Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. NMII: From Structure to Regulation and Function
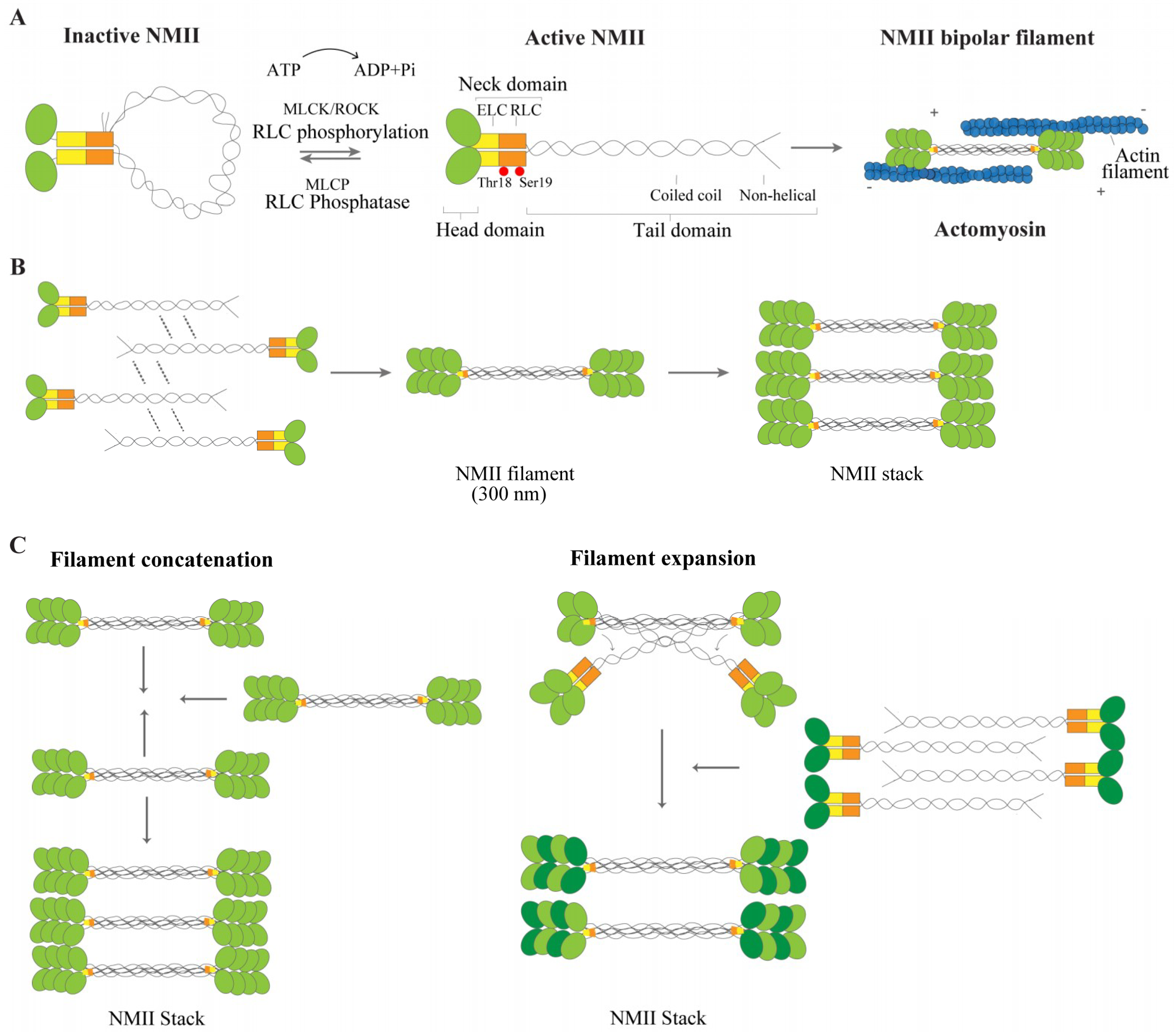
2.1. NMII Isoforms and Structure
2.2. NMII Regulation and Function
2.3. NMII: From Filament Formation to Stacks
3. Actomyosin in Neurons
3.1. NMII: From Neurite Formation during Neuronal Polarization to Axon Regeneration in the Adult
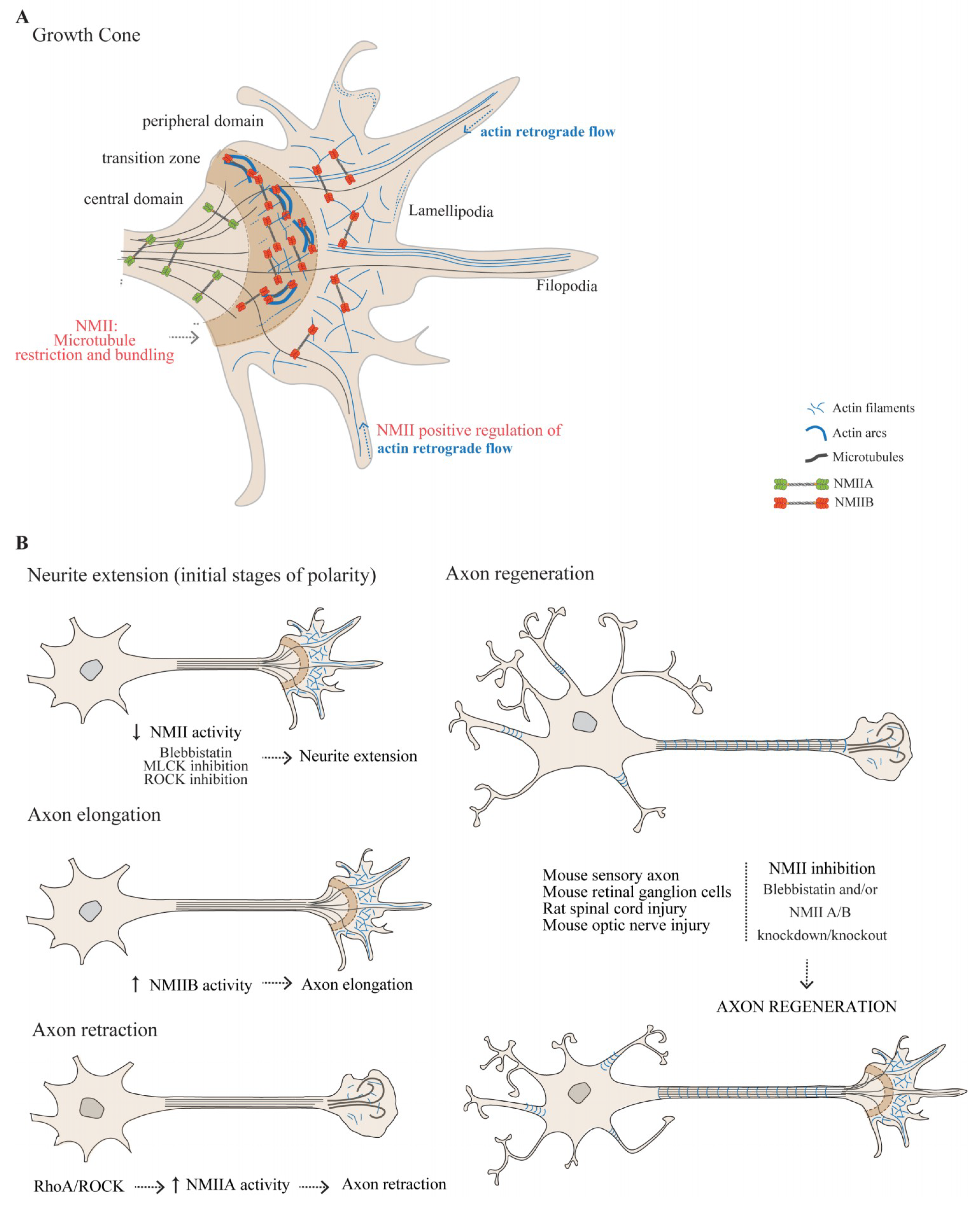
3.1.1. The Actomyosin Cytoskeleton in the Growth Cone
3.1.2. The Role of NMII in Growth Cone-Mediated Axon Elongation
3.1.3. NMIIA and NMIIB Play Central Roles in Axon Guidance
3.1.4. NMII as a Modulator of Axon Regeneration in the Adult
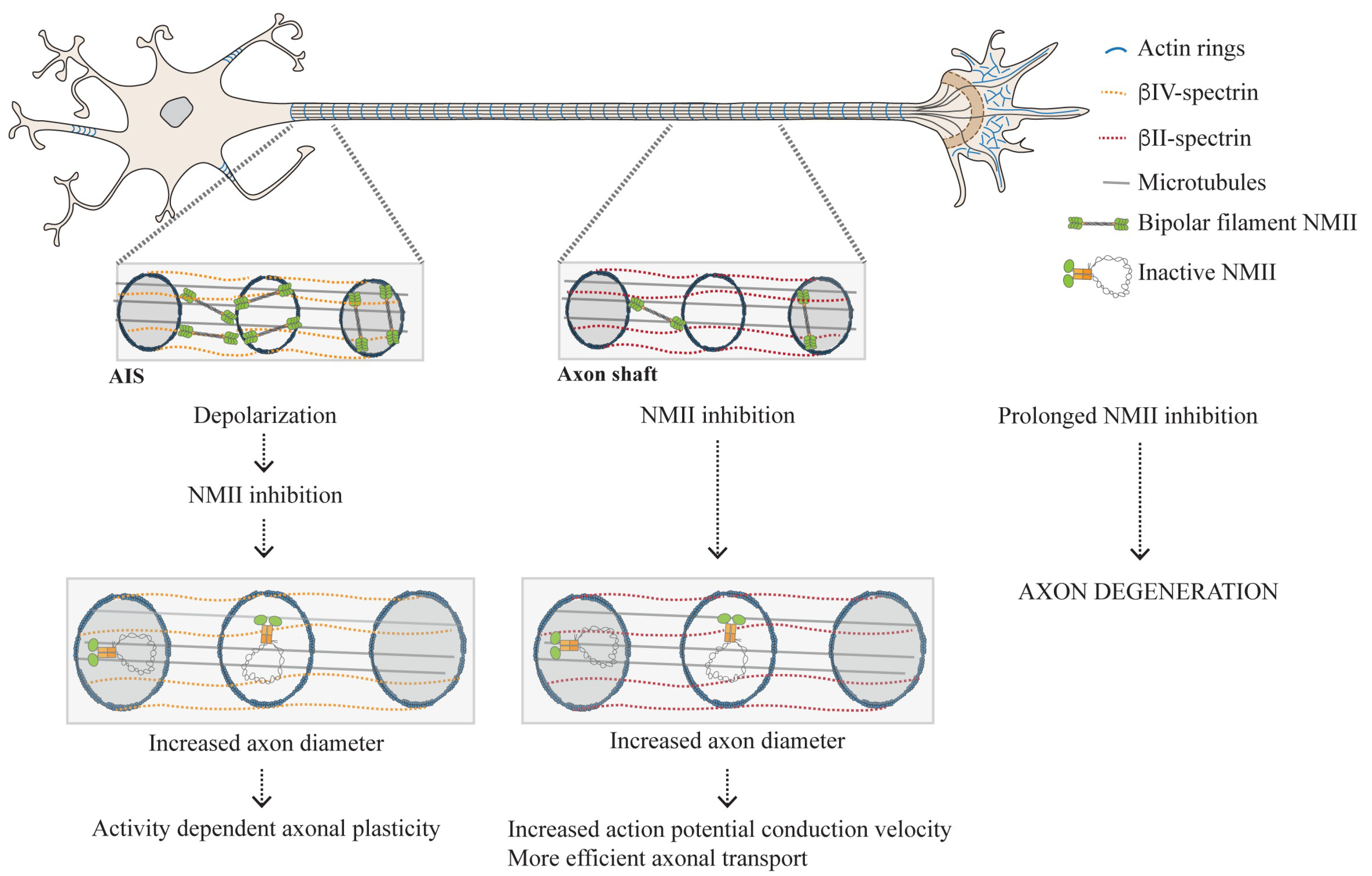
3.2. Distribution of NMII Throughout the Axon Shaft: From Enrichment in the AIS to Its Presence Throughout the Axon Shaft
3.2.1. Why Is Active NMII Enriched in the AIS?
3.2.2. The Actomyosin Cytoskeleton as a Key Regulator of Circumferential and Longitudinal Axonal Tension
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Odronitz, F.; Kollmar, M. Drawing the tree of eukaryotic life based on the analysis of 2,269 manually annotated myosins from 328 species. Genome Biol. 2007, 8, R196. [Google Scholar] [CrossRef]
- Sellers, J.R. Myosins: A diverse superfamily. Biochim. Biophys. Acta 2000, 1496, 3–22. [Google Scholar] [CrossRef]
- Pollard, T.D.; Weihing, R.R. Actin and myosin and cell movement. CRC Crit. Rev. Biochem. 1974, 2, 1–65. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Houdusse, A.; Cohen, C. Structure of the regulatory domain of scallop myosin at 2 å resolution: Implications for regulation. Structure 1996, 4, 21–32. [Google Scholar] [CrossRef]
- Houdusse, A.; Kalabokis, V.N.; Himmel, D.; Szent-Györgyi, A.G.; Cohen, C. Atomic Structure of Scallop Myosin Subfragment S1 Complexed with MgADP. Cell 1999, 97, 459–470. [Google Scholar] [CrossRef]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Base, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef]
- Xie, X.; Harrison, D.H.; Schlichting, I.; Sweet, R.M.; Kalabokis, V.N.; Szent-Gyorgyi, A.G.; Cohen, C. Structure of the regulatory domain of scallop myosin at 2.8 A resolution. Nature 1994, 368, 306–312. [Google Scholar] [CrossRef]
- Jordan, P.; Karess, R. Myosin light chain-activating phosphorylation sites are required for oogenesis in Drosophila. J. Cell Biol. 1997, 139, 1805–1819. [Google Scholar] [CrossRef]
- Burridge, K.; Bray, D. Purification and structural analysis of myosins from brain and other non-muscle tissues. J. Mol. Biol. 1975, 99, 1–14. [Google Scholar] [CrossRef]
- Golomb, E.; Ma, X.; Jana, S.S.; Preston, Y.A.; Kawamoto, S.; Shoham, N.G.; Goldin, E.; Conti, M.A.; Sellers, J.R.; Adelstein, R.S. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 2004, 279, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Katsuragawa, Y.; Yanagisawa, M.; Inoue, A.; Masaki, T. Two distinct nonmuscle myosin-heavy-chain mRNAs are differentially expressed in various chicken tissues. Identification of a novel gene family of vertebrate non-sarcomeric myosin heavy chains. Eur. J. Biochem. 1989, 184, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Adelstein, R.S. Chicken nonmuscle myosin heavy chains: Differential expression of two mRNAs and evidence for two different polypeptides. J. Cell Biol. 1991, 112, 915–924. [Google Scholar] [CrossRef]
