PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Isolation and Culture of Adult Mouse Cardiomyocytes
2.3. Viral Infections
2.4. Pharmacological Treatments
2.5. Immunoblot
2.6. Quantitative RT-PCR
- c-Kit Forward: ATTGTGCTGGATGGATGGAT
- c-Kit Reverse: GATCTGCTCTGCGTCCTGTT
- Co-Immunoprecipitation
2.7. Proximity Ligation Assay
2.8. Cell Death Assay
2.9. Statistical Analyses
3. Results
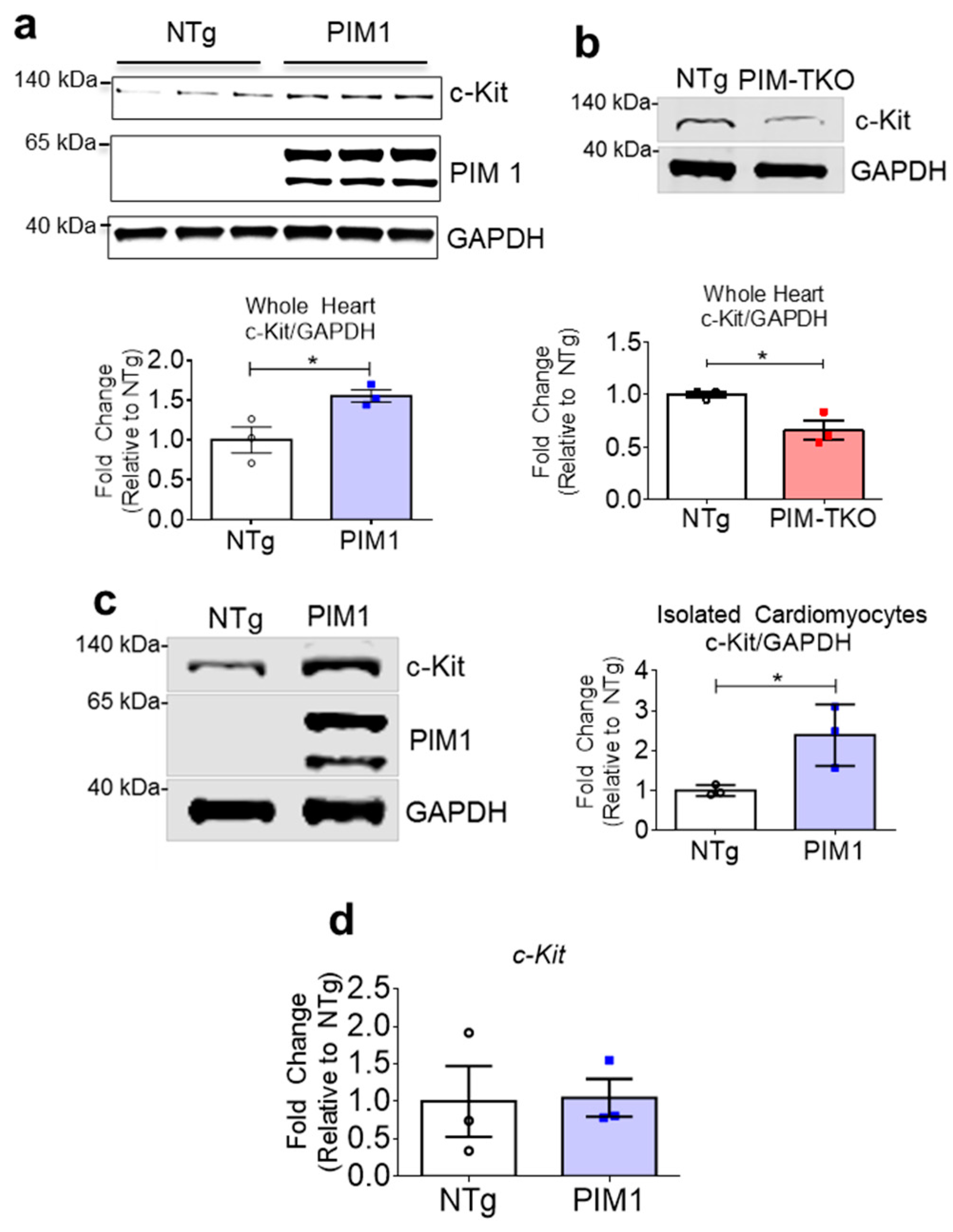
3.1. PIM1 Upregulates c-Kit Protein Expression
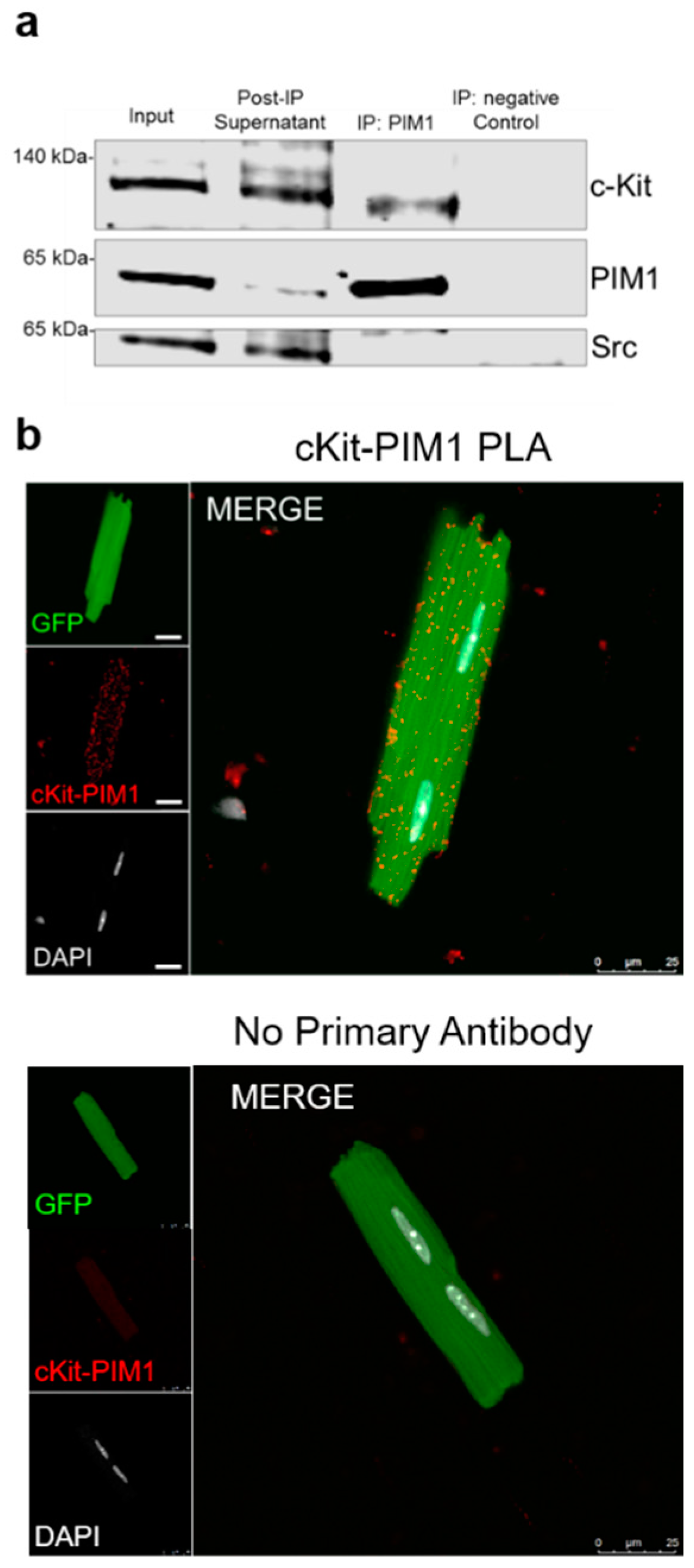
3.2. PIM1 Interacts with c-Kit
3.3. Cardioprotective Signaling Downstream of c-Kit is Elevated in PIM1 Cardiomyocytes
3.4. Elevated c-Kit Expression Enhances Resistance Against Oxidative Stress in Cardiomyocytes
3.5. PIM1 Confers Cardioprotection Independent of c-kit Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Engel, F.B. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 67–96. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Vergarajauregui, S.; Wu, C.C.; Piatkowski, T.; Becker, R.; Leone, M.; Hirth, S.; Ricciardi, F.; Falk, N.; Giessl, A.; et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife 2015, 4, e05563. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Kim, J.S.; Chae, S.W.; Koh, K.N.; Koh, G.Y. Cyclins and cyclin dependent kinases during cardiac development. Mol. Cells 1997, 7, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Poolman, R.A.; Gilchrist, R.; Brooks, G. Cell cycle profiles and expressions of p21(CIP1) and p27(KIP1) during myocyte development. Int. J. Cardiol. 1998, 67, 133–142. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef]
- Kulandavelu, S.; Karantalis, V.; Fritsch, J.; Hatzistergos, K.E.; Loescher, V.Y.; McCall, F.; Wang, B.; Bagno, L.; Golpanian, S.; Wolf, A.; et al. Pim1 Kinase Overexpression Enhances ckit+ Cardiac Stem Cell Cardiac Repair Following Myocardial Infarction in Swine. J. Am. Coll. Cardiol. 2016, 68, 2454–2464. [Google Scholar] [CrossRef]
- Muraski, J.A.; Fischer, K.M.; Wu, W.; Cottage, C.T.; Quijada, P.; Mason, M.; Din, S.; Gude, N.; Alvarez, R.; Rota, M.; et al. Pim-1 kinase antagonizes aspects of myocardial hypertrophy and compensation to pathological pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 13889–13894. [Google Scholar] [CrossRef] [Green Version]
- Muraski, J.A.; Rota, M.; Misao, Y.; Fransioli, J.; Cottage, C.; Gude, N.; Esposito, G.; Delucchi, F.; Arcarese, M.; Alvarez, R.; et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat. Med. 2007, 13, 1467–1475. [Google Scholar] [CrossRef]
