Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. MMRF Data and Gene Dataset
2.3. Recombinant Heparanase and INHIBITORY Treatment
2.4. siRNA
2.5. Chromatin, Nuclear-Cytoplasmic Fractioning
2.6. Chromatin Immunoprecipitation (CHIP)
2.7. Micrococcal Nuclease Assay
2.8. Western Blotting
2.9. RNA Isolation and RT-qPCR
2.10. Statistical Analyses
3. Results
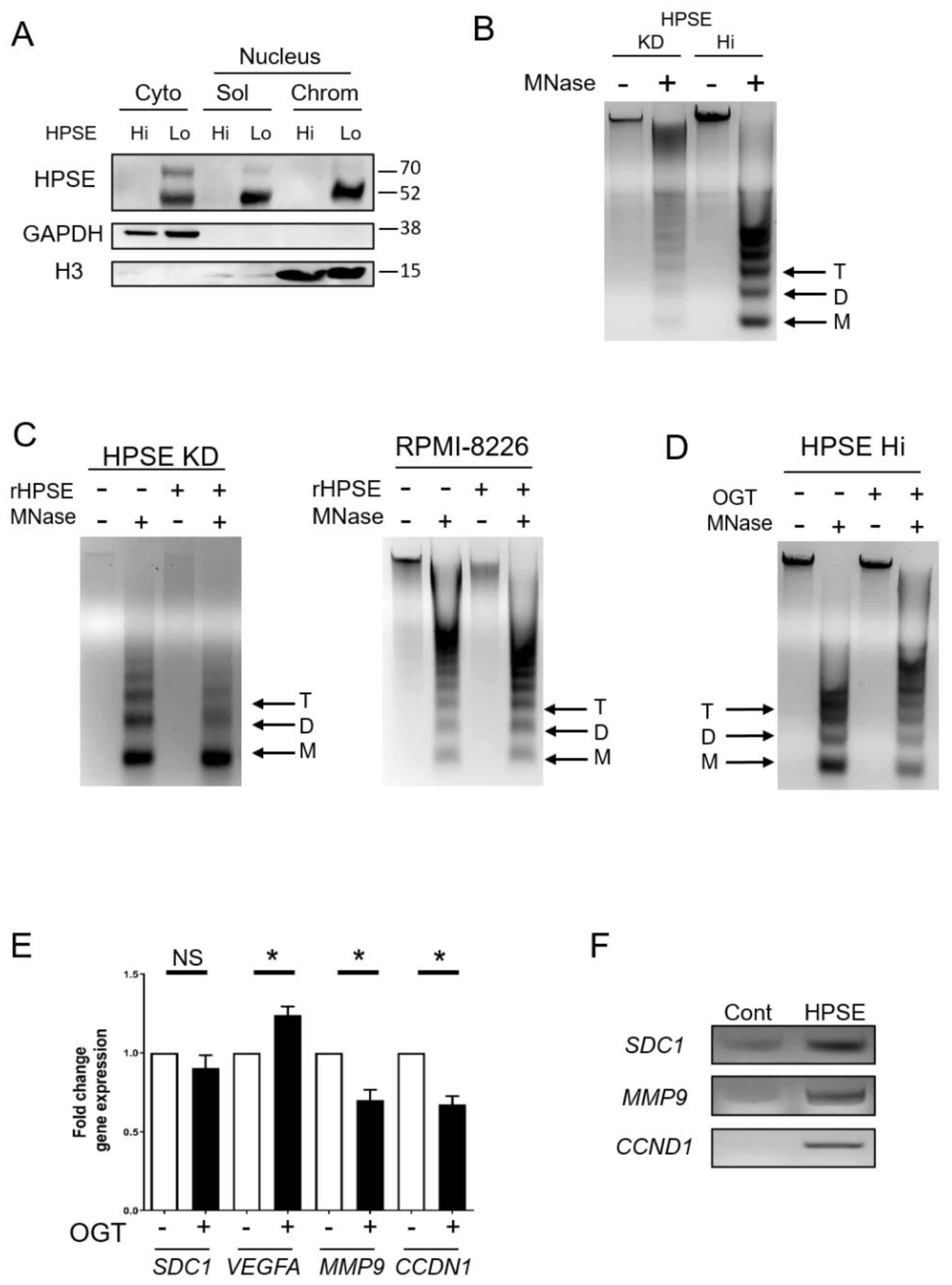
3.1. Heparanase Enhances Chromatin Accessibility
3.2. Elevation of Heparanase Is Associated with Increased Histone Acetylation and Upregulation of Genes that Promote an Aggressive Tumor Phenotype
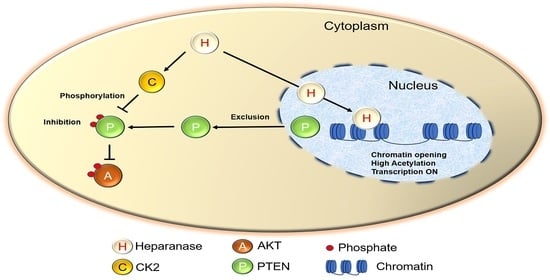
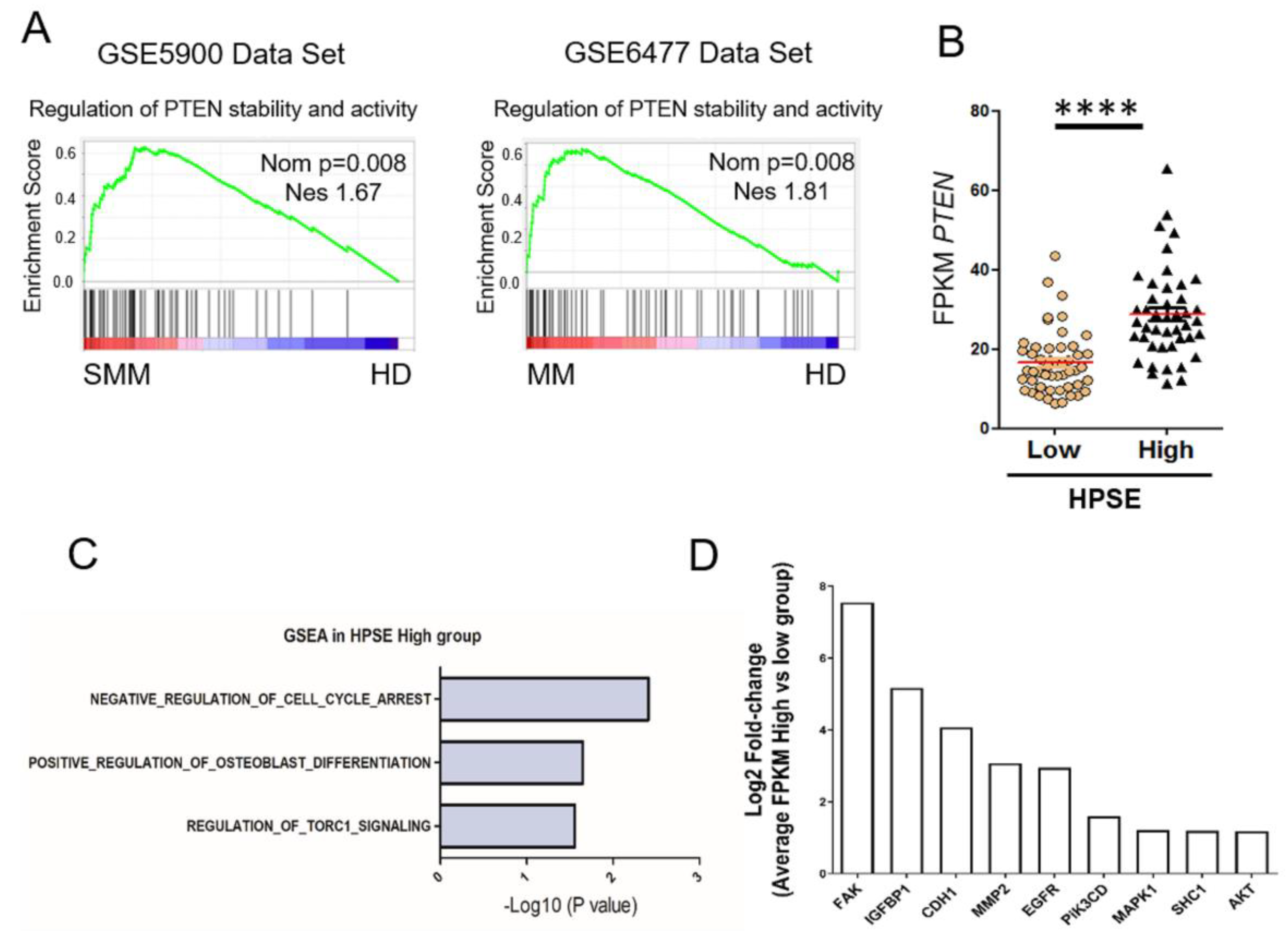
3.3. Heparanase Causes Loss of PTEN from the Nucleus and Upregulation of Genes Associated with an Aggressive Tumor Phenotype
3.4. Heparanase Increases PTEN Expression and Phosphorylation through CK2 to Promote PTEN Protein Stability Leading to Its Inactivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vlodavsky, I.; Ilan, N.; Sanderson, R.D. Forty years of basic and translational heparanase research. Adv. Exp. Med. Biol. 2020, 1221, 3–59. [Google Scholar] [PubMed]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [PubMed]
- Roucourt, B.; Meeussen, S.; Bao, J.; Zimmermann, P.; David, G. Heparanase activates the syndecan-syntenin-alix exosome pathway. Cell Res. 2015, 25, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.Y.; Ilan, N.; Shushy, M.; Ben-Izhak, O.; Vlodavsky, I.; Goldshmidt, O. Human heparanase nuclear localization and enzymatic activity. Lab. Investig. 2004, 84, 535–544. [Google Scholar] [CrossRef]
- Stewart, M.D.; Sanderson, R.D. Heparan sulfate in the nucleus and its control of cellular functions. Matrix Biol. 2014, 35, 56–59. [Google Scholar] [CrossRef]
- Purushothaman, A.; Hurst, D.R.; Pisano, C.; Mizumoto, S.; Sugahara, K.; Sanderson, R.D. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (hat) activity to promote expression of genes that drive an aggressive tumor phenotype. J. Biol. Chem. 2011, 286, 30377–30383. [Google Scholar] [CrossRef]
- Chen, L.; Sanderson, R.D. Heparanase regulates levels of syndecan-1 in the nucleus. PLoS ONE 2009, 4, e4947. [Google Scholar] [CrossRef]
- He, Y.Q.; Sutcliffe, E.L.; Bunting, K.L.; Li, J.; Goodall, K.J.; Poon, I.K.; Hulett, M.D.; Freeman, C.; Zafar, A.; McInnes, R.L.; et al. The endoglycosidase heparanase enters the nucleus of T lymphocytes and modulates h3 methylation at actively transcribed genes via the interplay with key chromatin modifying enzymes. Transcription 2012, 3, 130–145. [Google Scholar] [CrossRef]
- Parish, C.R.; Freeman, C.; Ziolkowski, A.F.; He, Y.Q.; Sutcliffe, E.L.; Zafar, A.; Rao, S.; Simeonovic, C.J. Unexpected new roles for heparanase in type 1 diabetes and immune gene regulation. Matrix Biol. 2013, 32, 228–233. [Google Scholar] [CrossRef]
