Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders


 and
and
Abstract
1. Human Neural Stem Cells: Introduction
2. Neural Stem Cells and Neurodevelopmental Disorders
2.1. Genetic and Environmental Insults to NSCs
2.2. TORCH and Microcephaly
2.3. TORCH Pathogens Effects on Self-Renewing NSCs
2.3.1. Zika Virus
2.3.2. Toxoplasma
2.3.3. Rubella Virus
2.3.4. Herpetic Viruses
Cytomegalovirus
Herpes Simplex Virus
2.3.5. Coxsackie Virus
3. TORCH Pathogens Effects on NSC Neuronal and Glial Derivatives
3.1. ZIKV
3.2. Toxoplasma
3.3. CMV
3.4. HSV
3.5. HSV-1 Infection and Alzheimer’s Disease Neurodegeneration
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diaz, A.L.; Gleeson, J.G. The Molecular and Genetic Mechanisms of Neocortex Development. Clin. Perinatol. 2009, 36, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed]
- Silbereis, J.C.; Pochareddy, S.; Zhu, Y.; Li, M.; Sestan, N. The Cellular and Molecular Landscapes of the Developing Human Central Nervous System. Neuron 2016, 89, 248. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.M.; Meyer, K.A.; Santpere, G.; Gulden, F.O.; Sestan, N. Evolution of the Human Nervous System Function, Structure, and Development. Cell 2017, 170, 226–247. [Google Scholar] [CrossRef] [PubMed]
- Kriegstein, A.; Alvarez-Buylla, A. The Glial Nature of Embryonic and Adult Neural Stem Cells. Annu. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef] [PubMed]
- Breunig, J.J.; Haydar, T.F.; Rakic, P. Neural Stem Cells: Historical Perspective and Future Prospects. Neuron 2011, 70, 614–625. [Google Scholar] [CrossRef]
- Fietz, S.A.; Kelava, I.; Vogt, J.; Wilsch-Bräuninger, M.; Stenzel, D.; Fish, J.L.; Corbeil, D.; Riehn, A.; Distler, W.; Nitsch, R.; et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 2010, 13, 690–699. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.L.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef]
- Reillo, I.; Borrell, V. Germinal zones in the developing cerebral cortex of ferret: Ontogeny, cell cycle kinetics, and diversity of progenitors. Cereb. Cortex 2012, 22, 2039–2054. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; Sandoval-Espinosa, C.; Kriegstein, A.R. Transformation of the Radial Glia Scaffold Demarcates Two Stages of Human Cerebral Cortex Development. Neuron 2016, 91, 1219–1227. [Google Scholar] [CrossRef]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.L.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Cattaneo, E. Neural stem cell systems: Physiological players or in vitro entities? Nat. Rev. Neurosci. 2010, 11, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (80-) 1992, 255, 1707–1710. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Pollard, S.M.; Gorba, T.; Reitano, E.; Toselli, M.; Biella, G.; Sun, Y.; Sanzone, S.; Ying, Q.L.; Cattaneo, E.; et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005, 3, 1594–1606. [Google Scholar] [CrossRef] [PubMed]
- Pollard, S.M.; Conti, L.; Sun, Y.; Goffredo, D.; Smith, A. Adherent Neural Stem (NS) Cells from Fetal and Adult Forebrain. Cereb. Cortex 2006, 16, i112–i120. [Google Scholar] [CrossRef]
- Goffredo, D.; Conti, L.; Di Febo, F.; Biella, G.; Tosoni, A.; Vago, G.; Biunno, I.; Moiana, A.; Bolognini, D.; Toselli, M.; et al. Setting the conditions for efficient, robust and reproducible generation of functionally active neurons from adult subventricular zone-derived neural stem cells. Cell Death Differ. 2008, 15, 1847–1856. [Google Scholar] [CrossRef]
- Albieri, I.; Onorati, M.; Calabrese, G.; Moiana, A.; Biasci, D.; Badaloni, A.; Camnasio, S.; Spiliotopoulos, D.; Ivics, Z.; Cattaneo, E.; et al. A DNA transposon-based approach to functional screening in neural stem cells. J. Biotechnol. 2010, 150, 11–21. [Google Scholar] [CrossRef]
- Onorati, M.; Camnasio, S.; Binetti, M.; Jung, C.B.; Moretti, A.; Cattaneo, E. Neuropotent self-renewing neural stem (NS) cells derived from mouse induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2010, 43, 287–295. [Google Scholar] [CrossRef]
- Onorati, M.; Binetti, M.; Conti, L.; Camnasio, S.; Calabrese, G.; Albieri, I.; Di Febo, F.; Toselli, M.; Biella, G.; Martynoga, B.; et al. Preservation of positional identity in fetus-derived neural stem (NS) cells from different mouse central nervous system compartments. Cell. Mol. Life Sci. 2011, 68, 1769–1783. [Google Scholar] [CrossRef]
- Sun, Y.; Pollard, S.; Conti, L.; Toselli, M.; Biella, G.; Parkin, G.; Willatt, L.; Falk, A.; Cattaneo, E.; Smith, A. Long-term tripotent differentiation capacity of human neural stem (NS) cells in adherent culture. Mol. Cell. Neurosci. 2008, 38, 245–258. [Google Scholar] [CrossRef]
- Hook, L.; Vives, J.; Fulton, N.; Leveridge, M.; Lingard, S.; Bootman, M.D.; Falk, A.; Pollard, S.M.; Allsopp, T.E.; Dalma-Weiszhausz, D.; et al. Non-immortalized human neural stem (NS) cells as a scalable platform for cellular assays. Neurochem. Int. 2011, 59, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Tailor, J.; Kittappa, R.; Leto, K.; Gates, M.; Borel, M.; Paulsen, O.; Spitzer, S.; Thora Karadottir, R.; Rossi, F.; Falk, A.; et al. Stem cells expanded from the human embryonic hindbrain stably retain regional specification and high neurogenic potency. J. Neurosci. 2013, 33, 12407–12422. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef]
- Dell’Anno, M.T.; Wang, X.; Onorati, M.; Li, M.; Talpo, F.; Sekine, Y.; Ma, S.; Liu, F.; Cafferty, W.B.J.; Sestan, N.; et al. Human neuroepithelial stem cell regional specificity enables spinal cord repair through a relay circuit. Nat. Commun. 2018, 9, 3419. [Google Scholar] [CrossRef] [PubMed]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef]
- Koch, P.; Opitz, T.; Steinbeck, J.A.; Ladewig, J.; Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 2009, 106, 3225–3230. [Google Scholar] [CrossRef]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef]
- Edri, R.; Yaffe, Y.; Ziller, M.J.; Mutukula, N.; Volkman, R.; David, E.; Jacob-Hirsch, J.; Malcov, H.; Levy, C.; Rechavi, G.; et al. Analysing human neural stem cell ontogeny by consecutive isolation of Notch active neural progenitors. Nat. Commun. 2015, 6, 6500. [Google Scholar] [CrossRef]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.N.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genomics 2015, 8, 1–11. [Google Scholar] [CrossRef]
