Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Lipid Profiling
2.3. RNA-Sequencing Analysis
2.4. Compilation of Transcriptomic Studies
2.5. Gene Set Enrichment Analysis
2.6. Experimental Design and Statistical Analyses
3. Results
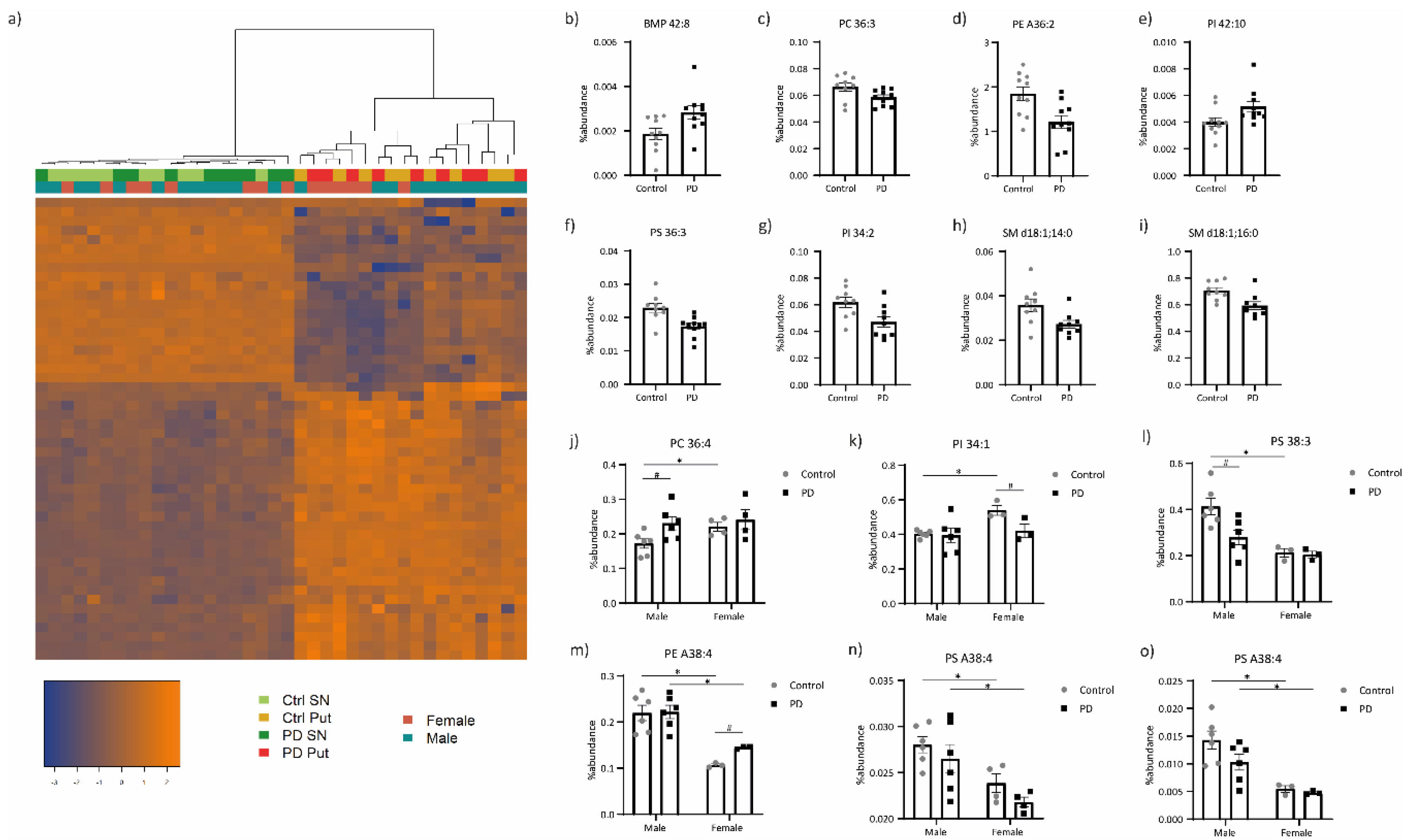
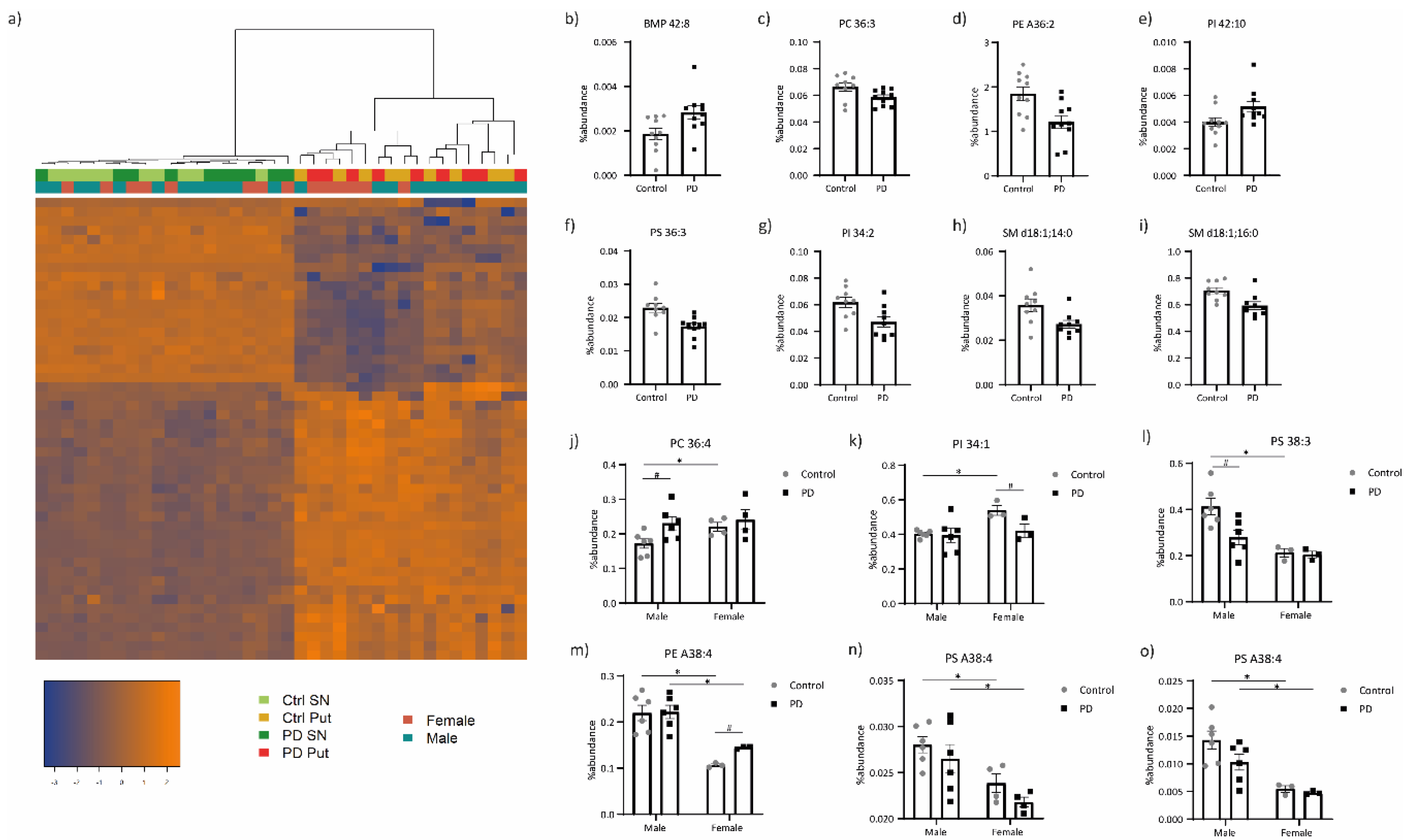
3.1. Lipid Profiling
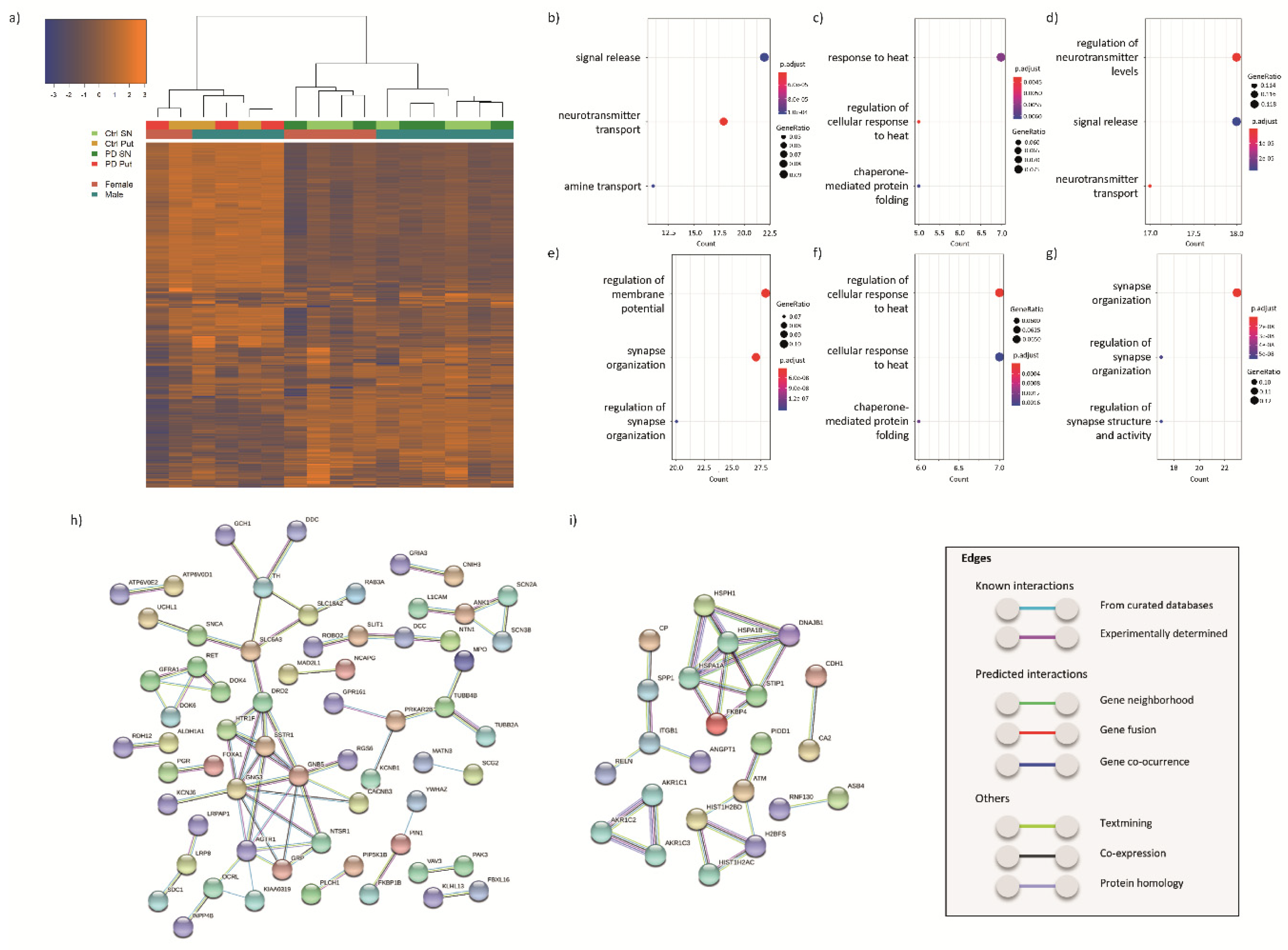
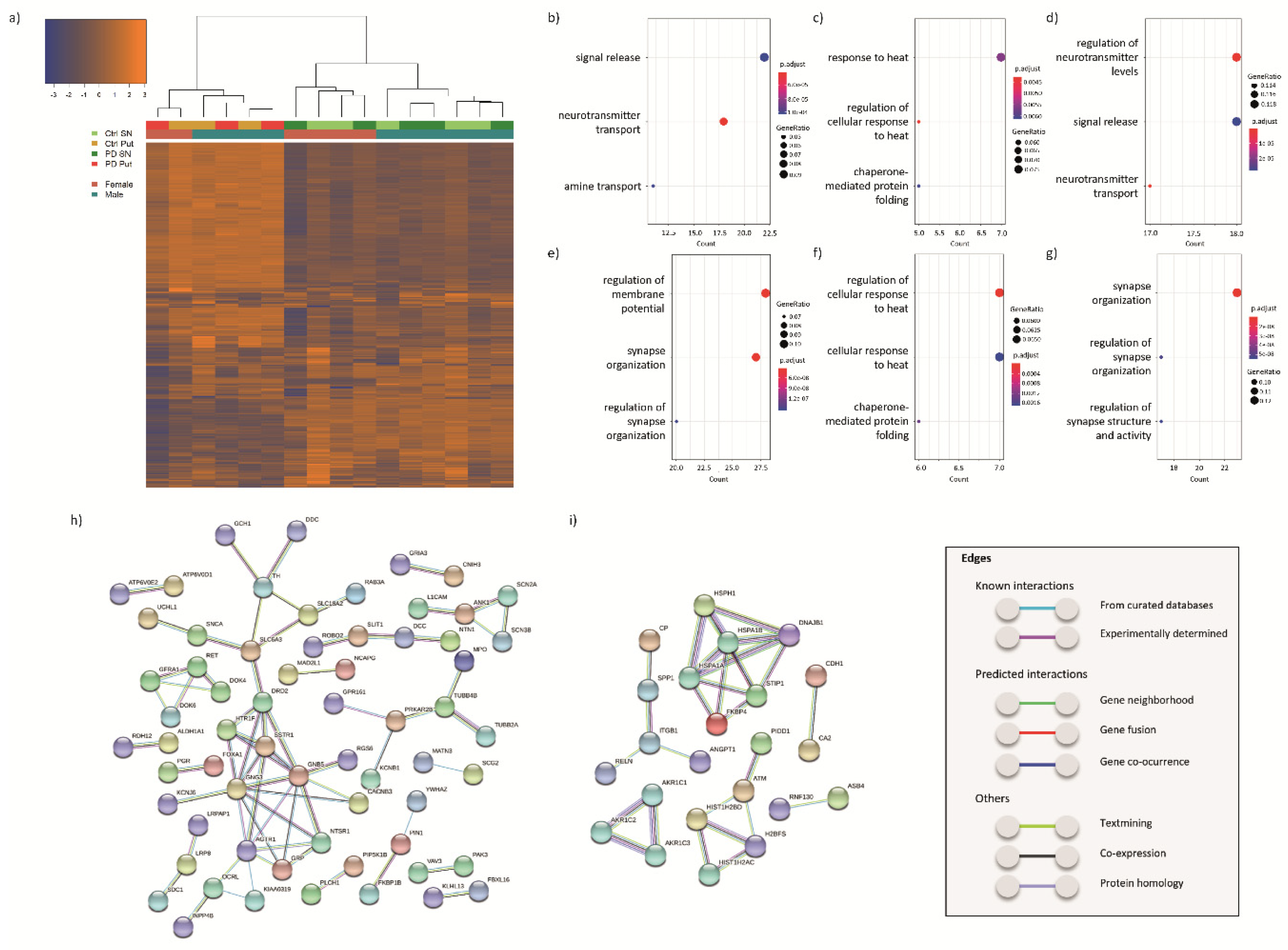
3.2. Transcriptomic Profiling
3.2.1. DEGs in the SN of PD Patients and Controls
3.2.2. DEGs in the Putamen of PD Patients and Controls
3.2.3. DEGs Common between SN and Putamen
3.3. Integration of the Lipid and Transcriptomic Profiling Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Lau, L.M.L.; Breteler, M.M.B. Epidemiology of Parkinson’s disease. Lancet. Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Xia, R.; Mao, Z.-H. Progression of motor symptoms in Parkinson’s disease. Neurosci. Bull. 2012, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Schapira, A.H. V Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Vila, M.; Przedborski, S. Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 365–375. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30. [Google Scholar] [CrossRef]
- Kumaran, R.; Cookson, M.R. Pathways to Parkinsonism Redux: Convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum. Mol. Genet. 2015, 24. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Bubacco, L.; Greggio, E.; Civiero, L. Ceramides in Parkinson’s Disease: From Recent Evidence to New Hypotheses. Front. Neurosci. 2019, 13, 330. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.L.; Lawson, V.A.; Finkelstein, D.I.; Hill, A.F. The role of lipids in α-synuclein misfolding and neurotoxicity. J. Biol. Chem. 2019, 294, 9016–9028. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lass, A.; Zimmermann, R.; Oberer, M.; Zechner, R. Lipolysis—A highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog. Lipid Res. 2011, 50, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Fernandis, A.Z.; Wenk, M.R. Membrane lipids as signaling molecules. Curr. Opin. Lipidol. 2007, 18, 121–128. [Google Scholar] [CrossRef]
- Bieberich, E. It’s a lipid’s world: Bioactive lipid metabolism and signaling in neural stem cell differentiation. Neurochem. Res. 2012, 37, 1208–1229. [Google Scholar] [CrossRef] [Green Version]
- Holthuis, J.C.M.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Fink, A.L. Lipid Binding Inhibits α-Synuclein Fibril Formation. J. Biol. Chem. 2003, 278, 16873–16877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivatt, R.M.; Whitworth, A.J. SREBF1 links lipogenesis to mitophagy and sporadic Parkinson disease. Autophagy 2014, 10, 1476–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454. [Google Scholar] [CrossRef] [PubMed]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Borrageiro, G.; Haylett, W.; Seedat, S.; Kuivaniemi, H.; Bardien, S. A review of genome-wide transcriptomics studies in Parkinson’s disease. Eur. J. Neurosci. 2018, 47, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mantione, K.J.; Kream, R.M.; Kuzelova, H.; Ptacek, R.; Raboch, J.; Samuel, J.M.; Stefano, G.B. Comparing bioinformatic gene expression profiling methods: Microarray and RNA-Seq. Med. Sci. Monit. Basic Res. 2014, 20, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Fung-Leung, W.-P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Thoma, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef] [Green Version]
