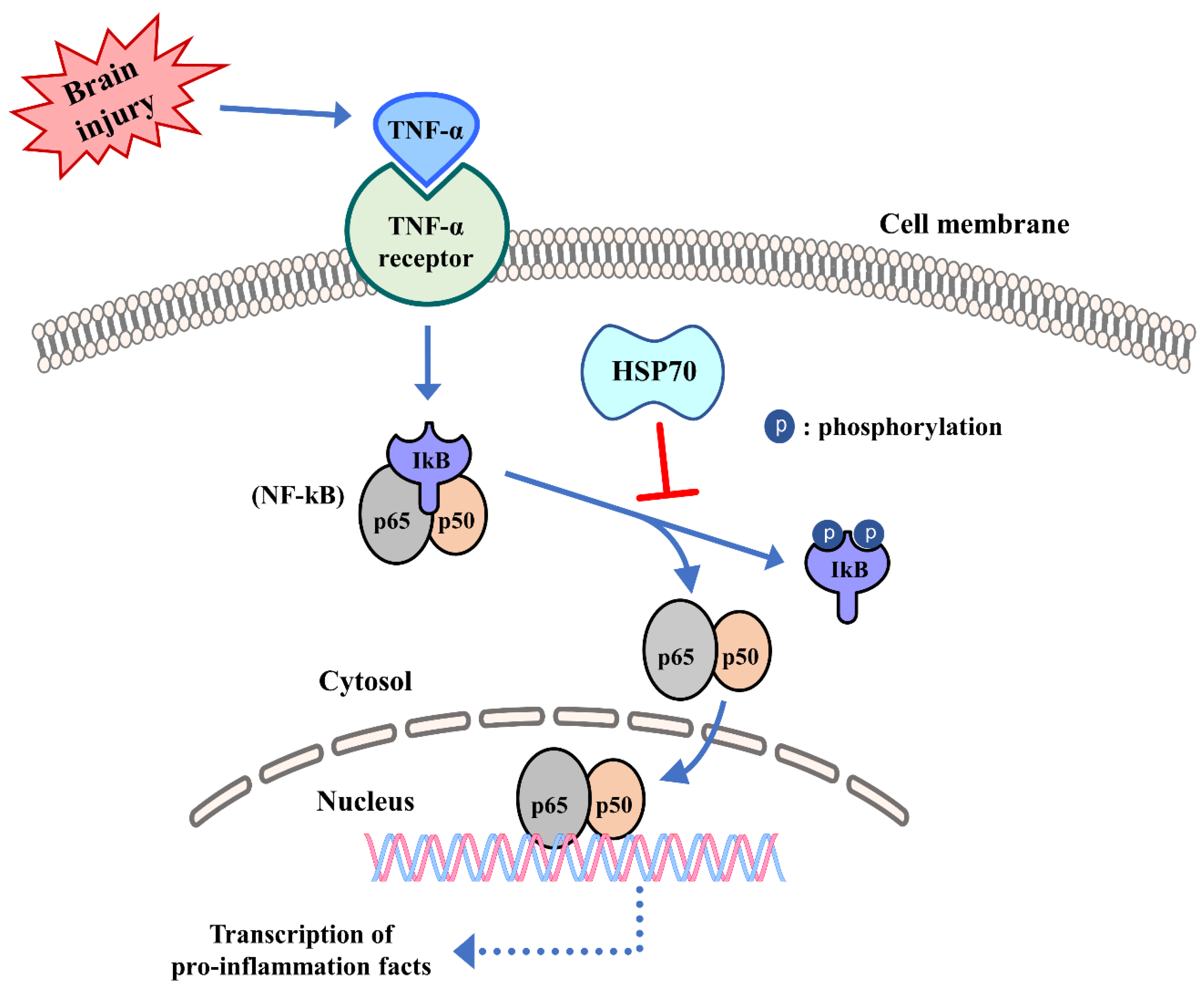
6. Inflammation Regulation of Heat Shock Protein 70
7. Heat Shock Protein 70 as a (Pharmacological) Therapeutic Target for Brain Injury
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Doberentz, E.; Genneper, L.; Wagner, R.; Madea, B. Expression times for hsp27 and hsp70 as an indicator of thermal stress during death due to fire. Int. J. Legal Med. 2017, 131, 1707–1718. [Google Scholar] [CrossRef]
- Mash, D.C.; Duque, L.; Pablo, J.; Qin, Y.; Adi, N.; Hearn, W.L.; Hyma, B.A.; Karch, S.B.; Druid, H.; Wetli, C.V. Brain biomarkers for identifying excited delirium as a cause of sudden death. Forensic Sci. Int. 2009, 190, e13–e19. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, P.R.; Hong, S.; Sharp, F.R. Molecular identification of the ischemic penumbra. Stroke 2004, 35, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- States, B.A.; Honkaniemi, J.; Weinstein, P.R.; Sharp, F.R. DNA fragmentation and hsp70 protein induction in hippocampus and cortex occurs in separate neurons following permanent middle cerebral artery occlusions. J. Cereb. Blood Flow Metab. 1996, 16, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Frebel, K.; Wiese, S. Signalling molecules essential for neuronal survival and differentiation. Biochem. Soc. Trans. 2006, 34, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- De Maio, A. Extracellular hsp70: Export and function. Curr. Protein Pept. Sci. 2014, 15, 225–231. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Zheng, Z.; Lee, J.E.; Yenari, M.A. The 70 kda heat shock protein protects against experimental traumatic brain injury. Neurobiol. Dis. 2013, 58, 289–295. [Google Scholar] [CrossRef]
- Kim, N.; Kim, J.Y.; Yenari, M.A. Anti-inflammatory properties and pharmacological induction of hsp70 after brain injury. Inflammopharmacology 2012, 20, 177–185. [Google Scholar] [CrossRef]
- Kacimi, R.; Yenari, M.A. Pharmacologic heat shock protein 70 induction confers cytoprotection against inflammation in gliovascular cells. Glia 2015, 63, 1200–1212. [Google Scholar] [CrossRef]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The hsp90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Zuehlke, A.; Johnson, J.L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef]
- Johnson, J.L.; Brown, C. Plasticity of the hsp90 chaperone machine in divergent eukaryotic organisms. Cell Stress Chaperones 2009, 14, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. Hsp90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef]
- Xu, Z.S.; Li, Z.Y.; Chen, Y.; Chen, M.; Li, L.C.; Ma, Y.Z. Heat shock protein 90 in plants: Molecular mechanisms and roles in stress responses. Int. J. Mol. Sci. 2012, 13, 15706–15723. [Google Scholar] [CrossRef] [PubMed]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Roux, J.; Lee, H.; Miyazawa, B.; Lee, J.W.; Gartland, B.; Howard, A.J.; Matthay, M.A.; Carles, M.; Pittet, J.F. Activation of the stress protein response inhibits the stat1 signalling pathway and inos function in alveolar macrophages: Role of hsp90 and hsp70. Thorax 2010, 65, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.R. Heat shock proteins and protection of the nervous system. Ann. N. Y. Acad. Sci. 2007, 1113, 147–158. [Google Scholar] [CrossRef]