- Murakami, N.; Elzinga, M. Immunohistochemical studies on the distribution of cellular myosin II isoforms in brain and aorta. Cell Motil. Cytoskeleton 1992, 22, 281–295. [Google Scholar] [CrossRef]
- Simons, M.; Wang, M.; McBride, O.W.; Kawamoto, S.; Yamakawa, K.; Gdula, D.; Adelstein, R.S.; Weir, L. Human nonmuscle myosin heavy chains are encoded by two genes located on different chromosomes. Circ. Res. 1991, 69, 530–539. [Google Scholar] [CrossRef]
- Sandquist, J.C.; Means, A.R. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell 2008, 19, 5156–5167. [Google Scholar] [CrossRef]
- Wylie, S.R.; Chantler, P.D. Myosin IIC: A third molecular motor driving neuronal dynamics. Mol. Biol. Cell 2008, 19, 3956–3968. [Google Scholar] [CrossRef][Green Version]
- Kolega, J. Cytoplasmic dynamics of myosin IIA and IIB: Spatial ‘sorting’ of isoforms in locomoting cells. J. Cell Sci. 1998, 111, 2085–2095. [Google Scholar]
- Maupin, P.; Phillips, C.L.; Adelstein, R.S.; Pollard, T.D. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J. Cell Sci. 1994, 107 Pt 11, 3077–3090. [Google Scholar]
- Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.; Choi, C.K.; Horwitz, A.F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007, 176, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Sousa, S. Non-Muscle Myosin 2A (NM2A): Structure, Regulation and Function. Cells 2020, 9, 1590. [Google Scholar] [CrossRef] [PubMed]
- Heissler, S.M.; Sellers, J.R. Various Themes of Myosin Regulation. J. Mol. Biol. 2016, 428, 1927–1946. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, R.S.; Conti, M.A. Phosphorylation of platelet myosin increases actin-activated myosin ATPase activity. Nature 1975, 256, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Scholey, J.M.; Taylor, K.A.; Kendrick-Jones, J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature 1980, 287, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Murata-Hori, M.; Suizu, F.; Iwasaki, T.; Kikuchi, A.; Hosoya, H. ZIP kinase identified as a novel myosin regulatory light chain kinase in HeLa cells. FEBS Lett. 1999, 451, 81–84. [Google Scholar] [CrossRef]
- Sellers, J.R.; Pato, M.D.; Adelstein, R.S. Reversible phosphorylation of smooth muscle myosin, heavy meromyosin, and platelet myosin. J. Biol. Chem. 1981, 256, 13137–13142. [Google Scholar]
- Tan, I.; Yong, J.; Dong, J.M.; Lim, L.; Leung, T. A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 2008, 135, 123–136. [Google Scholar] [CrossRef]
- Yamashiro, S.; Totsukawa, G.; Yamakita, Y.; Sasaki, Y.; Madaule, P.; Ishizaki, T.; Narumiya, S.; Matsumura, F. Citron kinase, a Rho-dependent kinase, induces di-phosphorylation of regulatory light chain of myosin II. Mol. Biol. Cell 2003, 14, 1745–1756. [Google Scholar] [CrossRef]
- Woodhead, J.L.; Zhao, F.Q.; Craig, R.; Egelman, E.H.; Alamo, L.; Padron, R. Atomic model of a myosin filament in the relaxed state. Nature 2005, 436, 1195–1199. [Google Scholar] [CrossRef]