- Agelidis, A.M.; Hadigal, S.R.; Jaishankar, D.; Shukla, D. Viral activation of heparanase drives pathogenesis of herpes simplex virus-1. Cell Rep. 2017, 20, 439–450. [Google Scholar] [CrossRef]
- Richardson, P.G.; Laubach, J.; Gandolfi, S.; Facon, T.; Weisel, K.; O’Gorman, P. Maintenance and continuous therapy for multiple myeloma. Expert Rev. Anticancer. Ther. 2018, 18, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.; Sanderson, R.D. Heparanase: A dynamic promoter of myeloma progression. Adv. Exp. Med. Biol. 2020, 1221, 331–349. [Google Scholar] [PubMed]
- Ramani, V.C.; Vlodavsky, I.; Ng, M.; Zhang, Y.; Barbieri, P.; Noseda, A.; Sanderson, R.D. Chemotherapy induces expression and release of heparanase leading to changes associated with an aggressive tumor phenotype. Matrix Biol. 2016, 55, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Ramani, V.C.; Bandari, S.K.; Amin, R.; Brown, E.E.; Ritchie, J.P.; Stewart, M.D.; Sanderson, R.D. Heparanase promotes myeloma stemness and in vivo tumorigenesis. Matrix Biol. 2020, 88, 53–68. [Google Scholar] [CrossRef]
- Kelly, T.; Miao, H.Q.; Yang, Y.; Navarro, E.; Kussie, P.; Huang, Y.; MacLeod, V.; Casciano, J.; Joseph, L.; Zhan, F.; et al. High heparanase activity in multiple myeloma is associated with elevated microvessel density. Cancer Res. 2003, 63, 8749–8756. [Google Scholar]
- Borset, M.; Hjertner, O.; Yaccoby, S.; Epstein, J.; Sanderson, R.D. Syndecan-1 is targeted to the uropods of polarized myeloma cells where it promotes adhesion and sequesters heparin-binding proteins. Blood 2000, 96, 2528–2536. [Google Scholar] [CrossRef]
- Purushothaman, A.; Chen, L.; Yang, Y.; Sanderson, R.D. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J. Biol. Chem. 2008, 283, 32628–32636. [Google Scholar] [CrossRef]
- Tong, A.W.; Lee, J.; Wang, R.M.; Dalton, W.S.; Tsuruo, T.; Fay, J.W.; Stone, M.J. Elimination of chemoresistant multiple myeloma clonogenic colony-forming cells by combined treatment with a plasma cell-reactive monoclonal antibody and a p-glycoprotein-reactive monoclonal antibody. Cancer Res. 1989, 49, 4829–4834. [Google Scholar]
- Gingis-Velitski, S.; Zetser, A.; Kaplan, V.; Ben-Zaken, O.; Cohen, E.; Levy-Adam, F.; Bashenko, Y.; Flugelman, M.Y.; Vlodavsky, I.; Ilan, N. Heparanase uptake is mediated by cell membrane heparan sulfate proteoglycans. J. Biol. Chem. 2004, 279, 44084–44092. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Yin, H.; Shi, C.X.; Que, Q.; Basu, G.; Azorsa, D.; Perkins, L.M.; Braggio, E.; et al. Kinome-wide rnai studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, grk6. Blood 2010, 115, 1594–1604. [Google Scholar] [CrossRef]