- Giffard, R.G.; Yenari, M.A. Many mechanisms for hsp70 protection from cerebral ischemia. J. Neurosurg. Anesthesiol. 2004, 16, 53–61. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuronal 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Pernet, L.; Faure, V.; Gilquin, B.; Dufour-Guerin, S.; Khochbin, S.; Vourc’h, C. Hdac6-ubiquitin interaction controls the duration of hsf1 activation after heat shock. Mol. Biol. Cell 2014, 25, 4187–4194. [Google Scholar] [CrossRef]
- Zhao, H.; Michaelis, M.L.; Blagg, B.S. Hsp90 modulation for the treatment of alzheimer’s disease. Adv. Pharmacol. 2012, 64, 1–25. [Google Scholar] [CrossRef]
- Hohfeld, J.; Minami, Y.; Hartl, F.U. Hip, a novel cochaperone involved in the eukaryotic hsc70/hsp40 reaction cycle. Cell 1995, 83, 589–598. [Google Scholar] [CrossRef]
- Frydman, J.; Hohfeld, J. Chaperones get in touch: The hip-hop connection. Trends Biochem. Sci. 1997, 22, 87–92. [Google Scholar] [CrossRef]
- Chen, S.; Smith, D.F. Hop as an adaptor in the heat shock protein 70 (hsp70) and hsp90 chaperone machinery. J. Biol. Chem. 1998, 273, 35194–35200. [Google Scholar] [CrossRef]
- Hernandez, M.P.; Sullivan, W.P.; Toft, D.O. The assembly and intermolecular properties of the hsp70-hop-hsp90 molecular chaperone complex. J. Biol. Chem. 2002, 277, 38294–38304. [Google Scholar] [CrossRef]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Hohfeld, J.; Patterson, C. The co-chaperone chip regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef]
- Murata, S.; Minami, Y.; Minami, M.; Chiba, T.; Tanaka, K. Chip is a chaperone-dependent e3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001, 2, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.B.; McDonough, H.; Boellmann, F.; Cyr, D.M.; Patterson, C. Chip-mediated stress recovery by sequential ubiquitination of substrates and hsp70. Nature 2006, 440, 551–555. [Google Scholar] [CrossRef]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an e3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The hsc70 co-chaperone chip targets immature cftr for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Esser, C.; Scheffner, M.; Hohfeld, J. The chaperone-associated ubiquitin ligase chip is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and chip selectively mediate ubiquitination and degradation of hypoxia-inducible factor (hif)-1alpha but not hif-2alpha. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef]
- Peng, H.M.; Morishima, Y.; Clapp, K.M.; Lau, M.; Pratt, W.B.; Osawa, Y. Dynamic cycling with hsp90 stabilizes neuronal nitric oxide synthase through calmodulin-dependent inhibition of ubiquitination. Biochemistry 2009, 48, 8483–8490. [Google Scholar] [CrossRef]
- Peng, H.M.; Morishima, Y.; Pratt, W.B.; Osawa, Y. Modulation of heme/substrate binding cleft of neuronal nitric-oxide synthase (nnos) regulates binding of hsp90 and hsp70 proteins and nnos ubiquitination. J. Biol. Chem. 2012, 287, 1556–1565. [Google Scholar] [CrossRef]
- Nowak, T.S., Jr.; Bond, U.; Schlesinger, M.J. Heat shock rna levels in brain and other tissues after hyperthermia and transient ischemia. J. Neurochem. 1990, 54, 451–458. [Google Scholar] [CrossRef]
- Abe, K.; Tanzi, R.E.; Kogure, K. Induction of hsp70 mrna after transient ischemia in gerbil brain. Neurosci. Lett. 1991, 125, 166–168. [Google Scholar] [CrossRef]
- Sharp, F.R.; Lu, A.; Tang, Y.; Millhorn, D.E. Multiple molecular penumbras after focal cerebral ischemia. J. Cereb. Blood Flow. Metab. 2000, 20, 1011–1032. [Google Scholar] [CrossRef] [PubMed]
