Low ARID1A Expression is Associated with Poor Prognosis in Hepatocellular Carcinoma


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Data and Analysis
2.2. Gene Ontology (GO) Term Enrichment Analysis
2.3. Immunohistochemistry on Liver Cancer Tissue
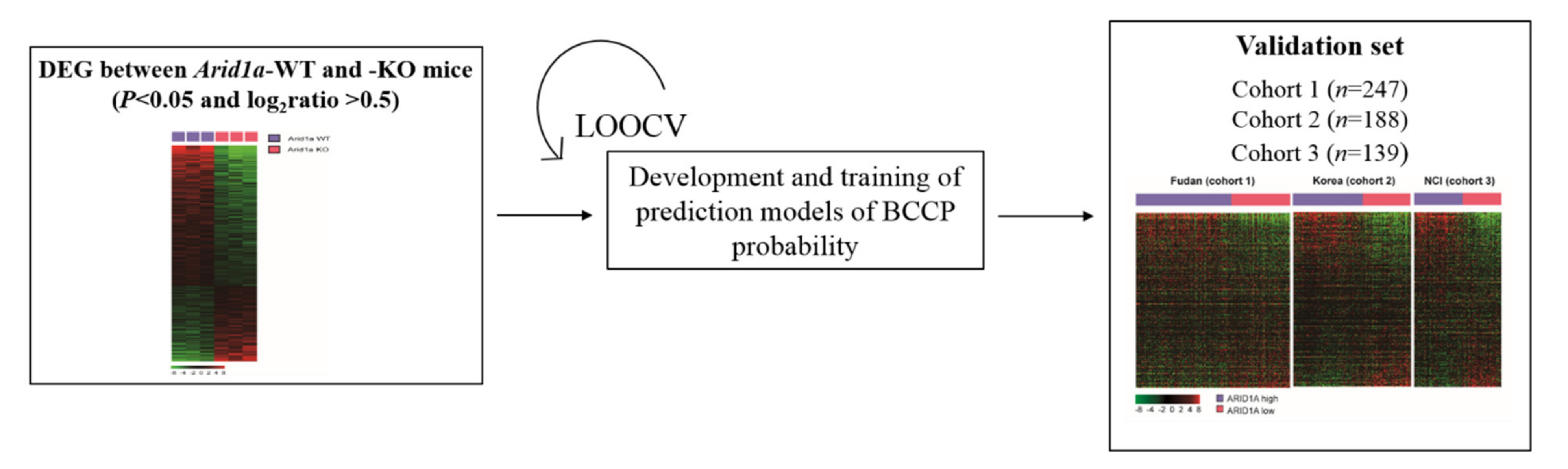
2.4. Validation of ARID1A Gene Signature in Three Independent HCC Patient Cohorts
2.5. TCGA Analysis Pipeline
2.6. Clinically Defined Molecular Subtypes of HCC and Associated Gene Signatures
2.7. Gene Network Analysis
3. Results
3.1. Development of ARID1A Gene Signature
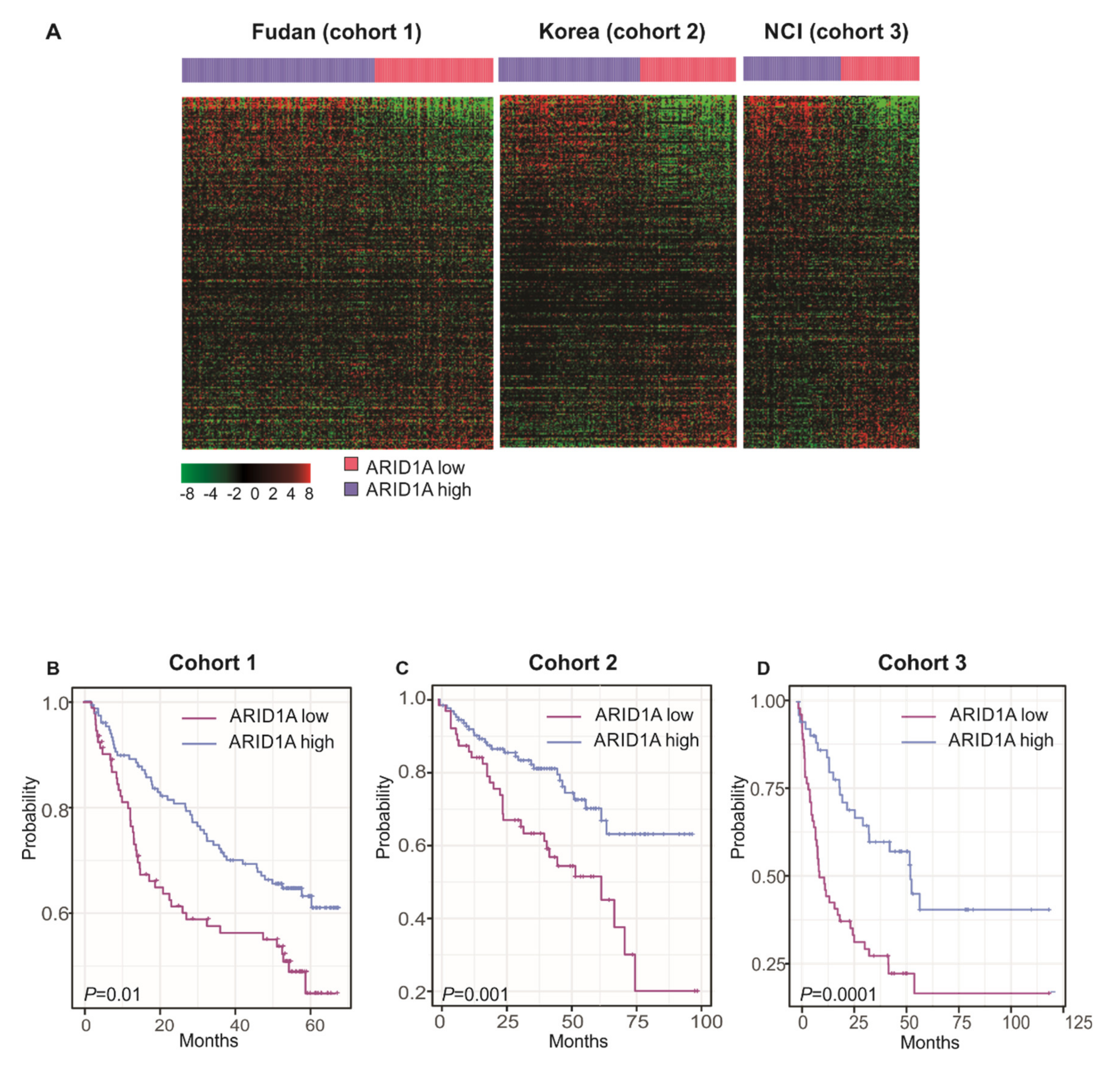
3.2. Clinical Significance ARID1A Gene Signature
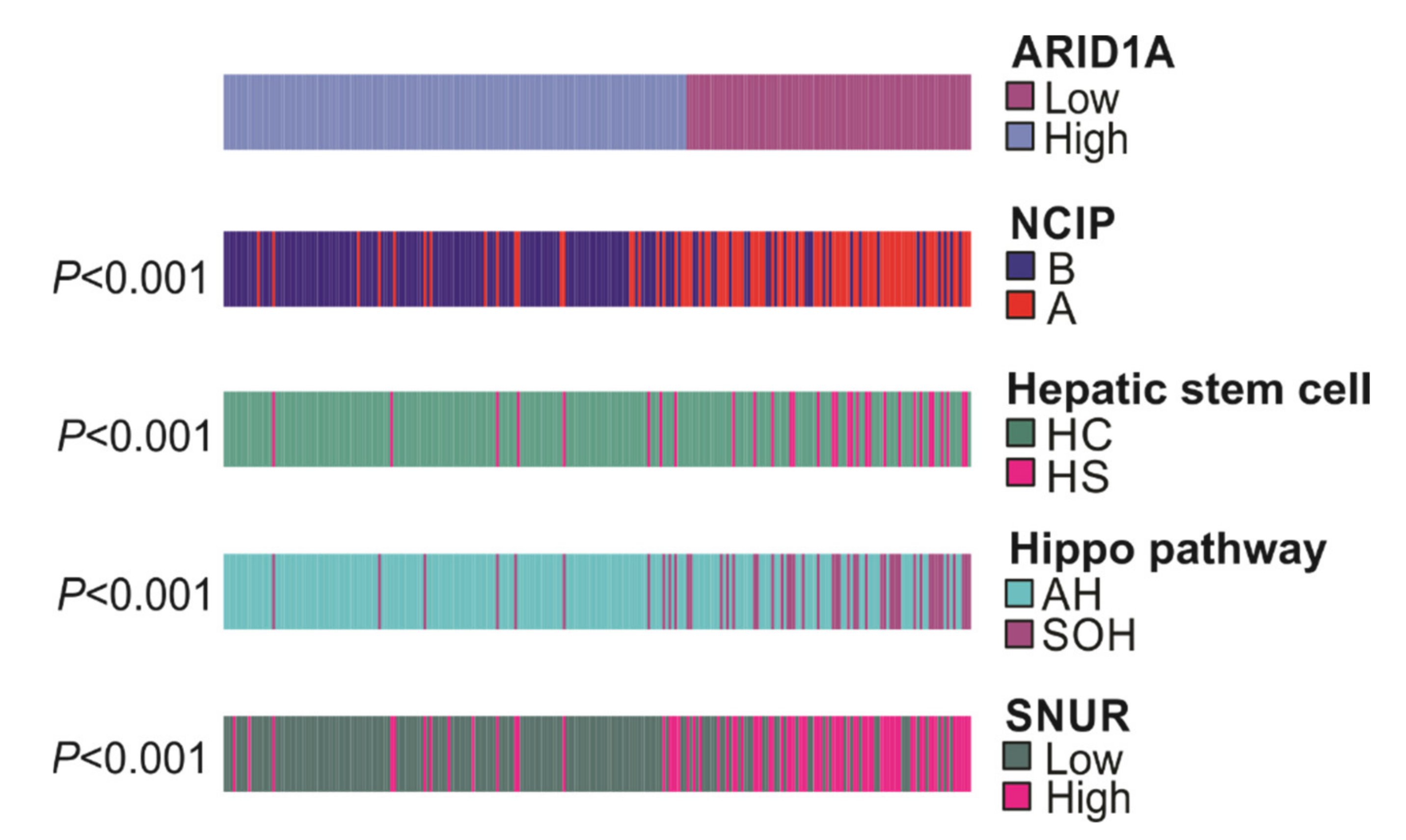
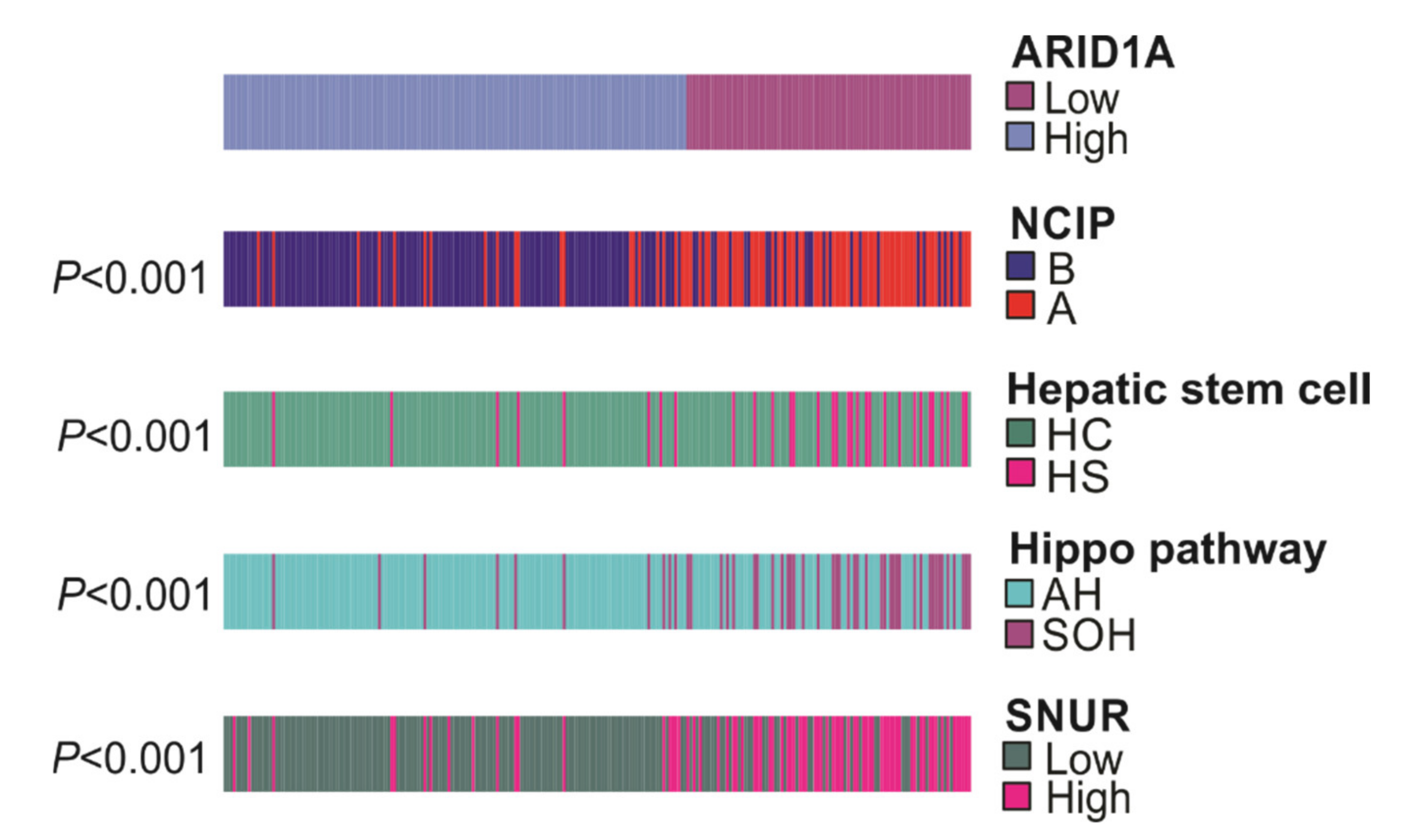
3.3. Association of ARID1A Gene Signature with Other Gene Signatures
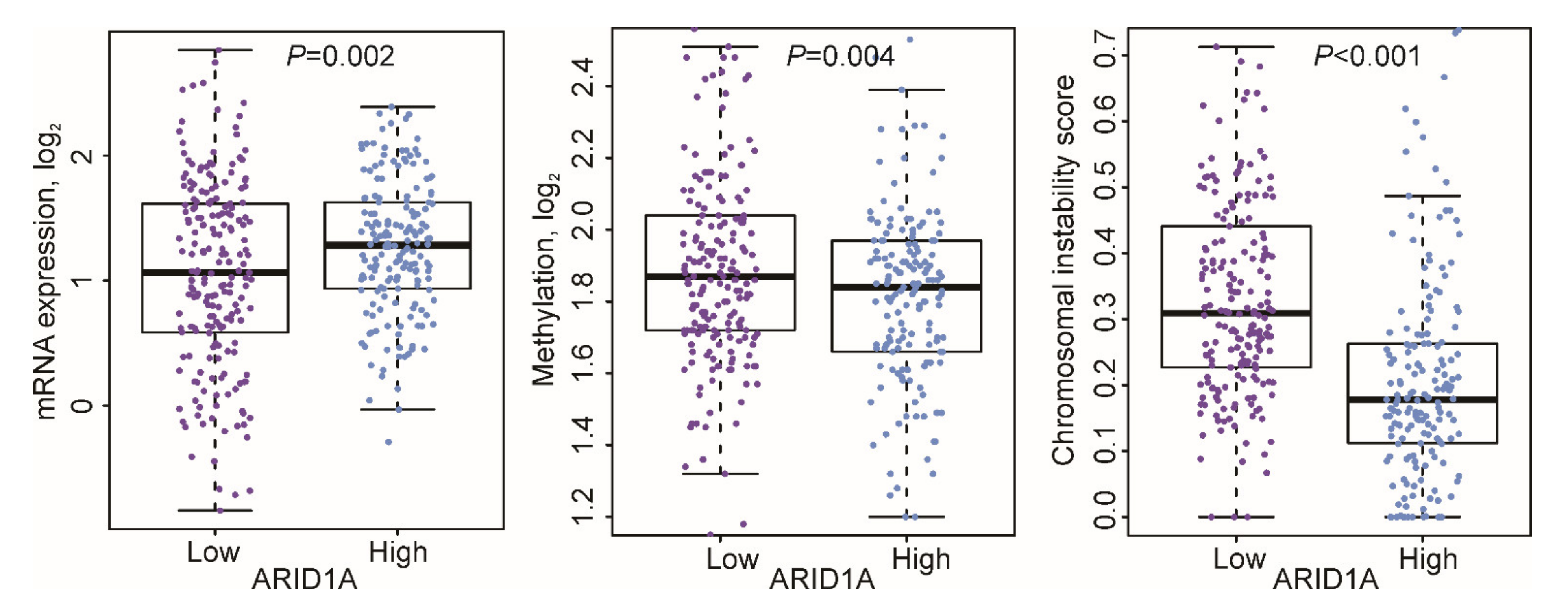
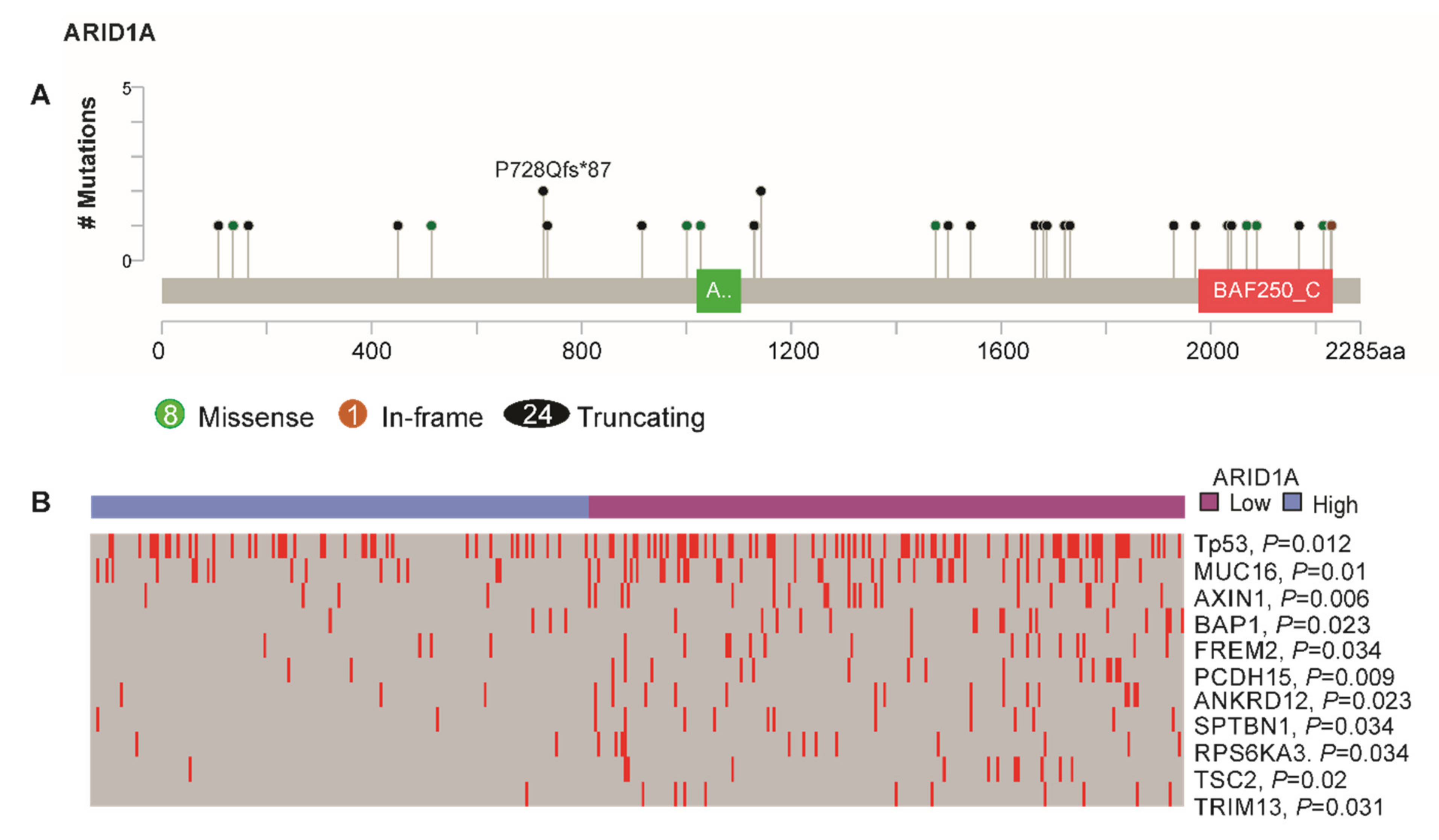
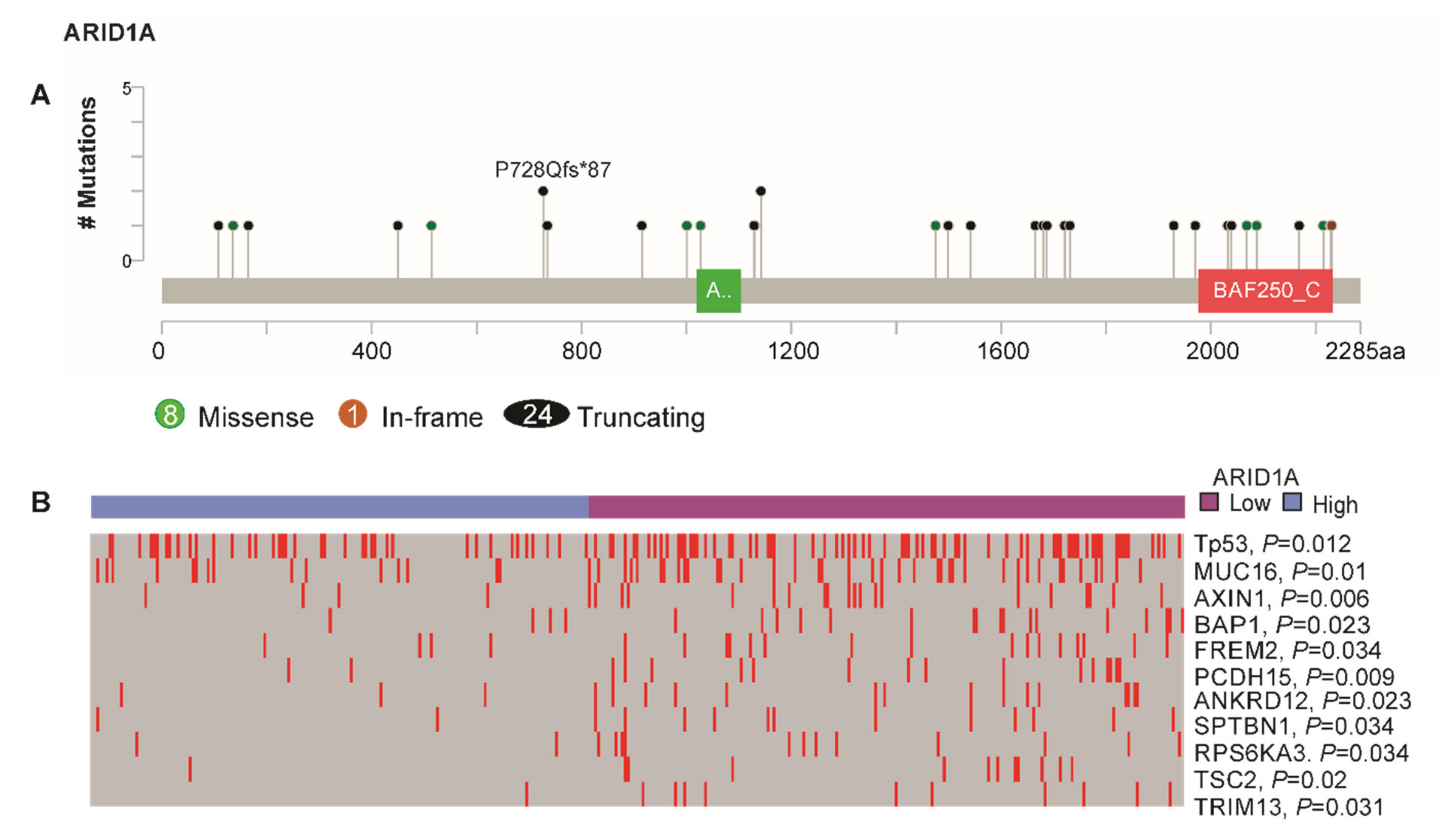