- Zhan, F.; Barlogie, B.; Arzoumanian, V.; Huang, Y.; Williams, D.R.; Hollmig, K.; Pineda-Roman, M.; Tricot, G.; van Rhee, F.; Zangari, M.; et al. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood 2007, 109, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Trotter, T.N.; Li, M.; Pan, Q.; Peker, D.; Rowan, P.D.; Li, J.; Zhan, F.; Suva, L.J.; Javed, A.; Yang, Y. Myeloma cell-derived runx2 promotes myeloma progression in bone. Blood 2015, 125, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chiu, A.P.; Neumaier, K.; Wang, F.; Zhang, D.; Hussein, B.; Lal, N.; Wan, A.; Liu, G.; Vlodavsky, I.; et al. Endothelial cell heparanase taken up by cardiomyocytes regulates lipoprotein lipase transfer to the coronary lumen after diabetes. Diabetes 2014, 63, 2643–2655. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, Y.; Zhang, D.; Puthanveetil, P.; Johnson, J.D.; Rodrigues, B. Fatty acid-induced nuclear translocation of heparanase uncouples glucose metabolism in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.C.; Lee, Y.H.; Lin, S.P.; Huangfu, W.C.; Liu, I.H. Cell-autonomous heparanase modulates self-renewal and migration in bone marrow-derived mesenchymal stem cells. J. Biomed. Sci. 2014, 21, 21. [Google Scholar] [CrossRef]
- Ross, S.; Cheung, E.; Petrakis, T.G.; Howell, M.; Kraus, W.L.; Hill, C.S. Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J. 2006, 25, 4490–4502. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Zhu, M.; Yang, J.; Liang, H.; He, J.; He, S.; Wang, P.; Kang, X.; McNutt, M.A.; Yin, Y.; et al. Pten interacts with histone h1 and controls chromatin condensation. Cell Rep. 2014, 8, 2003–2014. [Google Scholar] [CrossRef]
- Chang, H.; Qi, X.Y.; Claudio, J.; Zhuang, L.; Patterson, B.; Stewart, A.K. Analysis of pten deletions and mutations in multiple myeloma. Leuk. Res. 2006, 30, 262–265. [Google Scholar] [CrossRef]
- Liu, X.; Bruxvoort, K.J.; Zylstra, C.R.; Liu, J.; Cichowski, R.; Faugere, M.C.; Bouxsein, M.L.; Wan, C.; Williams, B.O.; Clemens, T.L. Lifelong accumulation of bone in mice lacking pten in osteoblasts. Proc. Natl. Acad. Sci. USA 2007, 104, 2259–2264. [Google Scholar] [CrossRef]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell cycle control by pten. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Matsumoto, C.S.; Almeida, L.O.; Guimaraes, D.M.; Martins, M.D.; Papagerakis, P.; Papagerakis, S.; Leopoldino, A.M.; Castilho, R.M.; Squarize, C.H. Pi3k-pten dysregulation leads to mtor-driven upregulation of the core clock gene bmal1 in normal and malignant epithelial cells. Oncotarget 2016, 7, 42393–42407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Liu, J.; Lei, S.; Zhang, J.; Zhou, W.; Yu, H.G. Pten inhibits the invasion and metastasis of gastric cancer via downregulation of fak expression. Cell. Signal. 