- Beretta, S.; Cuccione, E.; Versace, A.; Carone, D.; Riva, M.; Padovano, G.; Dell’Era, V.; Cai, R.; Monza, L.; Presotto, L.; et al. Cerebral collateral flow defines topography and evolution of molecular penumbra in experimental ischemic stroke. Neurobiol. Dis. 2015, 74, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, D.; Hong, S.; Matsumori, Y.; Kayama, T.; Swanson, R.A.; Dillman, W.H.; Liu, J.; Panter, S.S.; Weinstein, P.R. Overexpression of rat heat shock protein 70 reduces neuronal injury after transient focal ischemia, transient global ischemia, or kainic acid-induced seizures. Neurosurgery 2003, 53, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- DeGracia, D.J.; Jamison, J.T.; Szymanski, J.J.; Lewis, M.K. Translation arrest and ribonomics in post-ischemic brain: Layers and layers of players. J. Neurochem. 2008, 106, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Batulan, Z.; Taylor, D.M.; Aarons, R.J.; Minotti, S.; Doroudchi, M.M.; Nalbantoglu, J.; Durham, H.D. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 24, 213–225. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, Y.J.; Kim, J.Y.; Lee, W.T.; Yenari, M.A.; Giffard, R.G. The 70 kda heat shock protein suppresses matrix metalloproteinases in astrocytes. Neuroreport 2004, 15, 499–502. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, M.; Yoon, B.W.; Kim, Y.J.; Ma, S.J.; Roh, J.K.; Lee, J.S.; Seo, J.S. Targeted hsp70.1 disruption increases infarction volume after focal cerebral ischemia in mice. Stroke 2001, 32, 2905–2912. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Zheng, Z.; Lee, J.E.; Yenari, M.A. 70-kda heat shock protein downregulates dynamin in experimental stroke: A new therapeutic target? Stroke 2016, 47, 2103–2111. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Saito, A.; Hayashi, T.; Chan, P.H. The c-jun n-terminal protein kinase signaling pathway mediates bax activation and subsequent neuronal apoptosis through interaction with bim after transient focal cerebral ischemia. J. Neurosci. 2004, 24, 7879–7887. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Xu, A.; Mai, H.; Zhao, J.; Zhang, C.; Qi, R.; Wang, H.; Lu, D.; Zhu, L. The synergistic effects of heat shock protein 70 and ginsenoside rg1 against tert-butyl hydroperoxide damage model in vitro. Oxid. Med. Cell Longev. 2015, 2015, 437127. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, M.J.; Costa, C.; Eixarch, H.; Tepavcevic, V.; Castillo, M.; Martin, R.; Lubetzki, C.; Aigrot, M.S.; Montalban, X.; Espejo, C. Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLoS ONE 2014, 9, e105737. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Barreto-Chang, O.L.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J. Cereb. Blood Flow Metab. 2004, 24, 681–692. [Google Scholar] [CrossRef]
- Steel, R.; Doherty, J.P.; Buzzard, K.; Clemons, N.; Hawkins, C.J.; Anderson, R.L. Hsp72 inhibits apoptosis upstream of the mitochondria and not through interactions with apaf-1. J. Biol. Chem. 2004, 279, 51490–51499. [Google Scholar] [CrossRef]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.; Radicioni, S.M.; Mosser, D.D. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing bax translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar] [CrossRef]