- Xicoy, H.; Brouwers, J.F.; Kalnytska, O.; Wieringa, B.; Martens, G.J.M. Lipid Analysis of the 6-Hydroxydopamine-Treated SH-SY5Y Cell Model for Parkinson’s Disease. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Jeucken, A.; Brouwers, J. High-Throughput Screening of Lipidomic Adaptations in Cultured Cells. Biomolecules 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Jeucken, A.; Molenaar, M.R.; van de Lest, C.H.A.; Jansen, J.W.A.; Helms, J.B.; Brouwers, J.F. A Comprehensive Functional Characterization of Escherichia coli Lipid Genes. Cell Rep. 2019, 27, 1597–1606.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Lim, E.; Vaillant, F.; Asselin-Labat, M.-L.; Visvader, J.E.; Smyth, G.K. ROAST: Rotation gene set tests for complex microarray experiments. Bioinformatics 2010, 26, 2176–2182. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S.; Lomax, J.; Mungall, C.; Hitz, B.; Balakrishnan, R.; et al. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database J. Biol. Databases Curation 2016, 2016, baw100. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Andersen, S.L.; Briggs, F.B.S.; Winnike, J.H.; Natanzon, Y.; Maichle, S.; Knagge, K.J.; Newby, L.K.; Gregory, S.G. Metabolome-based signature of disease pathology in MS. Mult. Scler. Relat. Disord. 2019, 31, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Rothman, K.J. No adjustments are needed for multiple comparisons. Epidemiology 1990, 1, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–862. [Google Scholar] [CrossRef] [Green Version]
- Fabelo, N.; Martin, V.; González, C.; Alonso, A.; Diaz, M. Effects of oestradiol on brain lipid class and Fatty Acid composition: Comparison between pregnant and ovariectomised oestradiol-treated rats. J. Neuroendocrinol. 2012, 24, 292–309. [Google Scholar] [CrossRef]
- Riley, B.E.; Gardai, S.J.; Emig-Agius, D.; Bessarabova, M.; Ivliev, A.E.; Schüle, B.; Schüle, B.; Alexander, J.; Wallace, W.; Halliday, G.M.; et al. Systems-based analyses of brain regions functionally impacted in Parkinson’s disease reveals underlying causal mechanisms. PLoS ONE 2014, 9, e102909. [Google Scholar] [CrossRef]
- Cheng, D.; Jenner, A.M.; Shui, G.; Cheong, W.F.; Mitchell, T.W.; Nealon, J.R.; Kim, W.S.; McCann, H.; Wenk, M.R.; Halliday, G.M.; et al. Lipid pathway alterations in Parkinson’s disease primary visual cortex. PLoS ONE 2011, 6, e17299. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef] [Green Version]
- Taki, T. An approach to glycobiology from glycolipidomics: Ganglioside molecular scanning in the brains of patients with Alzheimer’s disease by TLC-blot/matrix assisted laser desorption/ionization-time of flight MS. Biol. Pharm. Bull. 2012, 35, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. V No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Riekkinen, P.; Rinne, U.K.; Pelliniemi, T.T.; Sonninen, V. Interaction between dopamine and phospholipids. Studies of the substantia nigra in Parkinson disease patients. Arch. Neurol. 1975, 32, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberger, T.A.; Villacreses, N.E.; Hovda, J.T.; Bosetti, F.; Weerasinghe, G.; Wine, R.N.; Harry, G.J.; Rapoport, S.I. Rat brain arachidonic acid metabolism is increased by a 6-day intracerebral ventricular infusion of bacterial lipopolysaccharide. J. Neurochem. 2004, 88, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.; Pawlosky, R.; Wegher, B.; Hibbeln, J. In vivo conversion of linoleic acid to arachidonic acid in human adults. Prostaglandins Leukot. Essent. Fatty Acids 1999, 60, 407–410. [Google Scholar] [CrossRef]
- Kobayashi, T.; Startchev, K.; Whitney, A.J.; Gruenber, J. Localization of lysobisphosphatidic acid-rich membrane domains in late endosomes. Biol. Chem. 2001, 382, 483–485. [Google Scholar] [CrossRef]
- Linke, T.; Wilkening, G.; Lansmann, S.; Moczall, H.; Bartelsen, O.; Weisgerber, J.; Sandhoff, K. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol. Chem. 2001, 382, 283–290. [Google Scholar] [CrossRef]
- Hein, L.K.; Duplock, S.; Fuller, M. Selective reduction of bis(monoacylglycero)phosphate ameliorates the storage burden in a THP-1 macrophage model of Gaucher disease. J. Lipid Res. 2013, 54, 1691–1697. [Google Scholar] [CrossRef] [Green Version]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Picillo, M.; Nicoletti, A.; Fetoni, V.; Garavaglia, B.; Barone, P.; Pellecchia, M.T. The relevance of gender in Parkinson’s disease: A review. J. Neurol. 2017, 264, 1583–1607. [Google Scholar] [CrossRef]