2014, 26, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Ammoun, S.; Schmid, M.C.; Zhou, L.; Ristic, N.; Ercolano, E.; Hilton, D.A.; Perks, C.M.; Hanemann, C.O. Insulin-like growth factor-binding protein-1 (igfbp-1) regulates human schwannoma proliferation, adhesion and survival. Oncogene 2012, 31, 1710–1722. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Li, Z.; Zhang, Y.; Li, D.; Zhang, G.; Luo, X.; Jie, Z.; Liu, Y.; Cao, Y.; Le, Z.; et al. Prl-3 promotes the peritoneal metastasis of gastric cancer through the pi3k/akt signaling pathway by regulating pten. Oncol. Rep. 2016, 36, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.R.; Maddika, S. Pten modulates egfr late endocytic trafficking and degradation by dephosphorylating rab7. Nat. Commun. 2016, 7, 10689. [Google Scholar] [CrossRef]
- Papakonstanti, E.A.; Ridley, A.J.; Vanhaesebroeck, B. The p110delta isoform of pi 3-kinase negatively controls rhoa and pten. EMBO J. 2007, 26, 3050–3061. [Google Scholar] [CrossRef]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and fak differentially regulate cell motility and directionality modulated by pten. J. Cell Biol. 1999, 146, 389–403. [Google Scholar] [CrossRef]
- Bononi, A.; Pinton, P. Study of pten subcellular localization. Methods 2015, 77–78, 92–103. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Yang, J.M.; Miyamoto, T.; Itoh, K.; Rho, E.; Zhang, Q.; Inoue, T.; Devreotes, P.N.; Sesaki, H.; Iijima, M. Opening the conformation is a master switch for the dual localization and phosphatase activity of pten. Sci. Rep. 2015, 5, 12600. [Google Scholar] [CrossRef]
- Miller, S.J.; Lou, D.Y.; Seldin, D.C.; Lane, W.S.; Neel, B.G. Direct identification of pten phosphorylation sites. FEBS Lett. 2002, 528, 145–153. [Google Scholar] [CrossRef]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin l is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.; Gunther, D.; Laube, F.; Wiederanders, B.; Kirschke, H. Hybridoma cells producing antibodies to cathepsin l have greatly reduced potential for tumour growth. J. Cancer Res. Clin. Oncol. 1994, 120, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H.; Eerola, R.; Hopsu-Havu, V.K.; Bromme, D.; Vuorio, E. Antisense rna inhibition of cathepsin l expression reduces tumorigenicity of malignant cells. Eur. J. Cancer 2000, 36, 787–795. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ramani, V.C.; Sanderson, R.D. Shed syndecan-1 translocates to the nucleus of cells delivering growth factors and inhibiting histone acetylation: A novel mechanism of tumor-host cross-talk. J. Biol. Chem. 2015, 290, 941–949. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of hats and hdacs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Peserico, A.; Simone, C. Physical and functional hat/hdac interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear pten in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Jiang, Y.; Chang, H.; Chen, G. Effects of microrna-20a on the proliferation, migration and apoptosis of multiple myeloma via the pten/pi3k/akt signaling pathway. Oncol. Lett. 2018, 15, 10001–10007. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Zhao, P.; Zang, L.; Zhang, Z.; Wang, X. Microrna-19a functions as an oncogene by regulating pten/akt/pakt pathway in myeloma. Leuk. Lymphoma 2017, 58, 932–940. [Google Scholar] [CrossRef]
- Piras, G.; Monne, M.; Palmas, A.D.; Calvisi, A.; Asproni, R.; Vacca, F.; Pilo, L.; Gabbas, A.; Latte, G. Methylation analysis of the phosphates and tensin homologue on chromosome 10 gene (pten) in multiple myeloma. Clin. Epigenetics 2014, 6, 16. [Google Scholar] [CrossRef]
- Georgescu, M.M. Pten tumor suppressor network in pi3k-akt pathway control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Hyun, T.; Yam, A.; Pece, S.; Xie, X.; Zhang, J.; Miki, T.; Gutkind, J.S.; Li, W. Loss of pten expression leading to high akt activation in human multiple myelomas. Blood 2000, 96, 3560–3568. [Google Scholar] [CrossRef] [PubMed]
- Riaz, A.; Ilan, N.; Vlodavsky, I.; Li, J.P.; Johansson, S. Characterization of heparanase-induced phosphatidylinositol 3-kinase-akt activation and its integrin dependence. J. Biol. Chem. 2013, 288, 12366–12375. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Galletti, S.; Garavelli, S.; Platonova, N.; Paoli, A.; Basile, A.; Taiana, E.; Neri, A.; Chiaramonte, R. Notch signaling deregulation in multiple myeloma: A rational molecular target. Oncotarget 2015, 6, 26826–26840. [Google Scholar] [CrossRef]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef]
- Vitale, D.; Kumar Katakam, S.; Greve, B.; Jang, B.; Oh, E.S.; Alaniz, L.; Gotte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Ashihara, E.; Yao, H.; Yokota, A.; Toda, Y.; Miura, Y.; Nakata, S.; Hirai, H.; Maekawa, T. Multiple myeloma cells adapted to long-exposure of hypoxia exhibit stem cell characters with tgf-beta/smad pathway activation. Biochem. Biophys. Res. Commun. 2018, 496, 490–496. [Google Scholar] [CrossRef]
- Du, J.; Liu, S.; He, J.; Liu, X.; Qu, Y.; Yan, W.; Fan, J.; Li, R.; Xi, H.; Fu, W.; et al. Microrna-451 regulates stemness of side population cells via pi3k/akt/mtor signaling pathway in multiple myeloma. Oncotarget 2015, 6, 14993–15007. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Duan, S.; Yuan, G.; Liu, X.; Ren, R.; Li, J.; Zhang, W.; Wu, J.; Xu, X.; Fu, L.; Li, Y.; et al. Pten deficiency reprogrammes human neural stem cells towards a glioblastoma stem cell-like phenotype. Nat. Commun. 2015, 6, 10068. [Google Scholar] [CrossRef]