- Ravagnan, L.; Gurbuxani, S.; Susin, S.A.; Maisse, C.; Daugas, E.; Zamzami, N.; Mak, T.; Jaattela, M.; Penninger, J.M.; Garrido, C.; et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol. 2001, 3, 839–843. [Google Scholar] [CrossRef]
- Lee, S.H.; Kwon, H.M.; Kim, Y.J.; Lee, K.M.; Kim, M.; Yoon, B.W. Effects of hsp70.1 gene knockout on the mitochondrial apoptotic pathway after focal cerebral ischemia. Stroke 2004, 35, 2195–2199. [Google Scholar] [CrossRef]
- Matsumori, Y.; Hong, S.M.; Aoyama, K.; Fan, Y.; Kayama, T.; Sheldon, R.A.; Vexler, Z.S.; Ferriero, D.M.; Weinstein, P.R.; Liu, J. Hsp70 overexpression sequesters aif and reduces neonatal hypoxic/ischemic brain injury. J. Cereb. Blood Flow Metab. 2005, 25, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Xiao, W.; Shi, Y.; Liu, M.; Xiao, X. Heat shock pretreatment inhibited the release of smac/diablo from mitochondria and apoptosis induced by hydrogen peroxide in cardiomyocytes and c2c12 myogenic cells. Cell Stress. Chaperones 2005, 10, 252–262. [Google Scholar] [CrossRef]
- Geissler, A.; Krimmer, T.; Bomer, U.; Guiard, B.; Rassow, J.; Pfanner, N. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b(2) modulates the deltapsi-dependence of translocation of the matrix-targeting sequence. Mol. Biol. Cell 2000, 11, 3977–3991. [Google Scholar] [CrossRef] [PubMed]
- Voloboueva, L.A.; Duan, M.; Ouyang, Y.; Emery, J.F.; Stoy, C.; Giffard, R.G. Overexpression of mitochondrial hsp70/hsp75 protects astrocytes against ischemic injury in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 1009–1016. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Xu, L.J.; Sun, Y.J.; Giffard, R.G. Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress. Cell Stress Chaperones 2006, 11, 180–186. [Google Scholar] [CrossRef]
- Xu, L.; Giffard, R.G. Hsp70 protects murine astrocytes from glucose deprivation injury. Neurosci. Lett. 1997, 224, 9–12. [Google Scholar] [CrossRef]
- Suzuki, K.; Murtuza, B.; Sammut, I.A.; Latif, N.; Jayakumar, J.; Smolenski, R.T.; Kaneda, Y.; Sawa, Y.; Matsuda, H.; Yacoub, M.H. Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia-reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 2002, 106, I270–I276. [Google Scholar]
- Kelly, S.; Yenari, M.A. Neuroprotection: Heat shock proteins. Curr. Med. Res. Opin. 2002, 18 (Suppl. 2), s55–s60. [Google Scholar] [CrossRef]
- Havasi, A.; Li, Z.; Wang, Z.; Martin, J.L.; Botla, V.; Ruchalski, K.; Schwartz, J.H.; Borkan, S.C. Hsp27 inhibits bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J. Biol. Chem. 2008, 283, 12305–12313. [Google Scholar] [CrossRef]
- Rane, M.J.; Pan, Y.; Singh, S.; Powell, D.W.; Wu, R.; Cummins, T.; Chen, Q.; McLeish, K.R.; Klein, J.B. Heat shock protein 27 controls apoptosis by regulating akt activation. J. Biol. Chem. 2003, 278, 27828–27835. [Google Scholar] [CrossRef] [PubMed]
- Schepers, H.; Geugien, M.; van der Toorn, M.; Bryantsev, A.L.; Kampinga, H.H.; Eggen, B.J.; Vellenga, E. Hsp27 protects aml cells against vp-16-induced apoptosis through modulation of p38 and c-jun. Exp. Hematol. 2005, 33, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Namura, S.; Zhu, J.; Fink, K.; Endres, M.; Srinivasan, A.; Tomaselli, K.J.; Yuan, J.; Moskowitz, M.A. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J. Neurosci. 1998, 18, 3659–3668. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef]
- Love, S. Apoptosis and brain ischaemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 267–282. [Google Scholar] [CrossRef]