3.4. ARID1A-Low Subtype Cancer in Multi-Platform Analysis
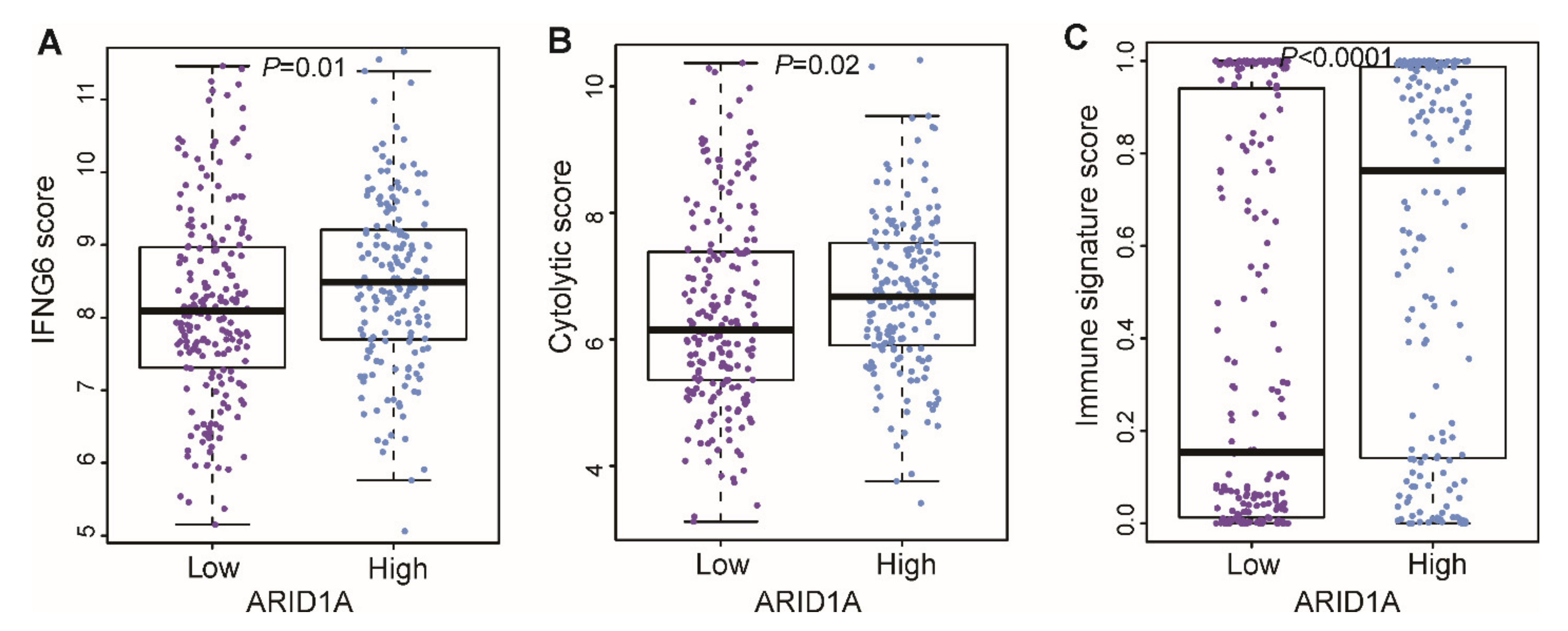
3.5. Correlation of ARID1A Gene Signature with Immune Signature
3.6. Functional Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, N.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, 359–386. [Google Scholar] [CrossRef]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Johnson, P.; Qin, S.; Park, J.; Poon, R.T.; Raoul, J.-L.; Philip, P.A.; Hsu, C.-H.; Hu, T.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.-L.; Kang, Y.-K.; Lin, D.-Y.; Park, J.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 436. [Google Scholar] [CrossRef]
- Best, J.; Schotten, C.; Lohmann, G.; Gerken, G.; Dechêne, A. Tivantinib for the treatment of hepatocellular carcinoma. Expert Opin. Pharmacother. 2017, 31, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Qin, S.; Huang, W.-T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.-K.; Chen, D.-S.; Toh, H.