- Haaxma, C.A.; Bloem, B.R.; Borm, G.F.; Oyen, W.J.G.; Leenders, K.L.; Eshuis, S.; Booij, J.; Dluzen, D.E.; Horstink, M.W.I.M. Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.N.; Cronin-Golomb, A. Gender Differences in Parkinson’s Disease: Clinical Characteristics and Cognition. Mov. Disord. 2010, 25, 2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiev, D.; Hamberg, K.; Hariz, M.; Forsgren, L.; Hariz, G.M. Gender Differences in Parkinson’s Disease: A Clinical Perspective; Blackwell Publishing Ltd.: Oxford, UK, 2017; Volume 136, pp. 570–584. [Google Scholar]
- Shulman, L.M. Gender differences in Parkinson’s disease. Gend. Med. 2007, 4, 8–18. [Google Scholar] [CrossRef]
- Morselli, E.; de Souza Santos, R.; Gao, S.; Ávalos, Y.; Criollo, A.; Palmer, B.F.; Clegg, D.J. Impact of estrogens and estrogen receptor-α in brain lipid metabolism. Am. J. Physiol. Metab. 2018, 315, E7–E14. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Navas, C.; Morselli, E.; Clegg, D.J. Sexually dimorphic brain fatty acid composition in low and high fat diet-fed mice. Mol. Metab. 2016, 5, 680–689. [Google Scholar] [CrossRef]
- Grison, A.; Zucchelli, S.; Urzì, A.; Zamparo, I.; Lazarevic, D.; Pascarella, G.; Roncaglia, P.; Giorgetti, A.; Garcia-Esparcia, P.; Vlachouli, C.; et al. Mesencephalic dopaminergic neurons express a repertoire of olfactory receptors and respond to odorant-like molecules. BMC Genom. 2014, 15, 729. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Esparcia, P.; Schlüter, A.; Carmona, M.; Moreno, J.; Ansoleaga, B.; Torrejón-Escribano, B.; Gustincich, S.; Pujol, A.; Ferrer, I. Functional genomics reveals dysregulation of cortical olfactory receptors in Parkinson disease: Novel putative chemoreceptors in the human brain. J. Neuropathol. Exp. Neurol. 2013, 72, 524–539. [Google Scholar] [CrossRef] [Green Version]
- Bodzioch, M.; Lapicka-Bodzioch, K.; Zapala, B.; Kamysz, W.; Kiec-Wilk, B.; Dembinska-Kiec, A. Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 2009, 94, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Mariani, E.; Frabetti, F.; Tarozzi, A.; Pelleri, M.C.; Pizzetti, F.; Casadei, R. Meta-Analysis of Parkinson’s Disease Transcriptome Data Using TRAM Software: Whole Substantia Nigra Tissue and Single Dopamine Neuron Differential Gene Expression. PLoS ONE 2016, 11, e0161567. [Google Scholar] [CrossRef]
- Dong, J.-T.; Chen, C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell. Mol. Life Sci. 2009, 66, 2691–2706. [Google Scholar] [CrossRef]
- Politis, M.; Niccolini, F. Serotonin in Parkinson’s disease. Behav. Brain Res. 2015, 277, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Benfante, R.; Asselta, R.; Marabini, L.; Cereda, E.; Siri, C.; Pezzoli, G.; Goldwurm, S.; Fornasari, D. Tryptophan hydroxylase type 2 variants modulate severity and outcome of addictive behaviors in Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 29, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yang, X.; Zhao, Q.; Tian, S.; Huang, H.; Chen, Y.; Xu, Y. Association between gene polymorphism and depression in Parkinson’s disease: A case-control study. J. Neurol. Sci. 2017, 375, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Weng, R.; Shen, S.; Burton, C.; Yang, L.; Nie, H.; Tian, Y.; Bai, Y.; Liu, H. Lipidomic profiling of tryptophan hydroxylase 2 knockout mice reveals novel lipid biomarkers associated with serotonin deficiency. Anal. Bioanal. Chem. 2016, 408, 2963–2973. [Google Scholar] [CrossRef]
- Haney, M.M.; Sinnott, J.; Osman, K.L.; Deninger, I.; Andel, E.; Caywood, V.; Mok, A.; Ballenger, B.; Cummings, K.; Thombs, L.; et al. Mice Lacking Brain-Derived Serotonin Have Altered Swallowing Function. Otolaryngol. Head. Neck Surg. 2019, 161, 468–471. [Google Scholar] [CrossRef]
- Szot, P.; White, S.S.; Greenup, J.L.; Leverenz, J.B.; Peskind, E.R.; Raskind, M.A. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J. Neurosci. 2006, 26, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Sommerauer, M.; Fedorova, T.D.; Hansen, A.K.; Knudsen, K.; Otto, M.; Jeppesen, J.; Frederiksen, Y.; Blicher, J.U.; Geday, J.; Nahimi, A.; et al. Evaluation of the noradrenergic system in Parkinson’s disease: An 11C-MeNER PET and neuromelanin MRI study. Brain 2018, 141, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1β, IL-2, IL-4, IL-6 and transforming growth factor-α levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef]