- Liu, X.; Billington, N.; Shu, S.; Yu, S.H.; Piszczek, G.; Sellers, J.R.; Korn, E.D. Effect of ATP and regulatory light-chain phosphorylation on the polymerization of mammalian nonmuscle myosin II. Proc. Natl. Acad. Sci. USA 2017, 114, E6516–E6525. [Google Scholar] [CrossRef]
- Craig, R.; Smith, R.; Kendrick-Jones, J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 1983, 302, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, T.Q.; Abramson, P.D.; Spudich, J.A. The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc. Natl. Acad. Sci. USA 1996, 93, 4459–4464. [Google Scholar] [CrossRef] [PubMed]
- Griffith, L.M.; Downs, S.M.; Spudich, J.A. Myosin light chain kinase and myosin light chain phosphatase from Dictyostelium: Effects of reversible phosphorylation on myosin structure and function. J. Cell Biol. 1987, 104, 1309–1323. [Google Scholar] [CrossRef]
- Grassie, M.E.; Moffat, L.D.; Walsh, M.P.; MacDonald, J.A. The myosin phosphatase targeting protein (MYPT) family: A regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch. Biochem. Biophys. 2011, 510, 147–159. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Zhenhuan, G.; Luxenburg, C. The contractome—A systems view of actomyosin contractility in non-muscle cells. J. Cell Sci. 2015, 128, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, C.B.; Tyska, M.J.; Mooseker, M.S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochim. Biophys. Acta 2007, 1773, 615–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kovacs, M.; Kawamoto, S.; Sellers, J.R.; Adelstein, R.S. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J. Biol. Chem. 2005, 280, 22769–22775. [Google Scholar] [CrossRef]
- Wang, F.; Kovacs, M.; Hu, A.; Limouze, J.; Harvey, E.V.; Sellers, J.R. Kinetic mechanism of non-muscle myosin IIB: Functional adaptations for tension generation and maintenance. J. Biol. Chem. 2003, 278, 27439–27448. [Google Scholar] [CrossRef]
- Tuzovic, L.; Yu, L.; Zeng, W.; Li, X.; Lu, H.; Lu, H.M.; Gonzalez, K.D.; Chung, W.K. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis. 2013, 1, e26144. [Google Scholar] [CrossRef]
- Billington, N.; Wang, A.; Mao, J.; Adelstein, R.S.; Sellers, J.R. Characterization of three full-length human nonmuscle myosin II paralogs. J. Biol. Chem. 2013, 288, 33398–33410. [Google Scholar] [CrossRef] [PubMed]
- Niederman, R.; Pollard, T.D. Human platelet myosin. II. In vitro assembly and structure of myosin filaments. J. Cell Biol. 1975, 67, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Beach, J.R.; Bruun, K.S.; Shao, L.; Li, D.; Swider, Z.; Remmert, K.; Zhang, Y.; Conti, M.A.; Adelstein, R.S.; Rusan, N.M.; et al. Actin dynamics and competition for myosin monomer govern the sequential amplification of myosin filaments. Nat. Cell Biol. 2017, 19, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Fenix, A.M.; Taneja, N.; Buttler, C.A.; Lewis, J.; Van Engelenburg, S.B.; Ohi, R.; Burnette, D.T.; Forscher, P. Expansion and concatenation of nonmuscle myosin IIA filaments drive cellular contractile system formation during interphase and mitosis. Mol. Biol. Cell 2016, 27, 1465–1478. [Google Scholar] [CrossRef]
- Beach, J.R.; Shao, L.; Remmert, K.; Li, D.; Betzig, E.; Hammer, J.A., 3rd. Nonmuscle myosin II isoforms coassemble in living cells. Curr. Biol. 2014, 24, 1160–1166. [Google Scholar] [CrossRef]