-C.; et al. Linifanib versus sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 2017, 32, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Chen, J.; Lin, H.; Jin, R.; Liu, J.; Liu, X.; Meng, N.; Cai, X. The clinicopathologic significance of BAF250a (ARID1A) expression in hepatocellular carcinoma. Pathol. Oncol. Res. 2015, 22, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, W.; Tian, F.; Jiang, K.; Liu, X.; Cen, J.; He, Q.; Qiu, Z.; Kienast, Y.; Wang, Z.; et al. Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J. Hepatol. 2018, 68, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chuang, J.-C.; Kanchwala, M.S.; Wu, L.; Celen, C.; Li, L.; Liang, H.; Zhang, S.; Maples, T.; Nguyen, L.H.; et al. Suppression of the SWI/SNF component Arid1a promotes mammalian regeneration. Cell Stem Cell 2016, 18, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Sohn, B.H.; Shim, J.-J.; Kim, S.-B.; Jang, K.Y.; Kim, S.M.; Kim, J.H.; Hwang, J.E.; Jang, H.-J.; Lee, H.-S.; Kim, S.-C.; et al. Inactivation of hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin. Cancer Res. 2015, 22, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Sohn, B.H.; Johnson, R.L.; Kang, M.; Kim, S.B.; Shim, J.J.; Mangala, L.S.; Kim, J.H.; Yoo, J.E.; Rodríguez-Aguayo, C.; et al. Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology 2015, 63, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Heo, J.; Libbrecht, L.; Chu, I.-S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef]
- Simon, R.; Lam, A.; Li, M.-C.; Ngan, M.; Menenzes, S.; Zhao, Y. Analysis of gene expression data using BRB-array tools. Cancer Inform. 2007, 3, 11–17. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Radmacher, M.D.; McShane, L.M.; Simon, R. A Paradigm for class prediction using gene expression profiles. J. Comput. Boil. 2002, 9, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Chu, I.