- Saura, J.; Parés, M.; Bové, J.; Pezzi, S.; Alberch, J.; Marin, C.; Tolosa, E.; Martí, M.J. Intranigral infusion of interleukin-1beta activates astrocytes and protects from subsequent 6-hydroxydopamine neurotoxicity. J. Neurochem. 2003, 85, 651–661. [Google Scholar] [CrossRef]
- Shulskaya, M.V.; Alieva, A.K.; Vlasov, I.N.; Zyrin, V.V.; Fedotova, E.Y.; Abramycheva, N.Y.; Usenko, T.S.; Yakimovsky, A.F.; Emelyanov, A.K.; Pchelina, S.N.; et al. Whole-Exome Sequencing in Searching for New Variants Associated With the Development of Parkinson’s Disease. Front. Aging Neurosci. 2018, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.R.; Liss, B. GDNF-Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease. FEBS Lett. 2015, 589, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Drinkut, A.; Tillack, K.; Meka, D.P.; Schulz, J.B.; Kügler, S.; Kramer, E.R. Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis. 2016, 7, e2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.M.M.; Wong, E.S.P.; Lim, K.-L. Protein Misfolding and Aggregation in Parkinson’s Disease. Antioxid. Redox Signal. 2009, 11, 2119–2134. [Google Scholar] [CrossRef]
- Sakharkar, M.K.; Singh, S.K.K.; Rajamanickam, K.; Essa, M.M.; Yang, J.; Chidambaram, S.B.; Kashmir Singh, S.K.; Rajamanickam, K.; Mohamed Essa, M.; Yang, J.; et al. A systems biology approach towards the identification of candidate therapeutic genes and potential biomarkers for Parkinson’s disease. PLoS ONE 2019, 14, e0220995. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Nagele, E.; DeMarshall, C.; Acharya, N.; Nagele, R. Diagnosis of Parkinson’s disease based on disease-specific autoantibody profiles in human sera. PLoS ONE 2012, 7, e32383. [Google Scholar] [CrossRef] [Green Version]
- Neuner, S.M.; Garfinkel, B.P.; Wilmott, L.A.; Ignatowska-Jankowska, B.M.; Citri, A.; Orly, J.; Lu, L.; Overall, R.W.; Mulligan, M.K.; Kempermann, G.; et al. Systems genetics identifies Hp1bp3 as a novel modulator of cognitive aging. Neurobiol. Aging 2016, 46, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, D.R.; Vallortigara, J.; Alghamdi, A.; Howlett, D.; Hortobágyi, T.; Johnson, M.; Attems, J.; Newhouse, S.; Ballard, C.; Thomas, A.J.; et al. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer’s disease: Association with cognitive impairment. Neurobiol. Aging 2014, 35, 2836–2844. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, D.R.; Vallortigara, J.; Alghamdi, A.; Hortobágyi, T.; Ballard, C.; Thomas, A.J.; O’Brien, J.T.; Aarsland, D.; Francis, P.T. Depression and synaptic zinc regulation in Alzheimer disease, dementia with lewy bodies, and Parkinson disease dementia. Am. J. Geriatr. Psychiatry 2015, 23, 141–148. [Google Scholar] [CrossRef]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef] [Green Version]
- Sheline, C.T.; Zhu, J.; Zhang, W.; Shi, C.; Cai, A.-L. Mitochondrial inhibitor models of Huntington’s disease and Parkinson’s disease induce zinc accumulation and are attenuated by inhibition of zinc neurotoxicity in vitro or in vivo. Neurodegener. Dis. 2013, 11, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, N.; Xiong, N.; You, Q.; Li, J.; Yu, J.; Qing, H.; Wang, T.; Cordell, H.J.; Isacson, O.; et al. Genetic Variants of Microtubule Actin Cross-linking Factor 1 (MACF1) Confer Risk for Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 2878–2888. [Google Scholar] [CrossRef] [PubMed]