- Zhu, H.H.; Wang, X.T.; Sun, Y.H.; He, W.K.; Liang, J.B.; Mo, B.H.; Li, L. Pim1 Overexpression Prevents Apoptosis in Cardiomyocytes after Exposure to Hypoxia and Oxidative Stress via Upregulating Cell Autophagy. Cell. Physiol. Biochem. 2018, 49, 2138–2150. [Google Scholar] [CrossRef]
- An, N.; Lin, Y.-W.; Mahajan, S.; Kellner, J.N.; Wang, Y.; Li, Z.; Kraft, A.S.; Kang, Y. Pim1 Serine/Threonine Kinase Regulates the Number and Functions of Murine Hematopoietic Stem Cells. Stem Cells 2013, 31, 1202–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, K.C.; Wang, L.; Hickey, E.R.; Studts, J.; Barringer, K.; Peng, C.; Kronkaitis, A.; Li, J.; White, A.; Mische, S.; et al. Structural basis of constitutive activity and a unique nucleotide binding mode of human Pim-1 kinase. J. Biol. Chem. 2005, 280, 6130–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Bhattacharya, N.; Weaver, M.; Petersen, K.; Meyer, M.; Gapter, L.; Magnuson, N.S. Pim-1: A serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J. Vet. Sci. 2001, 2, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borillo, G.A.; Mason, M.; Quijada, P.; Völkers, M.; Cottage, C.; McGregor, M.; Din, S.; Fischer, K.; Gude, N.; Avitabile, D.; et al. Pim-1 kinase protects mitochondrial integrity in cardiomyocytes. Circ. Res. 2010, 106, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Ellison, F.M.; McCoy, J.P.; Chen, J. c-Kit expression and stem cell factor-induced hematopoietic cell proliferation are up-regulated in aged B6D2F1 mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 448–456. [Google Scholar] [CrossRef]
- Fischer, K.M.; Cottage, C.T.; Wu, W.; Din, S.; Gude, N.A.; Avitabile, D.; Quijada, P.; Collins, B.L.; Fransioli, J.; Sussman, M.A. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing pim-1 kinase. Circulation 2009, 120, 2077–2087. [Google Scholar] [CrossRef]
- Gude, N.A.; Firouzi, F.; Broughton, K.M.; Ilves, K.; Nguyen, K.P.; Payne, C.R.; Sacchi, V.; Monsanto, M.M.; Casillas, A.R.; Khalafalla, F.G.; et al. Cardiac c-Kit biology revealed by inducible transgenesis. Circ. Res. 2018, 123, 57–72. [Google Scholar] [CrossRef]
- Linnekin, D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int. J. Biochem. Cell Biol. 1999, 31, 1053–1074. [Google Scholar] [CrossRef] [Green Version]
- Lips, D.J.; Bueno, O.F.; Wilkins, B.J.; Purcell, N.H.; Kaiser, R.A.; Lorenz, J.N.; Voisin, L.; Saba-El-Leil, M.K.; Meloche, S.; Pouysségur, J.; et al. MEK1-ERK2 Signaling Pathway Protects Myocardium From Ischemic Injury In Vivo. Circulation 2004, 109, 1938–1941. [Google Scholar] [CrossRef] [Green Version]