-S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.G.; Park, E.-S.; Cheon, J.H.; Kim, J.H.; Lee, J.-S.; Park, B.-J.; Kim, W.; Park, S.C.; Chung, Y.J.; Kim, B.G.; et al. Gene expression-based recurrence prediction of hepatitis B virus-related human hepatocellular carcinoma. Clin. Cancer Res. 2008, 14, 2056–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ock, C.-Y.; Hwang, J.-E.; Keam, B.; Kim, S.-B.; Shim, J.J.; Jang, H.-J.; Park, S.; Sohn, B.H.; Cha, M.; Ajani, J.A.; et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat. Commun. 2017, 8, 1050. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Rinaldi, C.; Jones, A.; Rozali, E.N.; Dick, I.M.; Khong, A.; Boon, L.; Robinson, B.W.; Nowak, A.K.; Bosco, A.; et al. Network analysis of immunotherapy-induced regressing tumours identifies novel synergistic drug combinations. Sci. Rep. 2015, 5, 12298. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2013, 30, 523–530. [Google Scholar] [CrossRef]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Boil. 2006, 7, 437–447. [Google Scholar] [CrossRef]
- Mathur, R.; Alver, B.H.; Roman, A.K.S.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W.M. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2016, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-J.M.; Li, J.; Wang, Y.; Akbani, R.; Lu, Y.; Mills, G.B.; Liang, H. TCPA v3.0: An integrative platform to explore the pan-cancer analysis of functional proteomic data. Mol. Cell. Proteom. 2019, 18, S15–S25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of gene and protein expression in normal and cancer tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sun, Q.; Shan, M.; Niu, M.; Liu, T.; Xia, B.; Liang, X.; Wei, W.; Sun, S.; Zhang, Y.; et al. Promoter Hypermethylation of ARID1A Gene is responsible for its low mrna expression in many invasive breast cancers. PLoS ONE 2013, 8, e53931. [Google Scholar] [CrossRef] [Green Version]