- Muddapu, V.R.; Mandali, A.; Chakravarthy, V.S.; Ramaswamy, S. A Computational Model of Loss of Dopaminergic Cells in Parkinson’s Disease Due to Glutamate-Induced Excitotoxicity. Front. Neural Circuits 2019, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, T.K.; Messenger, M.J.; Ward, M.A.; Woodhouse, S.; Osborne, D.J.; Duty, S.; O’Neill, M.J. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson’s disease. Pharmacol. Biochem. Behav. 2002, 73, 455–466. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, R.I.; Robertson, S.A.; Kastner, D.L. Neurodegenerative diseases have genetic hallmarks of autoinflammatory disease. Hum. Mol. Genet. 2018, 27, R108–R118. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Rockenstein, E.; Mante, M.; Mallory, M.; Masliah, E. beta-Synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 2001, 32, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Angelova, D.M.; Jones, H.B.L.; Brown, D.R. Levels of α- and β-synuclein regulate cellular susceptibility to toxicity from α-synuclein oligomers. FASEB J. 2018, 32, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Snyder, H.; Mensah, K.; Hsu, C.; Hashimoto, M.; Surgucheva, I.G.; Festoff, B.; Surguchov, A.; Masliah, E.; Matouschek, A.; Wolozin, B. beta-Synuclein reduces proteasomal inhibition by alpha-synuclein but not gamma-synuclein. J. Biol. Chem. 2005, 280, 7562–7569. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilman, S.; Low, P.A.; Quinn, N.; Albanese, A.; Ben-Shlomo, Y.; Fowler, C.J.; Kaufmann, H.; Klockgether, T.; Lang, A.E.; Lantos, P.L.; et al. Consensus statement on the diagnosis of multiple system atrophy. J. Neurol. Sci. 1999, 163, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.D.; Hess, W.A.; Calne, D.B. Olfactory impairment in Parkinson’s disease. Neurology 1983, 33, 943. [Google Scholar] [CrossRef]
- Ferrazza, R.; Cogo, S.; Melrose, H.; Bubacco, L.; Greggio, E.; Guella, G.; Civiero, L.; Plotegher, N. LRRK2 deficiency impacts ceramide metabolism in brain. Biochem. Biophys. Res. Commun. 2016, 478. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ravipati, A.; Nimmagadda, V.; Schubert, M.; Castellani, R.J.; Russell, J.W. Potential roles of PINK1 for increased PGC-1α-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 2014, 18, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, S.; Yamane, T.; Takahashi-Niki, K.; Kato, I.; Niki, T.; Goldberg, M.S.; Shen, J.; Ishimoto, K.; Doi, T.; Iguchi-Ariga, S.M.M.; et al. Transcriptional activation of low-density lipoprotein receptor gene by DJ-1 and effect of DJ-1 on cholesterol homeostasis. PLoS ONE 2012, 7, e38144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcos, A.L.; Corradi, G.R.; Mazzitelli, L.R.; Casali, C.I.; del Carmen Fernández Tome, M.; Adamo, H.P.; de Tezanos Pinto, F. The Parkinson-associated human P5B-ATPase ATP13A2 modifies lipid homeostasis. Biochim. Biophys. Acta Biomembr. 2019, 1861, 182993. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 leads to elevated mitochondrial lipid peroxidation and mitochondrial dysfunction. Brain 2015, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meierhofer, D.; Halbach, M.; Şen, N.E.; Gispert, S.; Auburger, G. Ataxin-2 (Atxn2)-Knock-Out Mice Show Branched Chain Amino Acids and Fatty Acids Pathway Alterations. Mol. Cell. Proteom. 2016, 15, 1728–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Diagnosis | Age | Gender | PMD | pH CSF | Braak Stage | Cause of Death |
|---|---|---|---|---|---|---|
| Control | 78 | M | 8:25 | 6.56 | 0 | Cardiac arrhythmia |
| Control | 69 | F | 8:30 | 0 | Myocardial infarction | |
| Control | 69 | F | 6:15 | 6.59 | 0 | Cardiogenic shock |
| Control | 84 | M | 7:05 | 5.90 | 0 | Exacerbation of COPD |
| Control | 73 | F | 6:40 | 0 | Respiratory failure | |
| Control | 84 | M | 5:35 | 6.98 | 0 | Heart failure |
| Control | 82 | M | 5:10 | 6.75 | 0 | Pneumonia |
| Control | 85 | F | 7:05 | 0 | Renal insufficiency | |
| Control | 79 | M | 5.45 | 6.38 | 0 | Euthanasia |
| Control | 80 | M | 4:25 | 6.59 | 0 | Euthanasia |
| PD | 86 | F | 4:08 | 6.32 | 5 | Cachexia and dehydration |
| PD | 86 | M | 7:25 | 6.26 | 4 | Cardiac arrest |
| PD | 74 | M | 4:35 | 6.58 | 6 | Respiratory insufficiency |
| PD | 68 | F | 4:05 | 6 | Euthanasia | |
| PD | 77 | M | 3:10 | 6.28 | 6 | Aspiration pneumonia |