- Li, X.M.; Ma, Y.T.; Yang, Y.N.; Liu, F.; Chen, B.D.; Han, W.; Zhang, J.F.; Gao, X.M. Downregulation of survival signalling pathways and increased apoptosis in the transition of pressure overload-induced cardiac hypertrophy to heart failure. Clin. Exp. Pharmacol. Physiol. 2009, 36, 1054–1061. [Google Scholar] [CrossRef]
- Song, Y.H.; Cai, H.; Zhao, Z.M.; Chang, W.J.; Gu, N.; Cao, S.P.; Wu, M.L. Icariin attenuated oxidative stress induced-cardiac apoptosis by mitochondria protection and ERK activation. Biomed. Pharmacother. 2016, 83, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.C.; Fan, Y.; Xu, Y.; Yang, Y.L.; Simpson, P.C.; Mann, M.J. Shift toward greater pathologic post-myocardial infarction remodeling with loss of the adaptive hypertrophic signaling of alpha1 adrenergic receptors in mice. PLoS ONE 2017, 12, e0188471. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Xu, T.; Masters, S.; Haian, F.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell- intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Cen, B.; Cai, H.; Song, J.H.; Kraft, A.; Kang, Y. Pim1 kinase regulates c-Kit gene translation. Exp. Hematol. Oncol. 2016, 5, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice Deficient for All PIM Kinases Display Reduced Body Size and Impaired Responses to Hematopoietic Growth Factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef] [Green Version]
- Moyzis, A.G.; Najor, R.H.; Cortez, M.Q.; Gustafsson, Å.B.; Kubli, D.A.; Lee, Y. PINK1 Is Dispensable for Mitochondrial Recruitment of Parkin and Activation of Mitophagy in Cardiac Myocytes. PLoS ONE 2015, 10, e0130707. [Google Scholar] [CrossRef]
- Ma, H.P.; Ma, X.N.; Ge, B.F.; Zhen, P.; Zhou, J.; Gao, Y.H.; Xian, C.J.; Chen, K.M. Icariin attenuates hypoxia-induced oxidative stress and apoptosis in osteoblasts and preserves their osteogenic differentiation potential in vitro. Cell Prolif. 2014, 47, 527–539. [Google Scholar] [CrossRef]
- Di Siena, S.; Gimmelli, R.; Nori, S.L.; Barbagallo, F.; Campolo, F.; Dolci, S.; Rossi, P.; Venneri, M.A.; Giannetta, E.; Gianfrilli, D.; et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis. 2016, 7, e2317. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.M.; Cottage, C.T.; Konstandin, M.H.; Völkers, M.; Khan, M.; Sussman, M.A. Pim-1 kinase inhibits pathological injury by promoting cardioprotective signaling. J. Mol. Cell. Cardiol. 2011, 51, 554–558. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Wang, B.J.; Broughton, K.M.; Alvarez, R.; Siddiqi, S.; Loaiza, R.; Nguyen, N.; Quijada, P.; Gude, N.; Sussman, M.A. PIM1-minicircle as a therapeutic treatment for myocardial infarction. PLoS ONE 2017, 12, e0173963. [Google Scholar] [CrossRef]