- Woods, C.G. Human microcephaly. Curr. Opin. Neurobiol. 2004, 14, 112–117. [Google Scholar] [CrossRef]
- Mochida, G.H.; Walsh, C.A. Molecular genetics of human microcephaly. Curr. Opin. Neurol. 2001, 14, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Kazmi, S.K.; Amin, M.; Asif, Z.; Islam, U.; Shahid, K.; Tehreem, S. Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genet. Res. (Camb.) 2018, 100, e7. [Google Scholar] [CrossRef] [PubMed]
- Schuler-Faccini, L.; Ribeiro, E.M.; Feitosa, I.M.L.; Horovitz, D.D.G.; Cavalcanti, D.P.; Pessoa, A.; Doriqui, M.J.R.; Neri, J.I.; de Pina Neto, J.M.; Wanderley, H.Y.C.; et al. Possible association between Zika virus infection and microcephaly — Brazil, 2015. Morb. Mortal. Wkly. Rep. 2016, 65, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Lazear, H.M. Zika virus-reigniting the TORCH. Nat. Rev. Microbiol. 2016, 14, 707–715. [Google Scholar] [CrossRef]
- Adams Waldorf, K.M.; McAdams, R.M. Influence of infection during pregnancy on fetal development. Reproduction 2013, 146, R151–R162. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Yuan, L.; Ross, D.; Johansen, A.; Sands, D.; Stanley, V.; Guemez-Gamboa, A.; Gregor, A.; Evans, T.; et al. Zika Virus Protease Cleavage of Host Protein Septin-2 Mediates Mitotic Defects in Neural Progenitors. Neuron 2019, 101, 1089–1098.e4. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugières, P.; Fourati, S.; De Langavant, L.C.; De Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. To the Editor. N. Engl. J. Med. 2016, 374, 1595–1597. [Google Scholar] [CrossRef]
- Pradhan, F.; Burns, J.D.; Agameya, A.; Patel, A.; Alfaqih, M.; Small, J.E.; Ooi, W. Case report: Zika virus meningoencephalitis and myelitis and associated magnetic resonance imaging findings. Am. J. Trop. Med. Hyg. 2017, 97, 340–343. [Google Scholar] [CrossRef]
- Bottari, N.B.; Schetinger, M.R.C.; Pillat, M.M.; Palma, T.V.; Ulrich, H.; Alves, M.S.; Morsch, V.M.; Melazzo, C.; de Barros, L.D.; Garcia, J.L.; et al. Resveratrol as a Therapy to Restore Neurogliogenesis of Neural Progenitor Cells Infected by Toxoplasma gondii. Mol. Neurobiol. 2019, 56, 2328–2338. [Google Scholar] [CrossRef]
- Wang, T.; Zhou, J.; Gan, X.; Wang, H.; Ding, X.; Chen, L.; Wang, Y.; Du, J.; Shen, J.; Yu, L. Toxoplasma gondii induce apoptosis of neural stem cells via endoplasmic reticulum stress pathway. Parasitology 2014, 141, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Parlog, A.; Schlüter, D.; Dunay, I.R. Toxoplasma gondii-induced neuronal alterations. Parasite Immunol. 2015, 37, 159–170. [Google Scholar] [CrossRef]
- Prandovszky, E.; Gaskell, E.; Martin, H.; Dubey, J.P.; Webster, J.P.; McConkey, G.A. The Neurotropic Parasite Toxoplasma Gondii Increases Dopamine Metabolism. PLoS ONE 2011, 6, e23866. [Google Scholar] [CrossRef] [PubMed]
- Dunn, A.J.; Wang, J. Cytokine effects on CNS biogenic amines. Neuroimmunomodulation 1995, 2, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Anisman, H.; Merali, Z.; Hayley, S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: Comorbidity between depression and neurodegenerative disorders. Prog. Neurobiol. 2008, 85, 1–74. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Psiquiatr. Biol. 2010, 17, 71–80. [Google Scholar] [CrossRef]
- Parlog, A.; Harsan, L.A.; Zagrebelsky, M.; Weller, M.; Von Elverfeldt, D.; Mawrin, C.; Korte, M.; Dunay, I.R. Chronic murine toxoplasmosis is defined by subtle changes in neuronal connectivity. DMM Dis. Model. Mech. 2014, 7, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Ngô, H.M.; Zhou, Y.; Lorenzi, H.; Wang, K.; Kim, T.K.; Zhou, Y.; Bissati, K.E.; Mui, E.; Fraczek, L.; Rajagopala, S.V.; et al. Toxoplasma Modulates Signature Pathways of Human Epilepsy, Neurodegeneration & Cancer. Sci. Rep. 2017, 7, 1–32. [Google Scholar] [CrossRef]
- Pedersen, M.G.; Mortensen, P.B.; Norgaard-Pedersen, B.; Postolache, T.T. Toxoplasma gondii infection and self-directed violence in mothers. Arch. Gen. Psychiatry 2012, 69, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, S.; Pinto, B.; Bruschi, F. Toxoplasmosis and neuropsychiatric diseases: Can serological studies establish a clear relationship? Neurol. Sci. 2013, 34, 417–425. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Pham, V.H.; Abe, K. Pathogenesis of Congenital Rubella Virus Infection in Human Fetuses: Viral Infection in the Ciliary Body Could Play an Important Role in Cataractogenesis. EBioMedicine 2015, 2, 59–63. [Google Scholar] [CrossRef]
- Rorke, L.B.; Spiro, A.J. Cerebral lesions in congenital rubella syndrome. J. Pediatr. 1967, 70, 243–255. [Google Scholar] [CrossRef]
- Rorke, L.B. Nervous System Lesions in the Congenital Rubella Syndrome. Arch. Otolaryngol. 1973, 98, 249–251. [Google Scholar] [CrossRef]
- Sun, G.; Chiuppesi, F.; Chen, X.; Wang, C.; Tian, E.; Nguyen, J.; Kha, M.; Trinh, D.; Zhang, H.; Marchetto, M.C.; et al. Modeling Human Cytomegalovirus-Induced Microcephaly in Human iPSC-Derived Brain Organoids. Cell Reports Med. 2020, 1, 100002. [Google Scholar] [CrossRef]
- McCarthy, M.; Auger, D.; Whittemore, S.R. Human cytomegalovirus causes productive infection and neuronal injury in differentiating fetal human central nervous system neuroepithelial precursor cells. J. Hum. Virol. 2000, 3, 215–228. [Google Scholar]
- Cheeran, M.C.J.; Hu, S.; Ni, H.T.; Sheng, W.; Palmquist, J.M.; Peterson, P.K.; Lokensgard, J.R. Neural precursor cell susceptibility to human cytomegalovirus diverges along glial or neuronal differentiation pathways. J. Neurosci. Res. 2005, 82, 839–850. [Google Scholar] [CrossRef]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, A.; Soderberg-Naucler, C. Human Cytomegalovirus Inhibits Neuronal Differentiation and Induces Apoptosis in Human Neural Precursor Cells. J. Virol. 2006, 80, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Lokensgard, J.R.; Cheeran, M.C.J.; Gekker, G.; Hu, S.; Chao, C.C.; Peterson, P.K. Human cytomegalovirus replication and of apoptosis in astrocytes. J. Hum. Virol. 1999, 2, 91–101. [Google Scholar] [PubMed]