- Shutova, M.S.; Spessott, W.A.; Giraudo, C.G.; Svitkina, T. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr. Biol. 2014, 24, 1958–1968. [Google Scholar] [CrossRef]
- Dasbiswas, K.; Hu, S.; Schnorrer, F.; Safran, S.A.; Bershadsky, A.D. Ordering of myosin II filaments driven by mechanical forces: Experiments and theory. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170114. [Google Scholar] [CrossRef]
- Hu, S.; Dasbiswas, K.; Guo, Z.; Tee, Y.H.; Thiagarajan, V.; Hersen, P.; Chew, T.L.; Safran, S.A.; Zaidel-Bar, R.; Bershadsky, A.D. Long-range self-organization of cytoskeletal myosin II filament stacks. Nat. Cell Biol. 2017, 19, 133–141. [Google Scholar] [CrossRef]
- Verkhovsky, A.B.; Svitkina, T.M.; Borisy, G.G. Myosin II filament assemblies in the active lamella of fibroblasts: Their morphogenesis and role in the formation of actin filament bundles. J. Cell Biol. 1995, 131, 989–1002. [Google Scholar] [CrossRef]
- Bradke, F.; Dotti, C.G. Establishment of neuronal polarity: Lessons from cultured hippocampal neurons. Curr. Opin. Neurobiol. 2000, 10, 574–581. [Google Scholar] [CrossRef]
- Kaech, S.; Banker, G. Culturing hippocampal neurons. Nat. Protoc. 2006, 1, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Schelski, M.; Bradke, F. Neuronal polarization: From spatiotemporal signaling to cytoskeletal dynamics. Mol. Cell Neurosci. 2017, 84, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Dupraz, S.; Hilton, B.J.; Husch, A.; Santos, T.E.; Coles, C.H.; Stern, S.; Brakebusch, C.; Bradke, F. RhoA Controls Axon Extension Independent of Specification in the Developing Brain. Curr. Biol. 2019, 29, 3874–3886.e9. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.W.; Kabir, N.; Forscher, P. Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J. Cell Biol. 2002, 158, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Bridgman, P.C. Growth cones contain myosin II bipolar filament arrays. Cell Motil. Cytoskeleton 2002, 52, 91–96. [Google Scholar] [CrossRef]
- Medeiros, N.A.; Burnette, D.T.; Forscher, P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 2006, 8, 215–226. [Google Scholar] [CrossRef]
- Rochlin, M.W.; Itoh, K.; Adelstein, R.S.; Bridgman, P.C. Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 1995, 108 Pt 12, 3661–3670. [Google Scholar]
- Turney, S.G.; Bridgman, P.C. Laminin stimulates and guides axonal outgrowth via growth cone myosin II activity. Nat. Neurosci. 2005, 8, 717–719. [Google Scholar] [CrossRef]
- Wylie, S.R.; Wu, P.J.; Patel, H.; Chantler, P.D. A conventional myosin motor drives neurite outgrowth. Proc. Natl. Acad. Sci. USA 1998, 95, 12967–12972. [Google Scholar] [CrossRef]
- Zhang, X.F.; Schaefer, A.W.; Burnette, D.T.; Schoonderwoert, V.T.; Forscher, P. Rho-dependent contractile responses in the neuronal growth cone are independent of classical peripheral retrograde actin flow. Neuron 2003, 40, 931–944. [Google Scholar] [CrossRef]
- Burnette, D.T.; Ji, L.; Schaefer, A.W.; Medeiros, N.A.; Danuser, G.; Forscher, P. Myosin II activity facilitates microtubule bundling in the neuronal growth cone neck. Dev. Cell 2008, 15, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.E.; Bridgman, P.C. Myosin function in nervous and sensory systems. J. Neurobiol. 2004, 58, 118–130. [Google Scholar] [CrossRef]