- Mohsin, S.; Khan, M.; Toko, H.; Bailey, B.; Cottage, C.T.; Wallach, K.; Nag, D.; Lee, A.; Siddiqi, S.; Lan, F.; et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J. Am. Coll. Cardiol. 2012, 60, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Vajravelu, B.N.; Hong, K.U.; Al-Maqtari, T.; Cao, P.; Keith, M.C.L.; Wysoczynski, M.; Zhao, J.; Moore, J.B.; Bolli, R. C-Kit promotes growth and migration of human cardiac progenitor cells via the PI3KAKT and MEK-ERK Pathways. PLoS ONE 2015, 10, e0140798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebeid, D.E.; Khalafalla, F.G.; Broughton, K.M.; Monsanto, M.M.; Esquer, C.Y.; Sacchi, V.; Hariharan, N.; Korski, K.I.; Moshref, M.; Emathinger, J.; et al. Pim1 maintains telomere length in mouse cardiomyocytes by inhibiting TGFβ signalling. Cardiovasc. Res. 2020, 116, cvaa066. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, S.; Khan, M.; Nguyen, J.; Alkatib, M.; Siddiqi, S.; Hariharan, N.; Wallach, K.; Monsanto, M.; Gude, N.; Dembitsky, W.; et al. Rejuvenation of human cardiac progenitor cells with pim-1 kinase. Circ. Res. 2013, 113, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quijada, P.; Toko, H.; Fischer, K.M.; Bailey, B.; Reilly, P.; Hunt, K.D.; Gude, N.A.; Avitabile, D.; Sussman, M.A. Preservation of myocardial structure is enhanced by pim-1 engineering of bone marrow cells. Circ. Res. 2012, 111, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Yan, G.; Xu, H.; He, W.; Liu, Z.; Ma, G. Hypoxic preconditioning increases survival of cardiac progenitor cells via the pim-1 kinase-mediated anti-apoptotic effect. Circ. J. 2014, 78, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Katare, R.; Caporali, A.; Zentilin, L.; Avolio, E.; Sala-Newby, G.; Oikawa, A.; Cesselli, D.; Beltrami, A.P.; Giacca, M.; Emanueli, C.; et al. Intravenous gene therapy with PIM-1 via a cardiotropic viral vector halts the progression of diabetic cardiomyopathy through promotion of prosurvival signaling. Circ. Res. 2011, 108, 1238–1251. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Cianflone, E.; Mancuso, T.; Aquila, I.; Agosti, V.; Torella, M.; Paolino, D.; Mollace, V.; Nadal-Ginard, B.; et al. Role of c-Kit in Myocardial Regeneration and Aging. Front. Endocrinol. 2019, 10, 371–385. [Google Scholar] [CrossRef]