- Van Den Pol, A.N.; Mocarski, E.; Saederup, N.; Vieira, J.; Meier, T.J. Cytomegalovirus cell tropism, replication, and gene transfer in brain. J. Neurosci. 1999, 19, 10948–10965. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Hannemann, H.; Kulkarni, A.S.; Schwartz, P.H.; O’Dowd, J.M.; Fortunato, E.A. Human Cytomegalovirus Infection Causes Premature and Abnormal Differentiation of Human Neural Progenitor Cells. J. Virol. 2010, 84, 3528–3541. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Di Maio, R.; Heath, B.; Raimondi, G.; Milosevic, J.; Watson, A.M.; Bamne, M.; Parks, W.T.; Yang, L.; Lin, B.; et al. Human Induced Pluripotent Stem Cell-Derived Models to Investigate Human Cytomegalovirus Infection in Neural Cells. PLoS ONE 2012, 7, 1–12. [Google Scholar] [CrossRef]
- Rotschafer, J.H.; Hu, S.; Little, M.; Erickson, M.; Low, W.C.; Cheeran, M.C.J. Modulation of neural stem/progenitor cell proliferation during experimental Herpes Simplex encephalitis is mediated by differential FGF-2 expression in the adult brain. Neurobiol. Dis. 2013, 58, 144–155. [Google Scholar] [CrossRef]
- Li Puma, D.D.; Piacentini, R.; Leone, L.; Gironi, K.; Marcocci, M.E.; De Chiara, G.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-β Protein Accumulation. Stem Cells 2019, 37, 1467–1480. [Google Scholar] [CrossRef]
- Cheung, N.S.; Carroll, F.Y.; Larm, J.A.; Beart, P.M.; Giardina, S.F. Kainate-induced apoptosis correlates with c-Jun activation in cultured cerebellar granule cells. J. Neurosci. Res. 1998, 52, 69–82. [Google Scholar] [CrossRef]
- Gill, J.S.; Windebank, A.J. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J. Clin. Investig. 1998, 101, 2842–2850. [Google Scholar] [CrossRef]
- Nuydens, R.; De Jong, M.; Van Den Kieboom, G.; Heers, C.; Dispersyn, G.; Cornelissen, F.; Nuyens, R.; Borgers, M.; Geerts, H. Okadaic acid-induced apoptosis in neuronal cells: Evidence for an abortive mitotic attempt. J. Neurochem. 1998, 70, 1124–1133. [Google Scholar] [CrossRef]
- Park, D.S.; Morris, E.J.; Padmanabhan, J.; Shelanski, M.L.; Geller, H.M.; Greene, L.A. Cyclin-dependent kinases participate in death of neurons evoked by DNA- damaging agents. J. Cell Biol. 1998, 143, 457–467. [Google Scholar] [CrossRef]
- Martin, C.; Leyton, L.; Hott, M.; Arancibia, Y.; Spichiger, C.; McNiven, M.A.; Court, F.A.; Concha, M.I.; Burgos, P.V.; Otth, C. Herpes simplex virus type 1 neuronal infection perturbs golgi apparatus integrity through activation of src tyrosine kinase and Dyn-2 GTPase. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, L.; Prasad, K.M.; Upton, C.H.; Viggiano, L.; Milosevic, J.; Raimondi, G.; McClain, L.; Chowdari, K.; Tischfield, J.; Sheldon, M.; et al. Persistent infection by HSV-1 is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance. Schizophr. Bull. 2015, 41, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Piacentini, R.; Li Puma, D.D.; Ripoli, C.; Elena Marcocci, M.; De Chiara, G.; Garaci, E.; Teresa Palamara, A.; Grassi, C. Herpes Simplex Virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-β protein accumulation. Sci. Rep. 2015, 5, 15444. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Itzhaki, R.F.; Shipley, S.J.; Dobson, C.B. Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci. Lett. 2007, 429, 95–100. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef]
- Piacentini, R.; Civitelli, L.; Ripoli, C.; Marcocci, M.E.; De Chiara, G.; Garaci, E.; Azzena, G.B.; Palamara, A.T.; Grassi, C. HSV-1 promotes Ca2+-mediated APP phosphorylation and Aβ accumulation in rat cortical neurons. Neurobiol. Aging 2011, 32, 2323.e13–2323.e26. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.; Mee, A.P.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Lerchundi, R.; Neira, R.; Valdivia, S.; Vio, K.; Concha, M.I.; Zambrano, A.; Otth, C. Tau cleavage at D421 by caspase-3 is induced in neurons and astrocytes infected with Herpes Simplex Virus Type 1. J. Alzheimer’s Dis. 2011, 23, 513–520. [Google Scholar] [CrossRef]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, 1–30. [Google Scholar] [CrossRef]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D human brain–like tissue model of herpes-induced Alzheimer’s disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef] [PubMed]
- Tsueng, G.; Tabor-Godwin, J.M.; Gopal, A.; Ruller, C.M.; Deline, S.; An, N.; Frausto, R.F.; Milner, R.; Crocker, S.J.; Whitton, J.L.; et al. Coxsackievirus Preferentially Replicates and Induces Cytopathic Effects in Undifferentiated Neural Progenitor Cells. J. Virol. 2011, 85, 5718–5732. [Google Scholar] [CrossRef]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B Exits the Host Cell in Shed Microvesicles Displaying Autophagosomal Markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Feuer, R.; Mena, I.; Pagarigan, R.R.; Harkins, S.; Hassett, D.E.; Whitton, J.L. Coxsackievirus B3 and the neonatal CNS: The roles of stem cells, developing neurons, and apoptosis in infection, viral dissemination, and disease. Am. J. Pathol. 2003, 163, 1379–1393. [Google Scholar] [CrossRef]
- Ruller, C.M.; Tabor-Godwin, J.M.; Van Deren, D.A.; Robinson, S.M.; MacIejewski, S.; Gluhm, S.; Gilbert, P.E.; An, N.; Gude, N.A.; Sussman, M.A.; et al. Neural stem cell depletion and CNS developmental defects after enteroviral infection. Am. J. Pathol. 2012, 180, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F.; Pistello, M. Zika virus: Implications for public health. Clin. Infect. Dis. 2016, 63, 227–233. [Google Scholar] [CrossRef]
- Vasudevan, J.; Skandhan, A.; Skandhan, A.K.P.; Balakrishnan, S.; Skandhand, K.P. Zika virus. Rev. Med. Microbiol. 2018, 29, 43–50. [Google Scholar] [CrossRef]
- Ming, G.L.; Tang, H.; Song, H. Advances in Zika Virus Research: Stem Cell Models, Challenges, and Opportunities. Cell Stem Cell 2016, 19, 690–702. [Google Scholar] [CrossRef]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef]
- Helgason, E.; Phung, Q.T.; Dueber, E.C. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: The emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013, 587, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Nguyen, J.; Johnson, J.; Haura, E.; Coppola, D.; Chellappan, S. Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun. 2015, 6, 10072. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Oshiumi, H.; Miyashita, M.; Aly, H.H.; Matsumoto, M.; Seya, T. Cell Type-Specific Subcellular Localization of Phospho-TBK1 in Response to Cytoplasmic Viral DNA. PLoS ONE 2013, 8, e83639. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Leon, W.R.M.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Nilles, E.J.; Cao-Lormeau, V.M. Rapid spread of emerging Zika virus in the Pacific area. Clin. Microbiol. Infect. 2014, 20, O595–O596. [Google Scholar] [CrossRef]