- Diefenbach, T.J.; Latham, V.M.; Yimlamai, D.; Liu, C.A.; Herman, I.M.; Jay, D.G. Myosin 1c and myosin IIB serve opposing roles in lamellipodial dynamics of the neuronal growth cone. J. Cell Biol. 2002, 158, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Espreafico, E.M.; Mooseker, M.S.; Forscher, P. Myosin drives retrograde F-actin flow in neuronal growth cones. Biol. Bull. 1997, 192, 183–185. [Google Scholar] [CrossRef]
- Brown, M.E.; Bridgman, P.C. Retrograde flow rate is increased in growth cones from myosin IIB knockout mice. J. Cell Sci. 2003, 116, 1087–1094. [Google Scholar] [CrossRef]
- Kollins, K.M.; Hu, J.; Bridgman, P.C.; Huang, Y.Q.; Gallo, G. Myosin-II negatively regulates minor process extension and the temporal development of neuronal polarity. Dev. Neurobiol. 2009, 69, 279–298. [Google Scholar] [CrossRef]
- Gomez, T.M.; Letourneau, P.C. Actin dynamics in growth cone motility and navigation. J. Neurochem. 2014, 129, 221–234. [Google Scholar] [CrossRef]
- Goldberg, D.J.; Burmeister, D.W. Stages in axon formation: Observations of growth of Aplysia axons in culture using video-enhanced contrast-differential interference contrast microscopy. J. Cell Biol. 1986, 103, 1921–1931. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Cytoskeletal dynamics and nerve growth. Neuron 1988, 1, 761–772. [Google Scholar] [CrossRef]
- Brown, J.; Bridgman, P.C. Role of myosin II in axon outgrowth. J. Histochem. Cytochem. 2003, 51, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.W.; Schoonderwoert, V.T.; Ji, L.; Mederios, N.; Danuser, G.; Forscher, P. Coordination of actin filament and microtubule dynamics during neurite outgrowth. Dev. Cell 2008, 15, 146–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.Q.; Cohan, C.S. Growth cone collapse through coincident loss of actin bundles and leading edge actin without actin depolymerization. J. Cell Biol. 2001, 153, 1071–1084. [Google Scholar] [CrossRef]
- Sayyad, W.A.; Amin, L.; Fabris, P.; Ercolini, E.; Torre, V. The role of myosin-II in force generation of DRG filopodia and lamellipodia. Sci. Rep. 2015, 5, 7842. [Google Scholar] [CrossRef]
- Myers, K.A.; Tint, I.; Nadar, C.V.; He, Y.; Black, M.M.; Baas, P.W. Antagonistic forces generated by cytoplasmic dynein and myosin-II during growth cone turning and axonal retraction. Traffic 2006, 7, 1333–1351. [Google Scholar] [CrossRef]
- Tullio, A.N.; Bridgman, P.C.; Tresser, N.J.; Chan, C.C.; Conti, M.A.; Adelstein, R.S.; Hara, Y. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J. Comp. Neurol. 2001, 433, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Chihara, K.; Nakamura, N.; Fukata, Y.; Yano, T.; Shibata, M.; Ikebe, M.; Kaibuchi, K. Myosin II activation promotes neurite retraction during the action of Rho and Rho-kinase. Genes Cells 1998, 3, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Loudon, R.P.; Silver, L.D.; Yee, H.F., Jr.; Gallo, G. RhoA-kinase and myosin II are required for the maintenance of growth cone polarity and guidance by nerve growth factor. J. Neurobiol. 2006, 66, 847–867. [Google Scholar] [CrossRef]