- Jin, K.; Graham, S.H.; Mao, X.; Nagayama, T.; Simon, R.P.; Greenberg, D.A. Fas (cd95) may mediate delayed cell death in hippocampal ca1 sector after global cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 1411–1421. [Google Scholar] [CrossRef]
- Guo, F.; Sigua, C.; Bali, P.; George, P.; Fiskus, W.; Scuto, A.; Annavarapu, S.; Mouttaki, A.; Sondarva, G.; Wei, S.; et al. Mechanistic role of heat shock protein 70 in bcr-abl-mediated resistance to apoptosis in human acute leukemia cells. Blood 2005, 105, 1246–1255. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Ferrari, D.; Los, M.; Wesselborg, S.; Peter, M.E. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998, 254, 439–459. [Google Scholar] [CrossRef]
- Gabai, V.L.; Mabuchi, K.; Mosser, D.D.; Sherman, M.Y. Hsp72 and stress kinase c-jun n-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 2002, 22, 3415–3424. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Ronai, Z.; Hei, T.K. Opposite roles of fap-1 and dynamin in the regulation of fas (cd95) translocation to the cell surface and susceptibility to fas ligand-mediated apoptosis. J. Biol. Chem. 2006, 281, 1840–1852. [Google Scholar] [CrossRef]
- Shan, Z.X.; Lin, Q.X.; Fu, Y.H.; Deng, C.Y.; Zhou, Z.L.; Zhu, J.N.; Liu, X.Y.; Zhang, Y.Y.; Li, Y.; Lin, S.G.; et al. Upregulated expression of mir-1/mir-206 in a rat model of myocardial infarction. Biochem. Biophys. Res. Commun. 2009, 381, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. Microrna-1 regulates cardiomyocyte apoptosis by targeting bcl-2. Int. Heart J. 2009, 50, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microrna mir-1 regulates cardiac arrhythmogenic potential by targeting gja1 and kcnj2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Lu, Y.; Pan, Z.; Chu, W.; Luo, X.; Lin, H.; Xiao, J.; Shan, H.; Wang, Z.; Yang, B. The muscle-specific micrornas mir-1 and mir-133 produce opposing effects on apoptosis by targeting hsp60, hsp70 and caspase-9 in cardiomyocytes. J. Cell Sci. 2007, 120, 3045–3052. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Lu, Y.; Yue, S.; Xu, L.J.; Xiong, X.X.; White, R.E.; Sun, X.; Giffard, R.G. Mir-181 regulates grp78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol. Dis. 2012, 45, 555–563. [Google Scholar] [CrossRef]
- Ouyang, Y.B.; Lu, Y.; Yue, S.; Giffard, R.G. Mir-181 targets multiple bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion 2012, 12, 213–219. [Google Scholar] [CrossRef]
- Wang, J.; Dore, S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 2007, 130, 1643–1652. [Google Scholar] [CrossRef]
- Davies, C.A.; Loddick, S.A.; Stroemer, R.P.; Hunt, J.; Rothwell, N.J. An integrated analysis of the progression of cell responses induced by permanent focal middle cerebral artery occlusion in the rat. Exp. Neurol. 1998, 154, 199–212. [Google Scholar] [CrossRef]
- Zheng, Z.; Yenari, M.A. Post-ischemic inflammation: Molecular mechanisms and therapeutic implications. Neurol. Res. 2004, 26, 884–892. [Google Scholar] [CrossRef]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Galea, E.; Aquino, D.A.; Li, G.C.; Xu, H.; Reis, D.J. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing nfkappab activation. J. Biol. Chem. 1996, 271, 17724–17732. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Blokhin, I.O.; Wilson, K.M.; Dhanesha, N.; Doddpattar, P.; Grumbach, I.M.; Chauhan, A.K.; Lentz, S.R. Protein methionine oxidation augments reperfusion injury in acute ischemic stroke. JCI Insight 2016, 1, e86460. [Google Scholar] [CrossRef] [PubMed]