- Chen, L.H.; Hamer, D.H. Zika Virus: Rapid spread in the western hemisphere. Ann. Intern. Med. 2016, 164, 613–615. [Google Scholar] [CrossRef]
- McAuley, J.B. Congenital Toxoplasmosis. J. Pediatric Infect. Dis. Soc. 2014, 3, S30–S35. [Google Scholar] [CrossRef]
- Di Genova, B.M.; Wilson, S.K.; Dubey, J.P.; Knoll, L.J. Intestinal delta-6-desaturase activity determines host range for Toxoplasma sexual reproduction. PLoS Biol. 2019, 17, e3000364. [Google Scholar] [CrossRef]
- Black, M.W.; Boothroyd, J.C. Lytic Cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 2000, 64, 607–623. [Google Scholar] [CrossRef]
- Hill, D.E.; Chirukandoth, S.; Dubey, J.P. Biology and epidemiology of Toxoplasma gondii in man and animals. Anim. Health Res. Rev. 2005, 6, 41–61. [Google Scholar] [CrossRef]
- Tanaka, N.; Ashour, D.; Dratz, E.; Halonen, S. Use of human induced pluripotent stem cell-derived neurons as a model for Cerebral Toxoplasmosis. Microbes Infect. 2016, 18, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gan, X.; Wang, Y.; Zhang, X.; Ding, X.; Chen, L.; Du, J.; Luo, Q.; Wang, T.; Shen, J.; et al. Toxoplasma gondii prevalent in China induce weaker apoptosis of neural stem cells C17.2 via endoplasmic reticulum stress (ERS) signaling pathways. Parasit. Vectors 2015, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Reef, S.E.; Cooper, L.Z.; Alford, C.A. Rubella. In Infectious Diseases of the Fetus and Newborn; Elsevier: Amsterdam, The Netherlands, 2011; pp. 861–898. ISBN 9781416064008. [Google Scholar]
- Kemper, T.L.; Lecours, A.R.; Gates, M.J.; Yakovlev, P.I. Retardation of the myelo- and cytoarchitectonic maturation of the brain in the congenital rubella syndrome. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1973, 51, 23–62. [Google Scholar] [PubMed]
- Lazar, M.; Perelygina, L.; Martines, R.; Greer, P.; Paddock, C.D.; Peltecu, G.; Lupulescu, E.; Icenogle, J.; Zaki, S.R. Immunolocalization and Distribution of Rubella Antigen in Fatal Congenital Rubella Syndrome. EBioMedicine 2016, 3, 86–92. [Google Scholar] [CrossRef]
- Chantler, J.K.; Smyrnis, L.; Tai, G. Selective infection of astrocytes in human glial cell cultures by rubella virus. Lab. Investig. 1995, 72, 334–340. [Google Scholar]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef]
- Naing, Z.W.; Scott, G.M.; Shand, A.; Hamilton, S.T.; Van Zuylen, W.J.; Basha, J.; Hall, B.; Craig, M.E.; Rawlinson, W.D. Congenital cytomegalovirus infection in pregnancy: A review of prevalence, clinical features, diagnosis and prevention. Aust. New Zeal. J. Obstet. Gynaecol. 2016, 56, 9–18. [Google Scholar] [CrossRef]
- Britt, W. Manifestations of Human Cytomegalovirus Infection: Proposed Mechanisms of Acute and Chronic Disease. In Current Topics in Microbiology and Immunology; Springer: Heidelberg, Germany, 2008; Volume 325, pp. 417–470. ISBN 9783540773481. [Google Scholar]
- Stinski, M.F.; Petrik, D.T. Functional Roles of the Human Cytomegalovirus Essential IE86 Protein. In Current Topics in Microbiology and Immunology; Springer: Heidelberg, Germany, 2008; Volume 325, pp. 133–152. ISBN 9783540773481. [Google Scholar]
- Marchini, A.; Liu, H.; Zhu, H. Human Cytomegalovirus with IE-2 (UL122) Deleted Fails To Express Early Lytic Genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef]
- Mercorelli, B.; Luganini, A.; Nannetti, G.; Tabarrini, O.; Palù, G.; Gribaudo, G.; Loregian, A. Drug Repurposing Approach Identifies Inhibitors of the Prototypic Viral Transcription Factor IE2 that Block Human Cytomegalovirus Replication. Cell Chem. Biol. 2016, 23, 340–351. [Google Scholar] [CrossRef]
- Mercorelli, B.; Luganini, A.; Celegato, M.; Palù, G.; Gribaudo, G.; Loregian, A. Repurposing the clinically approved calcium antagonist manidipine dihydrochloride as a new early inhibitor of human cytomegalovirus targeting the Immediate-Early 2 (IE2) protein. Antiviral Res. 2018, 150, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Sundtröm, E.; Seiger, Å.; Söderberg-Nauclér, C. Late human cytomegalovirus (HCMV) proteins inhibit differentiation of human neural precursor cells into astrocytes. J. Neurosci. Res. 2007, 85, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Schwartz, P.H.; Fortunato, E.A. Neonatal Neural Progenitor Cells and Their Neuronal and Glial Cell Derivatives Are Fully Permissive for Human Cytomegalovirus Infection. J. Virol. 2008, 82, 9994–10007. [Google Scholar] [CrossRef] [PubMed]
- Becroft, D.M. Prenatal cytomegalovirus infection: Epidemiology, pathology and pathogenesis. Perspect. Pediatr. Pathol. 1981, 6, 203–241. [Google Scholar]
- Bale, J.F.; Bray, P.F.; Bell, W.E. Neuroradiographic abnormalities in congenital cytomegalovirus infection. Pediatr. Neurol. 1985, 1, 42–47. [Google Scholar] [CrossRef]
- Perlman, J.M.; Argyle, C. Lethal cytomegalovirus infection in preterm infants: Clinical, radiological, and neuropathological findings. Ann. Neurol. 1992, 31, 64–68. [Google Scholar] [CrossRef]
- Sison, S.L.; O’Brien, B.S.; Johnson, A.J.; Seminary, E.R.; Terhune, S.S.; Ebert, A.D. Human Cytomegalovirus Disruption of Calcium Signaling in Neural Progenitor Cells and Organoids. J. Virol. 2019, 93, e00954-19. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron 1996, 17, 275–285. [Google Scholar] [CrossRef]
- Spitzer, N.C. Electrical activity in early neuronal development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef]
- Zheng, J.Q.; Poo, M. Calcium Signaling in Neuronal Motility. Annu. Rev. Cell Dev. Biol. 2007, 23, 375–404. [Google Scholar] [CrossRef]
- Liu, X.; Hashimoto-Torii, K.; Torii, M.; Ding, C.; Rakic, P. Gap junctions/hemichannels modulate interkinetic nuclear migration in the forebrain precursors. J. Neurosci. 2010, 30, 4197–4209. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.S.; Spitzer, N.C. Calcium signaling in neuronal development. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, A.; Shum, A.K.; McBride, H.J.; Kessler, J.A.; Feske, S.; Miller, R.J.; Prakriya, M. Store-operated CRAC channels regulate gene expression and proliferation in neural progenitor cells. J. Neurosci. 2014, 34, 9107–9123. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Irie, K.; Shimomura, T.; Umeshima, H.; Kushida, Y.; Kengaku, M.; Fujiyoshi, Y.; Hirano, T.; Tagawa, Y. Control of Spontaneous Ca2+ Transients Is Critical for Neuronal Maturation in the Developing Neocortex. Cereb. Cortex 2016, 26, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Toth, A.B.; Shum, A.K.; Prakriya, M. Regulation of neurogenesis by calcium signaling. Cell Calcium 2016, 59, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Chai, G.; Brewer, J.M.; Lovelace, L.L.; Lebioda, L. Fluoride inhibition of enolase: Crystal structure and thermodynamics. Biochemistry 2006, 45, 793–800. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Hear. Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef]