- Xu, Q.; Yuan, X.; Liu, G.; Black, K.L.; Yu, J.S. Hedgehog signaling regulates brain tumor-initiating cell proliferation and portends shorter survival for patients with pten-coexpressing glioblastomas. Stem Cells 2008, 26, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Wyatt, D.; Bocchetta, M.; Li, J.; Filipovic, A.; Green, A.; Peiffer, D.S.; Fuqua, S.; Miele, L.; Albain, K.S.; et al. Notch-1-pten-erk1/2 signaling axis promotes her2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018, 37, 4489–4504. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. The pten tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor kappa b activity. J. Biol. Chem. 2001, 276, 27740–27744. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.J.; Bae, E.; Hong, Y.K.; Hong, J.Y.; Kim, N.K.; Ahn, H.J.; Oh, J.J.; Park, D.S. Pten loss-mediated akt activation increases the properties of cancer stem-like cell populations in prostate cancer. Oncology 2014, 87, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Alvarez, U.; Hruska, K.A. Pten regulates rankl- and osteopontin-stimulated signal transduction during osteoclast differentiation and cell motility. J. Biol. Chem. 2003, 278, 5001–5008. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, Y.; Ramani, V.C.; Nan, L.; Suva, L.J.; Sanderson, R.D. Heparanase enhances local and systemic osteolysis in multiple myeloma by upregulating the expression and secretion of rankl. Cancer Res. 2010, 70, 8329–8338. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Tamura, M.; Yamada, K.M. Tumor suppressor pten inhibits integrin- and growth factor-mediated mitogen-activated protein (map) kinase signaling pathways. J. Cell Biol. 1998, 143, 1375–1383. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Z.; Yang, X.; Deng, K.; Cao, Y.; Chen, H.; Pan, L. Effect of wild type pten gene on proliferation and invasion of multiple myeloma. Int. J. Hematol. 2010, 92, 83–94. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, R.; Tripathi, K.; Sanderson, R.D. Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function. Cells 2020, 9, 2038. https://doi.org/10.3390/cells9092038
Amin R, Tripathi K, Sanderson RD. Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function. Cells. 2020; 9(9):2038. https://doi.org/10.3390/cells9092038
Chicago/Turabian StyleAmin, Rada, Kaushlendra Tripathi, and Ralph D. Sanderson. 2020. "Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function" Cells 9, no. 9: 2038. https://doi.org/10.3390/cells9092038
APA StyleAmin, R., Tripathi, K., & Sanderson, R. D. (2020). Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function. Cells, 9(9), 2038. https://doi.org/10.3390/cells9092038