- Van Molle, W.; Wielockx, B.; Mahieu, T.; Takada, M.; Taniguchi, T.; Sekikawa, K.; Libert, C. Hsp70 protects against tnf-induced lethal inflammatory shock. Immunity 2002, 16, 685–695. [Google Scholar] [CrossRef]
- Ding, X.Z.; Fernandez-Prada, C.M.; Bhattacharjee, A.K.; Hoover, D.L. Over-expression of hsp-70 inhibits bacterial lipopolysaccharide-induced production of cytokines in human monocyte-derived macrophages. Cytokine 2001, 16, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Manaenko, A.; Fathali, N.; Chen, H.; Suzuki, H.; Williams, S.; Zhang, J.H.; Tang, J. Heat shock protein 70 upregulation by geldanamycin reduces brain injury in a mouse model of intracerebral hemorrhage. Neurochem. Int. 2010, 57, 844–850. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polla, B.S.; Stubbe, H.; Kantengwa, S.; Maridonneau-Parini, I.; Jacquier-Sarlin, M.R. Differential induction of stress proteins and functional effects of heat shock in human phagocytes. Inflammation 1995, 19, 363–378. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yenari, M.A.; Lee, J.E. Regulation of inflammatory transcription factors by heat shock protein 70 in primary cultured astrocytes exposed to oxygen-glucose deprivation. Neuroscience 2015, 286, 272–280. [Google Scholar] [CrossRef]
- Guzhova, I.V.; Darieva, Z.A.; Melo, A.R.; Margulis, B.A. Major stress protein hsp70 interacts with nf-kb regulatory complex in human t-lymphoma cells. Cell Stress Chaperones 1997, 2, 132–139. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sharp, A.; Klockgether, T.; Gavrilyuk, V.; Feinstein, D.L. The heat shock response inhibits nf-kappab activation, nitric oxide synthase type 2 expression, and macrophage/microglial activation in brain. J. Cereb. Blood Flow Metab. 2000, 20, 800–811. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, J.Y.; Ma, H.; Lee, J.E.; Yenari, M.A. Anti-inflammatory effects of the 70 kda heat shock protein in experimental stroke. J. Cereb. Blood Flow Metab. 2008, 28, 53–63. [Google Scholar] [CrossRef]
- Ran, R.; Zhou, G.; Lu, A.; Zhang, L.; Tang, Y.; Rigby, A.C.; Sharp, F.R. Hsp70 mutant proteins modulate additional apoptotic pathways and improve cell survival. Cell Stress Chaperones 2004, 9, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Lu, A.; Zhang, L.; Tang, Y.; Zhu, H.; Xu, H.; Feng, Y.; Han, C.; Zhou, G.; Rigby, A.C.; et al. Hsp70 promotes tnf-mediated apoptosis by binding ikk gamma and impairing nf-kappa b survival signaling. Genes Dev. 2004, 18, 1466–1481. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.B.; Jiang, Y.; van Marle, G.; Mayne, M.B.; Ni, W.; Holden, J.; McArthur, J.C.; Power, C. Lentivirus infection in the brain induces matrix metalloproteinase expression: Role of envelope diversity. J. Virol. 2000, 74, 7211–7220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Giffard, R.G.; Han, R.Q.; Emery, J.F.; Duan, M.; Pittet, J.F. Regulation of apoptotic and inflammatory cell signaling in cerebral ischemia: The complex roles of heat shock protein 70. Anesthesiology 2008, 109, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Singh-Jasuja, H.; Toes, R.E.; Spee, P.; Munz, C.; Hilf, N.; Schoenberger, S.P.; Ricciardi-Castagnoli, P.; Neefjes, J.; Rammensee, H.G.; Arnold-Schild, D.; et al. Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class i molecules requires receptor-mediated endocytosis. J. Exp. Med. 2000, 191, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Haug, M.; Dannecker, L.; Schepp, C.P.; Kwok, W.W.; Wernet, D.; Buckner, J.H.; Kalbacher, H.; Dannecker, G.E.; Holzer, U. The heat shock protein hsp70 enhances antigen-specific proliferation of human cd4+ memory t cells. Eur. J. Immunol. 2005, 35, 3163–3172. [Google Scholar] [CrossRef]
- de Jong, P.R.; Schadenberg, A.W.; Jansen, N.J.; Prakken, B.J. Hsp70 and cardiac surgery: Molecular chaperone and inflammatory regulator with compartmentalized effects. Cell Stress Chaperones 2009, 14, 117–131. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Asea, A. Heat shock proteins and toll-like receptors. Handb. Exp. Pharmacol. 2008, 111–127. [Google Scholar] [CrossRef]
- Gaston, J.S. Heat shock proteins and innate immunity. Clin. Exp. Immunol. 2002, 127, 1–3. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Nagel, F.; Dietz, G.P.; Weise, J.; Tonges, L.; Schwarting, S.; Bahr, M. Tat-hsp70-mediated neuroprotection and increased survival of neuronal precursor cells after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2009, 29, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Ander, B.P.; Liao, I.H.; Hansen, J.E.; Kim, C.; Clements, D.; Weisbart, R.H.; Nishimura, R.N.; Sharp, F.R. Recombinant fv-hsp70 protein mediates neuroprotection after focal cerebral ischemia in rats. Stroke 2010, 41, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Ran, R.; Parmentier-Batteur, S.; Nee, A.; Sharp, F.R. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J. Neurochem. 2002, 81, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, F.; Guo, J.; Zhang, R.; Xing, X.; Qin, X. 17-aag post-treatment ameliorates memory impairment and hippocampal ca1 neuronal autophagic death induced by transient global cerebral ischemia. Brain Res. 2015, 1610, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Li, Z.; Lin, Z.; Wang, Y.; Wang, W.; Hu, B.; Wang, X.; Zhang, J.; Wang, Y.; Zhou, R.; et al. The hsp90 inhibitor 17-pag effectively inhibits the proliferation and migration of androgen-independent prostate cancer cells. Am. J. Cancer Res. 2015, 5, 3198–3209. [Google Scholar]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef]
- Porter, J.R.; Fritz, C.C.; Depew, K.M. Discovery and development of hsp90 inhibitors: A promising pathway for cancer therapy. Curr. Opin. Chem Biol. 2010, 14, 412–420. [Google Scholar] [CrossRef]
- Kim, N.; Kim, J.Y.; Yenari, M.A. Pharmacological induction of the 70-kda heat shock protein protects against brain injury. Neuroscience 2015, 284, 912–919. [Google Scholar] [CrossRef]
- Qi, J.; Han, X.; Liu, H.T.; Chen, T.; Zhang, J.L.; Yang, P.; Bo, S.H.; Lu, X.T.; Zhang, J. 17-dimethylaminoethylamino-17-demethoxygeldanamycin attenuates inflammatory responses in experimental stroke. Biol. Pharm. Bull 2014, 37, 1713–1718. [Google Scholar] [CrossRef]