- Shumilina, E.; Zemtsova, I.M.; Heise, N.; Schmid, E.; Eichenmüller, M.; Tyan, L.; Rexhepaj, R.; Lang, F. Phosphoinositide-dependent kinase PDK1 in the Regulation of Ca 2+ entry into mast cells. Cell. Physiol. Biochem. 2010, 26, 699–706. [Google Scholar] [CrossRef]
- Belzile, J.-P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human Cytomegalovirus Infection of Human Embryonic Stem Cell-Derived Primitive Neural Stem Cells Is Restricted at Several Steps but Leads to the Persistence of Viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef]
- Hobbs, W.E.; DeLuca, N.A. Perturbation of Cell Cycle Progression and Cellular Gene Expression as a Function of Herpes Simplex Virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar] [CrossRef]
- Xu, F.; Schillinger, J.A.; Sternberg, M.R.; Johnson, R.E.; Lee, F.K.; Nahmias, A.J.; Markowitz, L.E. Seroprevalence and Coinfection with Herpes Simplex Virus Type 1 and Type 2 in the United States, 1988–1994. J. Infect. Dis. 2002, 185, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Pebody, R.G.; Andrews, N.; Brown, D.; Gopal, R.; De Melker, H.; François, G.; Gatcheva, N.; Hellenbrand, W.; Jokinen, S.; Klavs, I.; et al. The seroepidemiology of herpes simplex virus type 1 and 2 in Europe. Sex. Transm. Infect. 2004, 80, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.A. Sexually Transmitted Diseases. In Molecular Pathology in Clinical Practice; Springer International Publishing: Cham, Switzerland, 2016; pp. 735–753. ISBN 9783319196749. [Google Scholar]
- Opstelten, W.; Neven, A.K.; Eekhof, J. Treatment and prevention of herpes labialis. Can. Fam. Phys. 2008, 54, 1683–1687. [Google Scholar]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grünewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar] [CrossRef]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Braun, E.; Zimmerman, T.; Hur, T.B.; Reinhartz, E.; Fellig, Y.; Panet, A.; Steiner, I. Neurotropism of herpes simplex virus type 1 in brain organ cultures. J. Gen. Virol. 2006, 87, 2827–2837. [Google Scholar] [CrossRef]
- Sun, X.; Shi, L.; Zhang, H.; Li, R.; Liang, R.; Liu, Z. Effects of toll-like receptor 3 on herpes simplex virus type-1-infected mouse neural stem cells. Can. J. Microbiol. 2015, 61, 201–208. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Z.; Shu, B.; Liu, X.; Zhang, Z.; Liu, Y.; Bai, B.; Hu, Q.; Mao, P.; Wang, H. Human Astrocytic Cells Support Persistent Coxsackievirus B3 Infection. J. Virol. 2013, 87, 12407–12421. [Google Scholar] [CrossRef]
- Muir, P.; Tilzey, A.J.; English, T.A.H.; Nicholson, F.; Signy, M.; Banatvala, J.E. Chronic Relapsing Pericarditis and Dilated Cardiomyopathy: Serological Evidence of Persistent Enterovirus Infection. Lancet 1989, 333, 804–807. [Google Scholar] [CrossRef]
- Chapman, N.M.; Kim, K.S. Persistent coxsackievirus infection: Enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr. Top. Microbiol. Immunol. 2008, 323, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.; Chia, A.; Voeller, M.; Lee, T.; Chang, R. Acute enterovirus infection followed by myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and viral persistence. J. Clin. Pathol. 2010, 63, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Tracy, S.; Tapprich, W.; Bailey, J.; Lee, C.-K.; Kim, K.; Barry, W.H.; Chapman, N.M. 5′-Terminal Deletions Occur in Coxsackievirus B3 during Replication in Murine Hearts and Cardiac Myocyte Cultures and Correlate with Encapsidation of Negative-Strand Viral RNA. J. Virol. 2005, 79, 7024–7041. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-S.; Chapman, N.M.; Tracy, S. Replication of Coxsackievirus B3 in Primary Cell Cultures Generates Novel Viral Genome Deletions. J. Virol. 2008, 82, 2033–2037. [Google Scholar] [CrossRef]
- Quaranta, P.; Lottini, G.; Chesi, G.; Contrafatto, F.; Russotto, R.; Macera, L.; Lai, M.; Spezia, P.G.; Brai, A.; Botta, M.; et al. DDX3 inhibitors show antiviral activity against positive-sense single-stranded RNA viruses but not against negative-sense single-stranded RNA viruses: The coxsackie B model. Antiviral Res. 2020, 178, 104750. [Google Scholar] [CrossRef]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastère, S.; Valour, F.; Baudouin, L.; Mallet, H.P.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barré syndrome—Case report, French Polynesia, December 2013. Eurosurveillance 2014, 19. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barré syndrome associated with Zika virus infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Petersen, E.; Wilson, M.E.; Touch, S.; McCloskey, B.; Mwaba, P.; Bates, M.; Dar, O.; Mattes, F.; Kidd, M.; Ippolito, G.; et al. Rapid Spread of Zika Virus in The Americas—Implications for Public Health Preparedness for Mass Gatherings at the 2016 Brazil Olympic Games. Int. J. Infect. Dis. 2016, 44, 11–15. [Google Scholar] [CrossRef]
- Barbi, L.; Coelho, A.V.C.; de Alencar, L.C.A.; Crovella, S. Prevalence of Guillain-Barré syndrome among Zika virus infected cases: A systematic review and meta-analysis. Brazilian J. Infect. Dis. 2018, 22, 137–141. [Google Scholar] [CrossRef]
- Carod-Artal, F.J. Neurological complications of Zika virus infection. Expert Rev. Anti. Infect. Ther. 2018, 16, 399–410. [Google Scholar] [CrossRef] [PubMed]
- de Araújo, P.S.R.; Silva Júnior, M.L.d.M.; Tenório, M.; dos Santos, F.G.T. Co-infection ZIKV and HSV-1 associated with meningoencephalitis: Case report and literature review. J. Infect. Public Health 2019, 12, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Tenter, A.M.; Heckeroth, A.R.; Weiss, L.M. Toxoplasma gondii: From animals to humans. Int. J. Parasitol. 2000, 30, 1217–1258. [Google Scholar] [CrossRef]
- Boothroyd, J.C.; Grigg, M.E. Population biology of Toxoplasma gondii and its relevance to human infection: Do different strains cause different disease? Curr. Opin. Microbiol. 2002, 5, 438–442. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 2012, 37, 137–162. [Google Scholar] [CrossRef]