| PD | 86 | M | 4:10 | 6.91 | 6 | Euthanasia |
| PD | 84 | M | 4:50 | 6.41 | 3 | Cachexia |
| PD | 76 | M | 9:15 | 6.33 | 6 | Ileus |
| PD | 77 | F | 6:05 | 3.20 | 5 | Stroke |
| PD | 65 | F | 7:35 | 6.55 | 6 | Cachexia and dehydration |
| Accession Number | Tissue | Control | PD Patient | Platform | Inclusion |
|---|---|---|---|---|---|
| GSE7621 | SN | 9 | 16 | [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | Yes |
| GSE42966 | SN | 6 | 9 | Agilent-014850 Whole Human Genome Microarray 4x44K G4112F | Yes |
| GSE43490 | SN | 5 | 8 | Agilent-014850 Whole Human Genome Microarray 4x44K G4112F | No (only overexpressed genes) |
| GSE49036 | SN | 8 | 15 | [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | Yes |
| GSE20164 | SN | 5 | 6 | [HG-U133A] Affymetrix Human Genome U133A Array | Yes |
| GSE20163 | SN | 9 | 8 | [HG-U133A] Affymetrix Human Genome U133A Array | Yes |
| GSE20292 | SN | 18 | 11 | [HG-U133A] Affymetrix Human Genome U133A Array | No (no DEGs) |
| GSE20333 | SN | 6 | 6 | [HG-Focus] Affymetrix Human HG-Focus Target Array | Yes |
| GSE8397 | SNm | 7 | 9 | [HG-U133A] Affymetrix Human Genome U133A Array; [HG-U133B] Affymetrix Human Genome U133B Array | No (two different SN tissues (medial and lateral)) |
| SNl | 8 | 15 | |||
| GSE54282 | SN | 3 | 3 | [HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array [HuGene10stv1_Hs_ENTREZG_15.0.0] | Yes |
| Put | 6 | 6 | Yes | ||
| GSE77666 | Put | 12 | 12 | NanoString nCounter gene expression system | Yes |
| GSE23290 | Put | 5 | 5 | [HuEx-1_0-st] Affymetrix Human Exon 1.0 ST Array | Yes |
| GSE20291 | Put | 20 | 15 | [HG-U133A] Affymetrix Human Genome U133A Array | No (no DEGs) |
| GSE7621 | GSE42966 | GSE49036 | GSE20164 | GSE54282 | GSE20163 | GSE20333 | Our Data | |
|---|---|---|---|---|---|---|---|---|
| DEGs | 932 | 64 | 872 | 242 | 24 | 432 | 133 | 304 |
| Overlap | 223 (24%) | 18 (28%) | 229 (26%) | 77 (32%) | 2 (8%) | 114 (26%) | 19 (14%) | 72 (24%) |
| Same direction | 206 (92%) | 14 (78%) | 218 (95%) | 71 (92%) | 1 (50%) | 103 (90%) | 12 (63%) | 66 (92%) |
| GSE54282 | GSE77666 | GSE23290 | Our Data | |
|---|---|---|---|---|
| DEGs | 56 | 4 | 2481 | 232 |
| Overlap | 7 (12%) | 3 (75%) | 35 (1.4%) | 25 (11%) |
| Same direction | 7 (100%) | 1 (33%) | 21 (60%) | 13 (52%) |
| ENSEMBL | Gene Symbol | Protein Name | Protein Function |
|---|---|---|---|
| Upregulated | |||
| ENSG00000089041 | P2RX7 | P2X purinoceptor 7 | ATP receptor that acts as a ligand-gated ion channel |
| ENSG00000119699 | TGFB3 | Transforming growth factor beta-3 proprotein | Embryogenesis and cell differentiation |
| ENSG00000120729 | MYOT | Myotilin | Myofibril assembly and stability |
| ENSG00000125551 | PLGLB2 | Plasminogen-like protein B2 | Unknown |
| ENSG00000127530 | OR7C1 | Olfactory receptor 7C1 | Odorant receptor |
| ENSG00000159842 | ABR | Active breakpoint cluster region-related protein | GTPase-activating protein for RAC and CDC42 |
| ENSG00000168209 | DDIT4 | DNA damage-inducible transcript 4 protein | Regulates cell growth, proliferation and survival in response to cellular energy levels and cellular stress |
| ENSG00000180015 | NA | ||
| ENSG00000186352 | ANKRD37 | Ankyrin repeat domain-containing protein 37 | Unknown |
| ENSG00000188269 | OR7A5 | Olfactory receptor 7A5 | Odorant receptor |
| ENSG00000214313 | AZGP1P1 | Pseudogene | |
| ENSG00000234769 | NA | ||
| ENSG00000244921 | NA | ||
| ENSG00000255823 | MTRNR2L8 | Humanin-like 8 | Unknown |
| ENSG00000272755 | NA | ||
| Downregulated | |||
| ENSG00000074317 | SNCB | Beta-synuclein | Regulator of SNCA |
| ENSG00000078401 | EDN1 | Endothelin-1 | Vasoconstrictor peptide |
| ENSG00000099957 | P2RX6 | P2X purinoceptor 6 | ATP receptor that acts as a ligand-gated ion channel |
| ENSG00000103199 | ZNF500 | Zinc finger protein 500 | Transcriptional regulation |
| ENSG00000137267 | TUBB2A | Tubulin beta-2A chain | Major constituent of microtubules |
| ENSG00000148803 | FUOM | Fucose mutarotase | Interconversion between alpha- and beta-L-fructose |