- Jones, Q.; Voegeli, T.S.; Li, G.; Chen, Y.; Currie, R.W. Heat shock proteins protect against ischemia and inflammation through multiple mechanisms. Inflamm. Allergy Drug Targets 2011, 10, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, K.; Zhang, H.; Brekken, J.; Huser, N.; Powell, R.E.; Timple, N.; Busch, D.J.; Neely, L.; Sensintaffar, J.L.; Yang, Y.C.; et al. Biib021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein hsp90. Mol. Cancer Ther. 2009, 8, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Boll, B.; Eltaib, F.; Reiners, K.S.; von Tresckow, B.; Tawadros, S.; Simhadri, V.R.; Burrows, F.J.; Lundgren, K.; Hansen, H.P.; Engert, A.; et al. Heat shock protein 90 inhibitor biib021 (cnf2024) depletes nf-kappab and sensitizes hodgkin’s lymphoma cells for natural killer cell-mediated cytotoxicity. Clin. Cancer Res. 2009, 15, 5108–5116. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shichinohe, H.; Kuroda, S.; Ishikawa, T.; Iwasaki, Y. Neuroprotective effect of a heat shock protein inducer, geranylgeranylacetone in permanent focal cerebral ischemia. Brain Res. 2005, 1032, 176–182. [Google Scholar] [CrossRef]
- Uchida, S.; Fujiki, M.; Nagai, Y.; Abe, T.; Kobayashi, H. Geranylgeranylacetone, a noninvasive heat shock protein inducer, induces protein kinase c and leads to neuroprotection against cerebral infarction in rats. Neurosci. Lett. 2006, 396, 220–224. [Google Scholar] [CrossRef]
- Zhao, Z.; Faden, A.I.; Loane, D.J.; Lipinski, M.M.; Sabirzhanov, B.; Stoica, B.A. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1897–1908. [Google Scholar] [CrossRef]
- Joshi, V.; Mishra, R.; Upadhyay, A.; Amanullah, A.; Poluri, K.M.; Singh, S.; Kumar, A.; Mishra, A. Polyphenolic flavonoid (myricetin) upregulated proteasomal degradation mechanisms: Eliminates neurodegenerative proteins aggregation. J. Cell Physiol. 2019, 234, 20900–20914. [Google Scholar] [CrossRef]
- Deane, C.A.; Brown, I.R. Induction of heat shock proteins in differentiated human neuronal cells following co-application of celastrol and arimoclomol. Cell Stress Chaperones 2016, 21, 837–848. [Google Scholar] [CrossRef]
- Petrovic, A.; Kaur, J.; Tomljanovic, I.; Nistri, A.; Mladinic, M. Pharmacological induction of heat shock protein 70 by celastrol protects motoneurons from excitotoxicity in rat spinal cord in vitro. Eur. J. Neurosci. 2019, 49, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Liu, X.; Zhang, D.; Wang, Y.; Hu, X.; Xu, F.; Jin, M.; Cao, F.; Xu, L. Celastrol treatment protects against acute ischemic stroke-induced brain injury by promoting an il-33/st2 axis-mediated microglia/macrophage m2 polarization. J. Neuroinflamm. 2018, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Maczynska, J.; Choromanska, A.; Kutkowska, J.; Kotulska, M.; Zalewski, M.; Zalewski, J.; Kulbacka, J.; Saczko, J. Effect of electrochemotherapy with betulinic acid or cisplatin on regulation of heat shock proteins in metastatic human carcinoma cells in vitro. Oncol. Rep. 2019, 41, 3444–3454. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. https://doi.org/10.3390/cells9092020
Kim JY, Barua S, Huang MY, Park J, Yenari MA, Lee JE. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells. 2020; 9(9):2020. https://doi.org/10.3390/cells9092020
Chicago/Turabian StyleKim, Jong Youl, Sumit Barua, Mei Ying Huang, Joohyun Park, Midori A. Yenari, and Jong Eun Lee. 2020. "Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury" Cells 9, no. 9: 2020. https://doi.org/10.3390/cells9092020
APA StyleKim, J. Y., Barua, S., Huang, M. Y., Park, J., Yenari, M. A., & Lee, J. E. (2020). Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells, 9(9), 2020. https://doi.org/10.3390/cells9092020