- Wylie, S.R.; Chantler, P.D. Myosin IIA drives neurite retraction. Mol. Biol. Cell 2003, 14, 4654–4666. [Google Scholar] [CrossRef]
- Yuan, X.B.; Jin, M.; Xu, X.; Song, Y.Q.; Wu, C.P.; Poo, M.M.; Duan, S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat. Cell Biol. 2003, 5, 38–45. [Google Scholar] [CrossRef]
- Hur, E.M.; Yang, I.H.; Kim, D.H.; Byun, J.; Xu, W.L.; Nicovich, P.R.; Cheong, R.; Levchenko, A.; Thakor, N.; Zhou, F.Q. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Endo, M.; Hata, K.; Taniguchi, J.; Kitajo, K.; Tomura, S.; Yamaguchi, A.; Mueller, B.K.; Yamashita, T. Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J. Neurochem. 2008, 105, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.R.; Chantler, P.D. Separate but linked functions of conventional myosins modulate adhesion and neurite outgrowth. Nat. Cell Biol. 2001, 3, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.E.; Schaffran, B.; Broguiere, N.; Meyn, L.; Zenobi-Wong, M.; Bradke, F. Axon Growth of CNS Neurons in Three Dimensions Is Amoeboid and Independent of Adhesions. Cell Rep. 2020, 32, 107907. [Google Scholar] [CrossRef]
- Kozma, R.; Sarner, S.; Ahmed, S.; Lim, L. Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell Biol. 1997, 17, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Ma, X.; Liu, C.; Adelstein, R.S. Replacement of nonmuscle myosin II-B with II-A rescues brain but not cardiac defects in mice. J. Biol. Chem. 2007, 282, 22102–22111. [Google Scholar] [CrossRef]
- Turney, S.G.; Ahmed, M.; Chandrasekar, I.; Wysolmerski, R.B.; Goeckeler, Z.M.; Rioux, R.M.; Whitesides, G.M.; Bridgman, P.C. Nerve growth factor stimulates axon outgrowth through negative regulation of growth cone actomyosin restraint of microtubule advance. Mol. Biol. Cell 2016, 27, 500–517. [Google Scholar] [CrossRef]
- Bridgman, P.C.; Dave, S.; Asnes, C.F.; Tullio, A.N.; Adelstein, R.S. Myosin IIB Is Required for Growth Cone Motility. J. Neurosci. 2001, 21, 6159–6169. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Szaro, B.G.; Schmidt, J.T. Myosin light chain kinase: Expression in neurons and upregulation during axon regeneration. J. Neurobiol. 1996, 31, 379–391. [Google Scholar] [CrossRef]
- Yu, P.; Santiago, L.Y.; Katagiri, Y.; Geller, H.M. Myosin II activity regulates neurite outgrowth and guidance in response to chondroitin sulfate proteoglycans. J. Neurochem. 2012, 120, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Wysolmerski, R.B.; Bridgman, P.C. Dorsal root ganglion neurons react to semaphorin 3A application through a biphasic response that requires multiple myosin II isoforms. Mol. Biol. Cell 2009, 20, 1167–1179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Javier-Torrent, M.; Marco, S.; Rocandio, D.; Pons-Vizcarra, M.; Janes, P.W.; Lackmann, M.; Egea, J.; Saura, C.A. Presenilin/gamma-secretase-dependent EphA3 processing mediates axon elongation through non-muscle myosin IIA. eLife 2019, 8, e43646. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Bradke, F. Axon Regeneration in the Central Nervous System: Facing the Challenges from the Inside. Annu. Rev. Cell Dev. Biol. 2018, 34, 495–521. [Google Scholar] [CrossRef] [PubMed]