- Gude, N.A.; Sussman, M.A. Chasing c-Kit through the heart: Taking a broader view. Pharmacol. Res. 2018, 127, 110–115. [Google Scholar] [CrossRef]
- Li, M.; Naqvi, N.; Yahiro, E.; Liu, K.; Powell, P.C.; Bradley, W.E.; Martin, D.I.K.; Graham, R.M.; Dell’Italia, L.J.; Husain, A. c-kit is required for cardiomyocyte terminal differentiation. Circ. Res. 2008, 102, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Hammerman, P.S.; Fox, C.J.; Birnbaum, M.J.; Thompson, C.B. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005, 105, 4477–4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, T.J.; Zhao, S.; Geiger, J.N.; Joneja, B.; Wojchowski, D.M. Pim-1 kinase protects hematopoietic FDC cells from genotoxin-induced death. Oncogene 2000, 19, 3684–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blume-Jensen, P.; Janknecht, R.; Hunter, T. The Kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. 2004, 8, 779–785. [Google Scholar] [CrossRef]
- Toko, H.; Konstandin, M.H.; Doroudgar, S.; Ormachea, L.; Joyo, E.; Joyo, A.Y.; Din, S.; Gude, N.A.; Collins, B.; Völkers, M.; et al. Regulation of cardiac hypertrophic signaling by prolyl isomerase Pin1. Circ. Res. 2013, 112, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Patten, R.D.; Force, T.; Kyriakis, J.M. Gene 33/RALT Is Induced by Hypoxia in Cardiomyocytes, Where It Promotes Cell Death by Suppressing Phosphatidylinositol 3-Kinase and Extracellular Signal-Regulated Kinase Survival Signaling. Mol. Cell. Biol. 2006, 26, 5043–5054. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Tokudome, T.; Horio, T.; Fukunaga, M.; Okumura, H.; Hino, J.; Mori, K.; Yoshihara, F.; Suga, S.I.; Kawano, Y.; Kohno, M.; et al. Ventricular nonmyocytes inhibit doxorubicin-induced myocyte apoptosis: Involvement of endogenous endothelin-1 as a paracrine factor. Endocrinology 2004, 145, 2458–2466. [Google Scholar] [CrossRef] [Green Version]
- Kühn, B.; Del Monte, F.; Hajjar, R.J.; Chang, Y.S.; Lebeche, D.; Arab, S.; Keating, M.T. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 2007, 13, 962–969. [Google Scholar] [CrossRef]
- Ni, Y.G.; Wang, N.; Cao, D.J.; Sachan, N.; Morris, D.J.; Gerard, R.D.; Kuro-o, M.; Rothermel, B.A.; Hill, J.A. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. USA 2007, 104, 20517–20522. [Google Scholar] [CrossRef] [Green Version]
- Bueno, O.F.; Molkentin, J.D. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ. Res. 2002, 91, 776–781. [Google Scholar] [CrossRef]