| ENSG00000155367 | PPM1J | Protein phosphatase 1J | Serine/threonine protein phosphatase |
| ENSG00000164082 | GRM2 | Metabotropic glutamate receptor 2 | Glutamate receptor |
| ENSG00000167306 | MYO5B | Unconventional myosin-Vb | Vesicular trafficking |
| ENSG00000171532 | NEUROD2 | Neurogenic differentiation factor 2 | Transcriptional regulation, implicated in neuronal determination |
| ENSG00000172794 | RAB37 | Ras-related protein Rab-37 | GTPase that regulates vesicle trafficking |
| ENSG00000174807 | CD248 | Endosialin | Angiogenesis |
| ENSG00000185567 | AHNAK2 | Protein AHNAK2 | Calcium signaling |
| ENSG00000196972 | SMIM10L2B | Small integral membrane protein 10-like protein 2B | Unknown |
| ENSG00000198563 | DDX39B | Spliceosome RNA helicase DDX39B | mRNA export from the nucleus to the cytoplasm; spliceosome assembly |
| ENSG00000234944 | LOC101929445 | Non protein coding—LINC02623 | |
| ENSG00000272789 | NA | ||
| ENSG00000277400 | MAFIP | MaFF-interacting protein | Coactivator of MAFF transcriptional activity |
| Name | Source | NGenes | Direction | p Value | FDR | p Value.Mixed | FDR.Mixed |
|---|---|---|---|---|---|---|---|
| SN | |||||||
| Phosphatidylcholines | GeneRIF Biological Term Annotations | 5 | Up | 0.034 | 0.246 | 0.095 | 0.151 |
| Phosphatidylcholine Biosynthesis | HumanCyc Pathways | 6 | Down | 0.070 | 0.246 | 0.114 | 0.151 |
| Phosphatidylcholine | Human Metabolome Database | 81 | Up | 0.074 | 0.246 | 0.124 | 0.151 |
| Phosphatidylcholines | CTD Gene-Chemical Interactions | 36 | Up | 0.084 | 0.246 | 0.189 | 0.206 |
| Phosphatidylcholines | dbGAP Gene-Trait Associations | 3 | Up | 0.124 | 0.246 | 0.126 | 0.151 |
| Phosphatidylcholine Biosynthesis Pathway | Biocarta Pathways | 3 | Down | 0.134 | 0.246 | 0.056 | 0.151 |
| GO:0031210 | phosphatidylcholine binding | 30 | Up | 0.144 | 0.246 | 0.111 | 0.151 |
| GO:0046470 | phosphatidylcholine metabolic process | 14 | Up | 0.414 | 0.622 | 0.060 | 0.151 |
| Phosphatidylcholinespecific | GeneRIF Biological Term Annotations | 5 | Up | 0.693 | 0.866 | 0.538 | 0.538 |
| GO:0034638 | phosphatidylcholine catabolic process | 5 | Up | 0.734 | 0.866 | 0.026 | 0.151 |
| GO:0006656 | phosphatidylcholine biosynthetic process | 45 | Down | 0.794 | 0.866 | 0.025 | 0.151 |
| Phosphatidylcholine | GeneRIF Biological Term Annotations | 31 | Down | 0.949 | 0.949 | 0.123 | 0.151 |
| GO:0008429 | phosphatidylethanolamine binding | 11 | Up | 0.096 | 0.390 | 0.254 | 0.355 |
| Phosphatidylethanolamine | CTD Gene-Chemical Interactions | 4 | Up | 0.130 | 0.390 | 0.480 | 0.480 |
| Phosphatidylethanolamine | Human Metabolome Database | 45 | Up | 0.344 | 0.687 | 0.272 | 0.355 |
| GO:0006646 | phosphatidylethanolamine biosynthetic process | 15 | Down | 0.577 | 0.704 | 0.025 | 0.120 |
| Phosphatidylethanolamine Biosynthesis | HumanCyc Pathways | 5 | Down | 0.587 | 0.704 | 0.040 | 0.120 |
| Phosphatidylethanolamine | GeneRIF Biological Term Annotations | 13 | Up | 0.822 | 0.822 | 0.296 | 0.355 |
| GO:0001786 | phosphatidylserine binding | 58 | Up | 0.046 | 0.457 | 0.064 | 0.214 |
| Phosphatidylserines | CTD Gene-Chemical Interactions | 9 | Up | 0.104 | 0.519 | 0.483 | 0.591 |
| Phosphatidylserineexpressing | GeneRIF Biological Term Annotations | 4 | Up | 0.308 | 0.955 | 0.532 | 0.591 |
| Phosphatidylserine | DrugBank Drug Targets | 10 | Up | 0.416 | 0.955 | 0.059 | 0.214 |
| Phosphatidylserine | GeneRIF Biological Term Annotations | 75 | Up | 0.611 | 0.955 | 0.202 | 0.337 |
| GO:0006659 | phosphatidylserine biosynthetic process | 5 | Down | 0.752 | 0.955 | 0.125 | 0.256 |
| GO:0006658 | phosphatidylserine metabolic process | 5 | Down | 0.777 | 0.955 | 0.514 | 0.591 |
| Phosphatidylserine | Human Metabolome Database | 45 | Up | 0.816 | 0.955 | 0.128 | 0.256 |
| GO:0006660 | phosphatidylserine catabolic process | 8 | Up | 0.940 | 0.955 | 0.006 | 0.060 |
| Phosphatidylserinebinding | GeneRIF Biological Term Annotations | 3 | Up | 0.955 | 0.955 | 0.988 | 0.988 |