- Mar, F.M.; Bonni, A.; Sousa, M.M. Cell intrinsic control of axon regeneration. EMBO Rep. 2014, 15, 254–263. [Google Scholar] [CrossRef]
- Ning, G.; Liu, Y.; Xu, H.; Li, Y.; Wu, H.; Wang, X.; Feng, S. Erratum: Gene silencing NMII promotes axonal regeneration against contusive spinal cord injury in rats. Int. J. Clin. Exp. Pathol. 2018, 11, 1840. [Google Scholar] [PubMed]
- Wang, X.W.; Yang, S.G.; Zhang, C.; Hu, M.W.; Qian, J.; Ma, J.J.; Zhang, Y.; Yang, B.B.; Weng, Y.L.; Ming, G.L.; et al. Knocking Out Non-muscle Myosin II in Retinal Ganglion Cells Promotes Long-Distance Optic Nerve Regeneration. Cell Rep. 2020, 31, 107537. [Google Scholar] [CrossRef]
- Berger, S.L.; Leo-Macias, A.; Yuen, S.; Khatri, L.; Pfennig, S.; Zhang, Y.; Agullo-Pascual, E.; Caillol, G.; Zhu, M.S.; Rothenberg, E.; et al. Localized Myosin II Activity Regulates Assembly and Plasticity of the Axon Initial Segment. Neuron 2018, 97, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Tufo, C.; Dumitrescu, A.S.; Grubb, M.S. Myosin II activity is required for structural plasticity at the axon initial segment. Eur. J. Neurosci. 2017, 46, 1751–1757. [Google Scholar] [CrossRef]
- Costa, A.R.; Sousa, S.C.; Pinto-Costa, R.; Mateus, J.C.; Lopes, C.D.; Costa, A.C.; Rosa, D.; Machado, D.; Pajuelo, L.; Wang, X.; et al. The membrane periodic skeleton is an actomyosin network that regulates axonal diameter and conduction. eLife 2020, 9, e55471. [Google Scholar] [CrossRef]
- Wang, T.; Li, W.; Martin, S.; Papadopulos, A.; Joensuu, M.; Liu, C.; Jiang, A.; Shamsollahi, G.; Amor, R.; Lanoue, V.; et al. Radial contractility of actomyosin rings facilitates axonal trafficking and structural stability. J. Cell Biol. 2020, 219, e201902001. [Google Scholar] [CrossRef]
- Grubb, M.S.; Burrone, J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 2010, 465, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 2013, 339, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Bhembre, N.; Bodas, S.; Veer, S.; Ghose, A.; Callan-Jones, A.; Pullarkat, P. The axonal actin-spectrin lattice acts as a tension buffering shock absorber. eLife 2020, 9, e51772. [Google Scholar] [CrossRef] [PubMed]
- Hammarlund, M.; Jorgensen, E.M.; Bastiani, M.J. Axons break in animals lacking beta-spectrin. J. Cell Biol. 2007, 176, 269–275. [Google Scholar] [CrossRef]
- Abouelezz, A.; Stefen, H.; Segerstrale, M.; Micinski, D.; Minkeviciene, R.; Lahti, L.; Hardeman, E.C.; Gunning, P.W.; Hoogenraad, C.C.; Taira, T.; et al. Tropomyosin Tpm3.1 Is Required to Maintain the Structure and Function of the Axon Initial Segment. iScience 2020, 23, 101053. [Google Scholar] [CrossRef]
- Fields, R.D. Signaling by neuronal swelling. Sci. Signal. 2011, 4, tr1. [Google Scholar] [CrossRef][Green Version]
- Costa, A.R.; Pinto-Costa, R.; Sousa, S.C.; Sousa, M.M. The Regulation of Axon Diameter: From Axonal Circumferential Contractility to Activity-Dependent Axon Swelling. Front. Mol. Neurosci. 2018, 11, 319. [Google Scholar] [CrossRef]
- Fan, A.; Tofangchi, A.; Kandel, M.; Popescu, G.; Saif, T. Coupled circumferential and axial tension driven by actin and myosin influences in vivo axon diameter. Sci. Rep. 2017, 7, 14188. [Google Scholar] [CrossRef]