- Ulm, S.; Liu, W.; Zi, M.; Tsui, H.; Chowdhury, S.K.; Endo, S.; Satoh, Y.; Prehar, S.; Wang, R.; Cartwright, E.J.; et al. Targeted deletion of ERK2 in cardiomyocytes attenuates hypertrophic response but provokes pathological stress induced cardiac dysfunction. J. Mol. Cell. Cardiol. 2014, 72, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Nihira, K.; Ando, Y.; Yamaguchi, T.; Kagami, Y.; Miki, Y.; Yoshida, K. Pim-1 controls NF-B signalling by stabilizing RelA/p65. Cell Death Differ. 2010, 17, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samse, K.; Emathinger, J.; Hariharan, N.; Quijada, P.; Ilves, K.; Völkers, M.; Ormachea, L.; De La Torre, A.; Orogo, A.M.; Alvarez, R.; et al. Functional effect of Pim1 depends upon intracellular localization in human cardiac progenitor cells. J. Biol. Chem. 2015, 290, 13935–13947. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Catalog Number | Dilution | Application |
|---|---|---|---|
| PIM1 | ThermoFisher 39-4600 | 1:500 | IB, Co-IP |
| PIM1 | ThermoFisher 710504 | 1:100 | PLA |
| c-Kit | R&D AF1356 | 1:200 1:100 | IB, PLA |
| GAPDH (Glyceraldehyde 3-phosphate) dehydrogenase | Millipore Sigma MAB374 | 1:5000 | IB |
| SRC | Abcam Ab47405 | 1:500 | IB |
| pERK1/2 Phosphorylated ERK1/2 | CST 9101 | 1:500 | IB |
| ERK1/2 | CST 9102 | 1:500 | IB |
| pAkt (Ser473) Phosphorylated Akt at serine 473 | CST 9271 | 1:500 | IB |
| Akt | CST 9272 | 1:500 | IB |
| GFP (Green Fluorescent Protein) | Rockland 600-101-215 | 1:1000 | IB |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebeid, D.E.; Firouzi, F.; Esquer, C.Y.; Navarrete, J.M.; Wang, B.J.; Gude, N.A.; Sussman, M.A. PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression. Cells 2020, 9, 2001. https://doi.org/10.3390/cells9092001
Ebeid DE, Firouzi F, Esquer CY, Navarrete JM, Wang BJ, Gude NA, Sussman MA. PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression. Cells. 2020; 9(9):2001. https://doi.org/10.3390/cells9092001
Chicago/Turabian StyleEbeid, David E., Fareheh Firouzi, Carolina Y. Esquer, Julian M. Navarrete, Bingyan J. Wang, Natalie A. Gude, and Mark A. Sussman. 2020. "PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression" Cells 9, no. 9: 2001. https://doi.org/10.3390/cells9092001
APA StyleEbeid, D. E., Firouzi, F., Esquer, C. Y., Navarrete, J. M., Wang, B. J., Gude, N. A., & Sussman, M. A. (2020). PIM1 Promotes Survival of Cardiomyocytes by Upregulating c-Kit Protein Expression. Cells, 9(9), 2001. https://doi.org/10.3390/cells9092001