- Saper, C.B.; Romanovsky, A.A.; Scammell, T.E. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat. Neurosci. 2012, 15, 1088–1095. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, X.; Kang, A.; Ran, C.; Wang, G.; Hao, H. Thinking outside the brain for cognitive improvement: Is peripheral immunomodulation on the way? Neuropharmacology 2015, 96, 94–104. [Google Scholar] [CrossRef]
- Fischer, H.G.; Nitzgen, B.; Reichmann, G.; Hadding, U. Cytokine responses induced by Toxoplasma gondii in astrocytes and microglial cells. Eur. J. Immunol. 1997, 27, 1539–1548. [Google Scholar] [CrossRef]
- Schlüter, D.; Kaefer, N.; Hof, H.; Wiestler, O.D.; Deckert-Schlüter, M. Expression pattern and cellular origin of cytokines in the normal and Toxoplasma gondii-infected murine brain. Am. J. Pathol. 1997, 150, 1021–1035. [Google Scholar] [PubMed]
- Jebbari, H.; Roberts, C.W.; Ferguson, D.J.P.; Bluethmann, H.; Alexander, J. A protective role for IL-6 during early infection with Toxoplasma gondii. Parasite Immunol. 1998, 20, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, D.; Deckert, M.; Hof, H.; Frei, K. Toxoplasma gondii infection of neurons induces neuronal cytokine and chemokine production, but gamma interferon- and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and growth of T. gondii. Infect. Immun. 2001, 69, 7889–7893. [Google Scholar] [CrossRef]
- Strack, A.; Asensio, V.C.; Campbell, I.L.; Schlüter, D.; Deckert, M. Chemokines are differentially expressed by astrocytes, microglia and inflammatory leukocytes in Toxoplasma encephalitis and critically regulated by interferon-γ. Acta Neuropathol. 2002, 103, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Händel, U.; Brunn, A.; Drögemüller, K.; Müller, W.; Deckert, M.; Schlüter, D. Neuronal gp130 expression is crucial to prevent neuronal loss, hyperinflammation, and lethal course of murine toxoplasma encephalitis. Am. J. Pathol. 2012, 181, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Money, K.M.; Stanwood, G.D. Developmental origins of brain disorders: Roles for dopamine. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.; Gross, U.; Lüder, C.G.K. Inhibition of host cell apoptosis by Toxoplasma gondii is accompanied by reduced activation of the caspase cascade and alterations of poly(ADP-ribose) polymerase expression. J. Cell Sci. 2001, 114, 3495–3505. [Google Scholar] [PubMed]
- Orlofsky, A.; Weiss, L.M.; Kawachi, N.; Prystowsky, M.B. Deficiency in the Anti-Apoptotic Protein A1-a Results in a Diminished Acute Inflammatory Response. J. Immunol. 2002, 168, 1840–1846. [Google Scholar] [CrossRef]
- Kim, L.; Denkers, E.Y. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J. Cell Sci. 2006, 119, 2119–2126. [Google Scholar] [CrossRef]
- Vutova, P.; Wirth, M.; Hippe, D.; Gross, U.; Schulze-Osthoff, K.; Schmitz, I.; Lüder, C.G.K. Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell. Microbiol. 2007, 9, 1556–1570. [Google Scholar] [CrossRef]
- Saito, K.; Markey, S.P.; Heyes, M.P. Chronic effects of γ-interferon on quinolinic acid and indoleamine-2,3-dioxygenase in brain of C57BL6 mice. Brain Res. 1991, 546, 151–154. [Google Scholar] [CrossRef]
- Catena-Dell’Osso, M.; Rotella, F.; Dell’Osso, A.; Fagiolini, A.; Marazziti, D. Inflammation, Serotonin and Major Depression. Curr. Drug Targets 2013, 14, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Neurauter, G.; Schrocksnadel, K.; Scholl-Burgi, S.; Sperner-Unterweger, B.; Schubert, C.; Ledochowski, M.; Fuchs, D. Chronic Immune Stimulation Correlates with Reduced Phenylalanine Turnover. Curr. Drug Metab. 2008, 9, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Kim, S.K.; Giacomini, N.; Boothroyd, J.C.; Sapolsky, R.M. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc. Natl. Acad. Sci. USA 2007, 104, 6442–6447. [Google Scholar] [CrossRef] [PubMed]
- Havlícek, J.; Gašová, Z.; Smith, A.P.; Zvára, K.; Flegr, J. Decrease of psychomotor performance in subjects with latent “asymptomatic” toxoplasmosis. Parasitology 2001, 122, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Stock, A.-K.; Heintschel von Heinegg, E.; Köhling, H.-L.; Beste, C. Latent Toxoplasma gondii infection leads to improved action control. Brain. Behav. Immun. 2014, 37, 103–108. [Google Scholar] [CrossRef]
- Jung, B.K.; Pyo, K.H.; Shin, K.Y.; Hwang, Y.S.; Lim, H.; Lee, S.J.; Moon, J.H.; Lee, S.H.; Suh, Y.H.; Chai, J.Y.; et al. Toxoplasma gondii infection in the brain inhibits neuronal degeneration and learning and memory impairments in a murine model of alzheimer’s disease. PLoS ONE 2012, 7, e33312. [Google Scholar] [CrossRef]
- Brown, A.S. Prenatal infection as a risk factor for schizophrenia. Schizophr. Bull. 2006, 32, 200–202. [Google Scholar] [CrossRef]
- Torrey, E.F.; Bartko, J.J.; Lun, Z.R.; Yolken, R.H. Antibodies to Toxoplasma gondii in patients with schizophrenia: A meta-analysis. Schizophr. Bull. 2007, 33, 729–736. [Google Scholar] [CrossRef]
- Hinze-Selch, D.; Däubener, W.; Eggert, L.; Erdag, S.; Stoltenberg, R.; Wilms, S. A controlled prospective study of Toxoplasma gondii infection in individuals with schizophrenia: Beyond seroprevalence. Schizophr. Bull. 2007, 33, 782–788. [Google Scholar] [CrossRef]
- Mortensen, P.B.; Nørgaard-Pedersen, B.; Waltoft, B.L.; Sørensen, T.L.; Hougaard, D.; Yolken, R.H. Early infections of Toxoplasma gondii and the later development of schizophrenia. Schizophr. Bull. 2007, 33, 741–744. [Google Scholar] [CrossRef]
- Zhu, S. Psychosis may be associated with toxoplasmosis. Med. Hypotheses 2009, 73, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Pellicciari, R. Manipulation of brain kynurenines: Glial targets, neuronal effects, and clinical opportunities. J. Pharmacol. Exp. Ther. 2002, 303, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.; Resnick, L.; Taub, F.; Stewart, R.V.; Dix, R.D. Infection of human neural cell aggregate cultures with a clinical isolate of cytomegalovirus. J. Neuropathol. Exp. Neurol. 1991, 50, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Lecointe, D.; Héry, C.; Janabi, N.; Dussaix, E.; Tardieu, M. Differences in kinetics of human cytomegalovirus cell-free viral release after in vitro infection of human microglial cells, astrocytes and monocyte-derived macrophages. J. Neurovirol. 1999, 5, 308–313. [Google Scholar] [CrossRef]