| Phosphatidylinositolbinding | GeneRIF Biological Term Annotations | 4 | Up | 0.032 | 0.380 | 0.521 | 0.556 |
| Phosphatidylinositols | CTD Gene-Chemical Interactions | 17 | Up | 0.134 | 0.493 | 0.052 | 0.304 |
| Phosphatidylinositol4 | GeneRIF Biological Term Annotations | 3 | Up | 0.158 | 0.493 | 0.166 | 0.304 |
| GO:0035091 | phosphatidylinositol binding | 99 | Down | 0.164 | 0.493 | 0.174 | 0.304 |
| Phosphatidylinositol Signaling System | KEGG Pathways | 72 | Up | 0.344 | 0.675 | 0.406 | 0.541 |
| Phosphatidylinositol | GeneRIF Biological Term Annotations | 331 | Up | 0.407 | 0.675 | 0.105 | 0.304 |
| GO:0046488 | phosphatidylinositol metabolic process | 24 | Up | 0.479 | 0.675 | 0.203 | 0.304 |
| GO:0006661 | phosphatidylinositol biosynthetic process | 78 | Up | 0.559 | 0.675 | 0.198 | 0.304 |
| GO:0046854 | phosphatidylinositol phosphorylation | 53 | Down | 0.641 | 0.675 | 0.147 | 0.304 |
| GO:0046856 | phosphatidylinositol dephosphorylation | 23 | Up | 0.648 | 0.675 | 0.557 | 0.557 |
| Phosphatidylinositol3 | GeneRIF Biological Term Annotations | 24 | Down | 0.655 | 0.675 | 0.523 | 0.556 |
| Phosphatidylinositol | Human Metabolome Database | 85 | Up | 0.676 | 0.676 | 0.183 | 0.304 |
| Putamen | |||||||
| GO:0035091 | phosphatidylinositol binding | 99 | Down | 0.010 | 0.124 | 0.500 | 0.883 |
| Phosphatidylinositols | CTD Gene-Chemical Interactions | 17 | Up | 0.049 | 0.296 | 0.279 | 0.883 |
| Phosphatidylinositol | Human Metabolome Database | 85 | Down | 0.179 | 0.715 | 0.813 | 0.883 |
| GO:0046854 | phosphatidylinositol phosphorylation | 53 | Down | 0.624 | 0.981 | 0.582 | 0.883 |
| GO:0046856 | phosphatidylinositol dephosphorylation | 23 | Down | 0.723 | 0.981 | 0.883 | 0.883 |
| GO:0046488 | phosphatidylinositol metabolic process | 24 | Down | 0.727 | 0.981 | 0.710 | 0.883 |
| Phosphatidylinositolbinding | GeneRIF Biological Term Annotations | 78 | Down | 0.748 | 0.981 | 0.726 | 0.883 |
| GO:0006661 | phosphatidylinositol biosynthetic process | 4 | Down | 0.756 | 0.981 | 0.645 | 0.883 |
| Phosphatidylinositol3 | GeneRIF Biological Term Annotations | 24 | Down | 0.759 | 0.981 | 0.749 | 0.883 |
| Phosphatidylinositol | GeneRIF Biological Term Annotations | 331 | Down | 0.919 | 0.981 | 0.495 | 0.883 |
| Phosphatidylinositol4 | GeneRIF Biological Term Annotations | 3 | Up | 0.975 | 0.981 | 0.410 | 0.883 |
| Phosphatidylinositol Signaling System | KEGG Pathways | 72 | Down | 0.981 | 0.981 | 0.631 | 0.883 |
| Sphingomyelin Metabolism/Ceramide Salvage | HumanCyc Pathways | 8 | Down | 0.028 | 0.220 | 0.104 | 0.623 |
| Sphingomyelins | dbGAP Gene-Trait Associations | 3 | Up | 0.134 | 0.393 | 0.402 | 0.623 |
| GO:0006685 | sphingomyelin catabolic process | 7 | Down | 0.147 | 0.393 | 0.167 | 0.623 |
| Sphingomyelin | Human Metabolome Database | 52 | Down | 0.232 | 0.464 | 0.764 | 0.764 |
| Sphingomyelins | CTD Gene-Chemical Interactions | 4 | Down | 0.334 | 0.535 | 0.467 | 0.623 |
| GO:0006686 | sphingomyelin biosynthetic process | 6 | Down | 0.443 | 0.590 | 0.291 | 0.623 |
| GO:0006684 | sphingomyelin metabolic process | 4 | Down | 0.556 | 0.635 | 0.716 | 0.764 |
| Sphingomyelin | GeneRIF Biological Term Annotations | 29 | Up | 0.900 | 0.900 | 0.339 | 0.623 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xicoy, H.; Brouwers, J.F.; Wieringa, B.; Martens, G.J.M. Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease. Cells 2020, 9, 1966. https://doi.org/10.3390/cells9091966
Xicoy H, Brouwers JF, Wieringa B, Martens GJM. Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease. Cells. 2020; 9(9):1966. https://doi.org/10.3390/cells9091966
Chicago/Turabian StyleXicoy, Helena, Jos F. Brouwers, Bé Wieringa, and Gerard J. M. Martens. 2020. "Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease" Cells 9, no. 9: 1966. https://doi.org/10.3390/cells9091966
APA StyleXicoy, H., Brouwers, J. F., Wieringa, B., & Martens, G. J. M. (2020). Explorative Combined Lipid and Transcriptomic Profiling of Substantia Nigra and Putamen in Parkinson’s Disease. Cells, 9(9), 1966. https://doi.org/10.3390/cells9091966