- Allain, C.; Angenard, G.; Clément, B.; Coulouarn, C. Integrative genomic analysis identifies the core transcriptional hallmarks of human hepatocellular carcinoma Running title: Transcriptional hallmarks of liver cancer. Cancer Res. 2016, 76, 6374–6381. [Google Scholar] [CrossRef] [Green Version]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; Elgindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.A.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Johnson, B.J.; Costelloe, E.O.; Fitzpatrick, D.R.; Haanen, J.B.A.G.; Schumacher, T.N.M.; Brown, L.E.; Kelso, A. Single-cell perforin and granzyme expression reveals the anatomical localization of effector CD8+ T cells in influenza virus-infected mice. Proc. Natl. Acad. Sci. USA 2003, 100, 2657–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.G.; Roberts, C.W.M. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yim, S.Y.; Kang, S.H.; Shin, J.-H.; Jeong, Y.S.; Sohn, B.H.; Um, S.H.; Lee, J.-S. Low ARID1A Expression is Associated with Poor Prognosis in Hepatocellular Carcinoma. Cells 2020, 9, 2002. https://doi.org/10.3390/cells9092002
Yim SY, Kang SH, Shin J-H, Jeong YS, Sohn BH, Um SH, Lee J-S. Low ARID1A Expression is Associated with Poor Prognosis in Hepatocellular Carcinoma. Cells. 2020; 9(9):2002. https://doi.org/10.3390/cells9092002
Chicago/Turabian StyleYim, Sun Young, Sang Hee Kang, Ji-Hyun Shin, Yun Seong Jeong, Bo Hwa Sohn, Soon Ho Um, and Ju-Seog Lee. 2020. "Low ARID1A Expression is Associated with Poor Prognosis in Hepatocellular Carcinoma" Cells 9, no. 9: 2002. https://doi.org/10.3390/cells9092002
APA StyleYim, S. Y., Kang, S. H., Shin, J.-H., Jeong, Y. S., Sohn, B. H., Um, S. H., & Lee, J.-S. (2020). Low ARID1A Expression is Associated with Poor Prognosis in Hepatocellular Carcinoma. Cells, 9(9), 2002. https://doi.org/10.3390/cells9092002