- Isomura, H.; Stinski, M.F. The Human Cytomegalovirus Major Immediate-Early Enhancer Determines the Efficiency of Immediate-Early Gene Transcription and Viral Replication in Permissive Cells at Low Multiplicity of Infection. J. Virol. 2003, 77, 3602–3614. [Google Scholar] [CrossRef]
- Kasparov, S. Suitability of hCMV for viral gene expression in the brain. Nat. Methods 2007, 4, 379. [Google Scholar] [CrossRef]
- Zimmer, B.; Ewaleifoh, O.; Harschnitz, O.; Lee, Y.-S.; Peneau, C.; McAlpine, J.L.; Liu, B.; Tchieu, J.; Steinbeck, J.A.; Lafaille, F.; et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc. Natl. Acad. Sci. USA 2018, 115, E8775–E8782. [Google Scholar] [CrossRef]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef]
- Iwasawa, C.; Tamura, R.; Sugiura, Y.; Suzuki, S.; Kuzumaki, N.; Narita, M.; Suematsu, M.; Nakamura, M.; Yoshida, K.; Toda, M.; et al. Increased cytotoxicity of herpes simplex virus thymidine kinase expression in human induced pluripotent stem cells. Int. J. Mol. Sci. 2019, 20, 810. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Bloom, D.C.; Naciri, J.N.; Smith, A.; Edwards, T.G.; McClain, L.; Callio, J.A.; Jessup, M.; Wood, J.; Chowdari, K.; et al. Modeling Herpes Simplex Virus 1 Infections in Human Central Nervous System Neuronal Cells Using Two- and Three-Dimensional Cultures Derived from Induced Pluripotent Stem Cells. J. Virol. 2019, 93, e00111-19. [Google Scholar] [CrossRef]
- Rosato, P.C.; Leib, D.A. Neurons versus herpes simplex virus: The innate immune interactions that contribute to a host-pathogen standoff. Future Virol. 2015, 10, 699–714. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.E.; Greene, L.A.; Ziff, E.B. Nerve growth factor and epidermal growth factor induce rapid transient changes in proto-oncogene transcription in PC12 cells. J. Biol. Chem. 1985, 260, 14101–14110. [Google Scholar] [PubMed]
- Cole, A.J.; Saffen, D.W.; Baraban, J.M.; Worley, P.F. Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature 1989, 340, 474–476. [Google Scholar] [CrossRef]
- Shirvan, A.; Ziv, I.; Zilkha-Falb, R.; Machlyn, T.; Barzilai, A.; Melamed, E. Expression of cell cycle-related genes during neuronal apoptosis: Is there a distinct pattern? Neurochem. Res. 1998, 23, 767–777. [Google Scholar] [CrossRef]
- Cai, W.; Schaffer, P.A. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a transactivating protein of herpes simplex virus type 1. J. Virol. 1991, 65, 4078–4090. [Google Scholar] [CrossRef]
- Ralph, W.M.; Cabatingan, M.S.; Schaffer, P.A. Induction of herpes simplex virus type 1 immediate-early gene expression by a cellular activity expressed in Vero and NB41A3 cells after growth arrest-release. J. Virol. 1994, 68, 6871–6882. [Google Scholar] [CrossRef]
- Bauerfeind, R.; Huttner, W.B. Biogenesis of constitutive secretory vesicles, secretory granules and synaptic vesicles. Curr. Opin. Cell Biol. 1993, 5, 628–635. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Prim. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Montine, T.J. The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124, 305–323. [Google Scholar] [CrossRef]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and mGluR5. In Advances in Pharmacology; Academic Press: Cambridge, MA, USA, 2018; Volume 82, pp. 293–323. ISBN 9780128140871. [Google Scholar]
- Guerreiro, R.J.; Gustafson, D.R.; Hardy, J. The genetic architecture of Alzheimer’s disease: Beyond APP, PSENS and APOE. Neurobiol. Aging 2012, 33, 437–456. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Grenier-Boley, B.; Harold, D.; Zelenika, D.; Chouraki, V.; Kamatani, Y.; Sleegers, K.; Ikram, M.A.; Hiltunen, M.; Reitz, C.; et al. Genome-wide haplotype association study identifies the FRMD4A gene as a risk locus for Alzheimer’s disease. Mol. Psychiatry 2013, 18, 461–470. [Google Scholar] [CrossRef]
- Huang, K.L.; Marcora, E.; Pimenova, A.A.; Di Narzo, A.F.; Kapoor, M.; Jin, S.C.; Harari, O.; Bertelsen, S.; Fairfax, B.P.; Czajkowski, J.; et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1052–1061. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Mez, J.; Vardarajan, B.N.; Staley, L.; Chung, J.; Zhang, X.; Farrell, J.J.; Rynkiewicz, M.J.; Cannon-Albright, L.A.; Teerlink, C.C.; et al. Association of Rare Coding Mutations With Alzheimer Disease and Other Dementias Among Adults of European Ancestry. JAMA Netw. Open 2019, 2, e191350. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Chartier-hariln, M.C.; Parfitt, M.; Legrain, S.; Pérez-tur, J.; Brousseau, T.; Evans, A.; Berr, C.; Vldal, O.; Roques, P.; Gourlet, V.; et al. Apolipoprotein e, ɛ4 allele as a major risk factor for sporadic early and late-onset forms of alzheimer’s disease: Analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet. 1994, 3, 569–574. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Kosugi, I.; Shinmura, Y.; Kawasaki, H.; Arai, Y.; Li, R.Y.; Baba, S.; Tsutsui, Y. Cytomegalovirus infection of the central nervous system stem cells from mouse embryo: A model for developmental brain disorders induced by cytomegalovirus. Lab. Investig. 2000, 80, 1373–1383. [Google Scholar] [CrossRef]
- Feuer, R.; Pagarigan, R.R.; Harkins, S.; Liu, F.; Hunziker, I.P.; Whitton, J.L. Coxsackievirus targets proliferating neuronal progenitor cells in the neonatal CNS. J. Neurosci. 2005, 25, 2434–2444. [Google Scholar] [CrossRef]
- Mayer, D.; Fischer, H.; Schneider, U.; Heimrich, B.; Schwemmle, M. Borna Disease Virus Replication in Organotypic Hippocampal Slice Cultures from Rats Results in Selective Damage of Dentate Granule Cells. J. Virol. 2005, 79, 11716–11723. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.I.; Kang, Y.J.; Brechtel, C.W.; Siviglia, E.; Russo, R.; Clemente, A.; Harrop, A.; McKercher, S.; Kaul, M.; Lipton, S.A. HIV/gp120 Decreases Adult Neural Progenitor Cell Proliferation via Checkpoint Kinase-Mediated Cell-Cycle Withdrawal and G1 Arrest. Cell Stem Cell 2007, 1, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, G.; Zimmerman, M.N.; Denk, L.L.; Porter, D.D.; Prince, G.A. Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice. Arch. Virol. 2002, 147, 167–179. [Google Scholar] [CrossRef]
- Steiner, I. Herpes simplex virus encephalitis: New infection or reactivation? Curr. Opin. Neurol. 2011, 24, 268–274. [Google Scholar] [CrossRef]