- Vassilopoulos, S.; Gibaud, S.; Jimenez, A.; Caillol, G.; Leterrier, C. Ultrastructure of the axonal periodic scaffold reveals a braid-like organization of actin rings. Nat. Commun. 2019, 10, 5803. [Google Scholar] [CrossRef]
- Tofangchi, A.; Fan, A.; Saif, M.T.A. Mechanism of Axonal Contractility in Embryonic Drosophila Motor Neurons In Vivo. Biophys. J. 2016, 111, 1519–1527. [Google Scholar] [CrossRef]
- Mutalik, S.P.; Joseph, J.; Pullarkat, P.A.; Ghose, A. Cytoskeletal Mechanisms of Axonal Contractility. Biophys. J. 2018, 115, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M.L. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef] [PubMed]
- Argellati, F.; Domenicotti, C.; Passalacqua, M.; Janda, E.; Melloni, E.; Marinari, U.M.; Pronzato, M.A.; Ricciarelli, R. Protein kinase C-dependent alpha-secretory processing of the amyloid precursor protein is mediated by phosphorylation of myosin II-B. FASEB J. 2009, 23, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Massone, S.; Argellati, F.; Passalacqua, M.; Armirotti, A.; Melone, L.; d’Abramo, C.; Marinari, U.M.; Domenicotti, C.; Pronzato, M.A.; Ricciarelli, R. Downregulation of myosin II-B by siRNA alters the subcellular localization of the amyloid precursor protein and increases amyloid-beta deposition in N2a cells. Biochem. Biophys. Res. Commun. 2007, 362, 633–638. [Google Scholar] [CrossRef]
- Wang, X.; Williams, D.; Muller, I.; Lemieux, M.; Dukart, R.; Maia, I.B.L.; Wang, H.; Woerman, A.L.; Schmitt-Ulms, G. Tau interactome analyses in CRISPR-Cas9 engineered neuronal cells reveal ATPase-dependent binding of wild-type but not P301L Tau to non-muscle myosins. Sci. Rep. 2019, 9, 16238. [Google Scholar] [CrossRef]
- Zhao, L.; Ma, Q.L.; Calon, F.; Harris-White, M.E.; Yang, F.; Lim, G.P.; Morihara, T.; Ubeda, O.J.; Ambegaokar, S.; Hansen, J.E.; et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat. Neurosci. 2006, 9, 234–242. [Google Scholar] [CrossRef]
- Unsain, N.; Bordenave, M.D.; Martinez, G.F.; Jalil, S.; von Bilderling, C.; Barabas, F.M.; Masullo, L.A.; Johnstone, A.D.; Barker, P.A.; Bisbal, M.; et al. Remodeling of the Actin/Spectrin Membrane-associated Periodic Skeleton, Growth Cone Collapse and F-Actin Decrease during Axonal Degeneration. Sci. Rep. 2018, 8, 3007. [Google Scholar] [CrossRef]
- Wang, G.; Simon, D.J.; Wu, Z.; Belsky, D.M.; Heller, E.; O’Rourke, M.K.; Hertz, N.T.; Molina, H.; Zhong, G.; Tessier-Lavigne, M.; et al. Structural plasticity of actin-spectrin membrane skeleton and functional role of actin and spectrin in axon degeneration. eLife 2019, 8, e38730. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, A.R.; Sousa, M.M. Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment. Cells 2020, 9, 1961. https://doi.org/10.3390/cells9091961
Costa AR, Sousa MM. Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment. Cells. 2020; 9(9):1961. https://doi.org/10.3390/cells9091961
Chicago/Turabian StyleCosta, Ana Rita, and Monica M. Sousa. 2020. "Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment" Cells 9, no. 9: 1961. https://doi.org/10.3390/cells9091961
APA StyleCosta, A. R., & Sousa, M. M. (2020). Non-Muscle Myosin II in Axonal Cell Biology: From the Growth Cone to the Axon Initial Segment. Cells, 9(9), 1961. https://doi.org/10.3390/cells9091961