- Ball, M.J. Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved? Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1982, 9, 303–306. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Yates, C.M.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J. Pathol. 1992, 167, 365–368. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Itabashi, S.; Arai, H.; Matsui, T.; Higuchi, S.; Sasaki, H. Herpes simplex virus and risk of Alzheimer’s disease. Lancet 1997, 349, 1102. [Google Scholar] [CrossRef]
- Pierrot, N.; Ferrao Santos, S.; Feyt, C.; Morel, M.; Brion, J.P.; Octave, J.N. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-β accumulation. J. Biol. Chem. 2006, 281, 39907–39914. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Gage, F.H. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Leone, L.; Colussi, C.; Gironi, K.; Longo, V.; Fusco, S.; Li Puma, D.D.; D’Ascenzo, M.; Grassi, C. Altered Nup153 Expression Impairs the Function of Cultured Hippocampal Neural Stem Cells Isolated from a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 5934–5949. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-G.; Jang, D.-J.; Son, J.; Kwak, C.; Choi, J.-H.; Ji, Y.-H.; Lee, Y.-S.; Son, H.; Kaang, B.-K. Effect of ablated hippocampal neurogenesis on the formation and extinction of contextual fear memory. Mol. Brain 2009, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, N.-S.; Chung, C.-H.; Lin, F.-H.; Chiang, C.-P.; Yeh, C.-B.; Huang, S.-Y.; Lu, R.-B.; Chang, H.-A.; Kao, Y.-C.; Yeh, H.-W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections—A Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
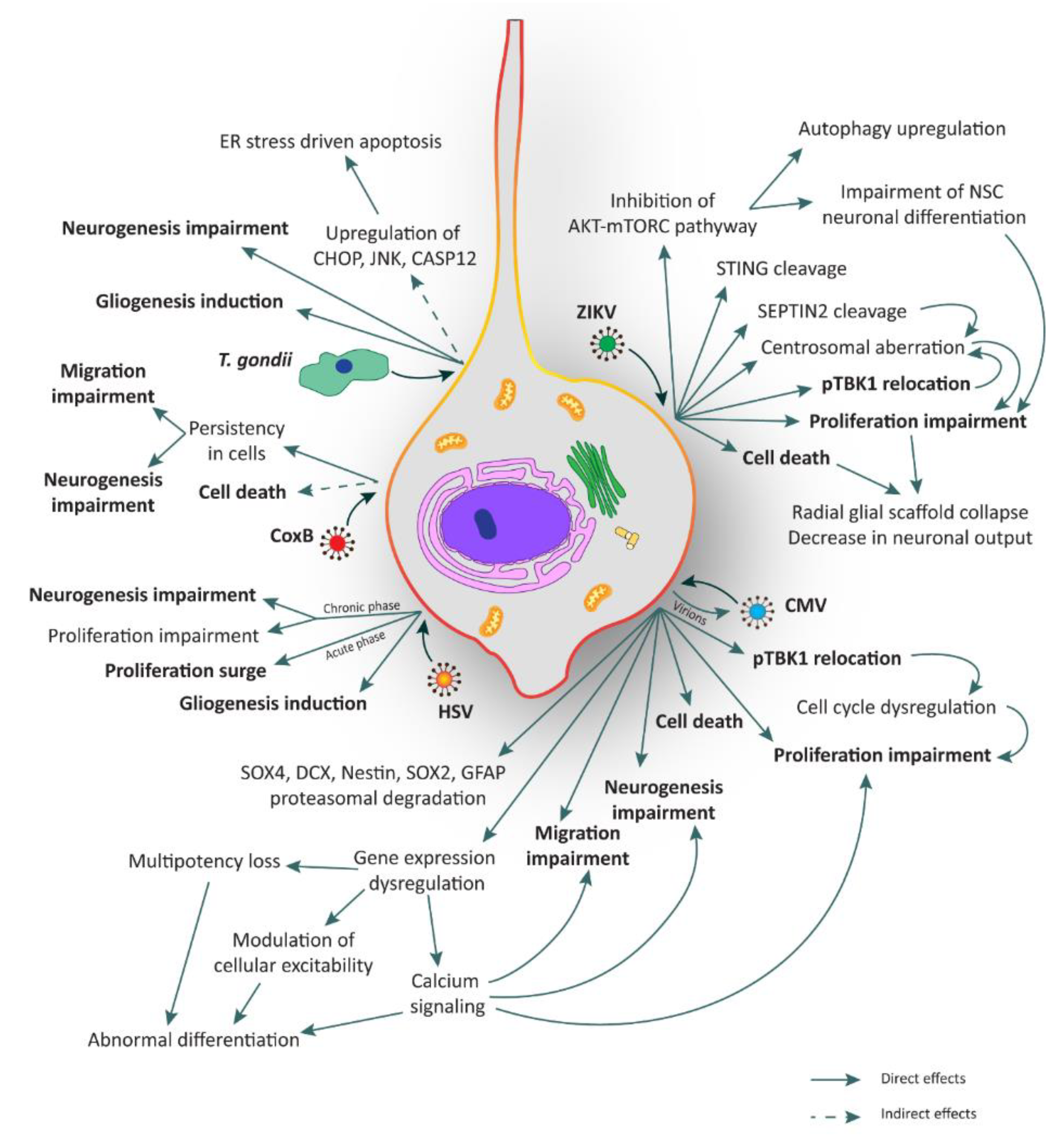
| Pathogen | Targets | Effects | Models | References |
|---|---|---|---|---|
| ZIKV | NPCs | AKT-mTORC pathway inhibition | Human fetal NSCs | [36] |
| STING cleavage | Human fibroblasts | [37] | ||
| SEPTIN2 cleavage | NPCs obtained from human H9 ESC-derived embryoid bodies | [38] | ||
| Centrosomal aberration | Human neocortical NES cells | [23] | ||
| pTBK1 relocation | Human neocortical NES cells | [23] | ||
| Proliferation impairment | Human iPSC-derived forebrain organoids | [39,40] | ||
| Radial scaffold disorganization and architectural impairment | Human organotypic fetal brain slices, post-mortem fetal brain samples | [23] | ||
| Cell death | Human iPSC-derived forebrain organoids | [39,40] | ||
| Neuronal progeny/Immune cells | Acute flaccid paralysis: (i) damage of motor neurons, (ii) Guillan–Barrè syndrome | / | [41] | |
| Meningoencephalitis | / | [42,43] | ||
| T. gondii | NPCs | Gliogenesis induction | Mouse NPCs | [44] |
| Neurogenesis impairment | Mouse NPCs | [44] | ||
| ER stress dependent apoptosis | Mouse NSCs | [45] | ||
| Neuronal progeny | Apoptosis | / | [46] | |
| Neurotransmitter metabolism alteration | Neural cells in mouse brain tissue | [46,47] | ||
| Neuroinflammation | / | [48,49,50] | ||
| Synaptic modification | Mouse model | [46,51] | ||
| Behavioral alterations and psychiatric diseases | Primary human temporal-lobe NSC lines | [46,52,53,54] | ||
| Rubella Virus | NPCs | Cell death | Autoptic fetal tissue | [55] |
| Neuronal progeny | Cell death | Autoptic fetal tissue | [55,56,57] | |
| CMV | NPCs | Neurogenesis impairment | Human iPSC-derived brain organoids | [58] |
| Migration impairment | Human iPSC-derived brain organoids | [58] | ||
| pTBK1 relocation | Human neocortical NES cells | [23] | ||
| Proliferation impairment | Human fetal brain-derived NPCs; human fetal NES cells; hNPCs | [59,60,61] | ||
| Cell death | Primary human neuronal cell cultures; human fetal NES cells; hNPCs; human iPSC-derived brain organoids | [58,59,61,62,63] | ||
| Dysregulation of genes involved in multipotency, modulation of cellular excitability and calcium signaling | Human iPSC-derived NSCs; hNPCs; human iPSC-derived brain organoids | [58,64,65] | ||
| SOX4, DCX, Nestin, SOX2 and GFAP proteasomal degradation | Human fetal brain-derived NPCs; hNPCs | [60,64] | ||
| Neuronal progeny | Apoptosis | Human iPSC-derived NSCs | [65] | |
| Downregulation of NMDA receptor | Human iPSC-derived NSCs | [65] | ||
| HSV | NPCs | Proliferation surge (in acute phase) | Mouse NSCs | [66] |
| Proliferation impairment (in chronic phase) | Mouse NSCs | [66] | ||
| Neurogenesis impairment (in chronic phase) | Mouse NSCs | [66] | ||
| APP fragmentation | Mouse adult hippocampal NSCs | [67] | ||
| Gliogenesis induction | Mouse adult hippocampal NSCs | [67] | ||
| Neuronal progeny | G1 re-entry stimulated apoptosis | Cerebellar granule cells; rat dorsal root ganglion neurons; human neuronal cell line; rat sympathetic neurons | [68,69,70,71] | |
| Golgi apparatus remodeling | Mouse cortical neurons | [72] | ||
| Changes in architecture and functional activity | Human iPSC-derived neurons | [73] | ||
| Decrease of synaptic transmission | Mouse cortical neurons | [74] | ||
| Accumulation of APP fragments | Primary cultures of cortical neurons from rat embryos; mouse brains | [75,76,77] | ||
| Increase of intracellular calcium | Rat cortical neurons | [77] | ||
| Increase of Tau phosphorylation and cleavage | Mouse fetal neurons; AD brain specimens | [78,79] | ||
| Neuroinflammation | Human brain organoids; mouse models | [80,81] | ||
| CoxB | NPCs | Cell death | Neonatal mice brain; mouse cortical NPCs | [82,83,84] |
| Migration and neurogenesis impairment | Neonatal mice central nervous system | [85] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baggiani, M.; Dell’Anno, M.T.; Pistello, M.; Conti, L.; Onorati, M. Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders. Cells 2020, 9, 1893. https://doi.org/10.3390/cells9081893
Baggiani M, Dell’Anno MT, Pistello M, Conti L, Onorati M. Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders. Cells. 2020; 9(8):1893. https://doi.org/10.3390/cells9081893
Chicago/Turabian StyleBaggiani, Matteo, Maria Teresa Dell’Anno, Mauro Pistello, Luciano Conti, and Marco Onorati. 2020. "Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders" Cells 9, no. 8: 1893. https://doi.org/10.3390/cells9081893
APA StyleBaggiani, M., Dell’Anno, M. T., Pistello, M., Conti, L., & Onorati, M. (2020). Human Neural Stem Cell Systems to Explore Pathogen-Related Neurodevelopmental and Neurodegenerative Disorders. Cells, 9(8), 1893. https://doi.org/10.3390/cells9081893