Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System †


 ,
,  ,
,
Abstract
1. Introduction
2. Development of Normal Pulmonary Vasculature and BPD-PH from the Basic Scientific Point-of-View
2.1. Normal Development of Pulmonary Vasculature
2.1.1. Lung Vasculature Formation and Maturation
2.1.2. Pulmonary Vascular Development Needs Multicellular Crosstalks
2.2. Pathologic Development of Pulmonary Vasculature Leading to PH in BPD
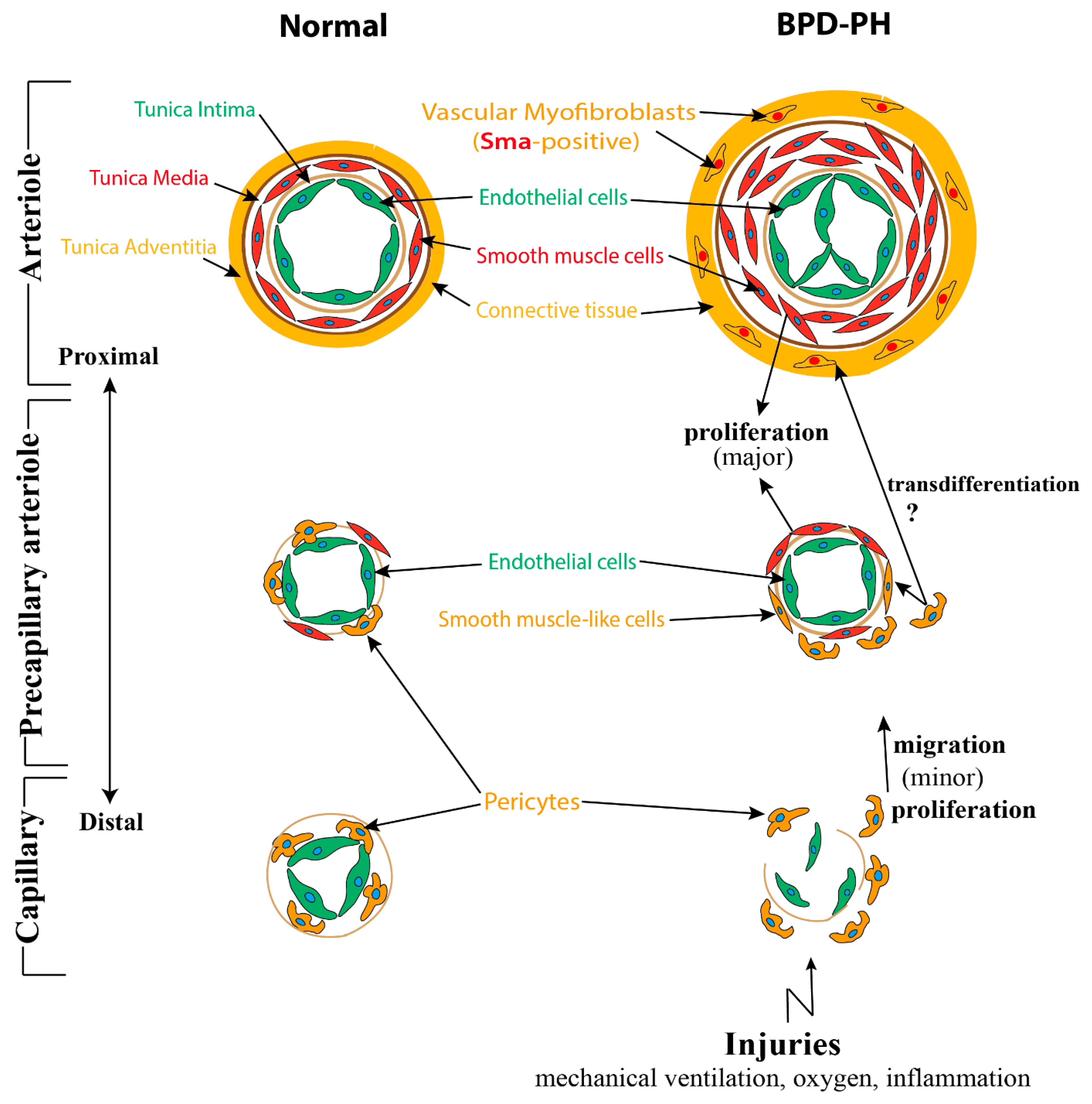
2.2.1. Abnormalities of Pulmonary Vasculature Observed in BPD-PH
2.2.2. Impaired Multicellular Interactions Disrupt Pulmonary Vascular Development in BPD-PH
2.3. MicroRNAs (miRs) May Be Associated with the Pathologic Vascular Development of BPD-PH
3. Role of FGF/FGF10 Signaling in Pulmonary Vascular Formation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blencowe, H.; Cousens, S.; Oestergaard, M.Z.; Chou, D.; Moller, A.B.; Narwal, R.; Adler, A.; Vera Garcia, C.; Rohde, S.; Say, L.; et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: A systematic analysis and implications. Lancet 2012, 379, 2162–2172. [Google Scholar] [CrossRef]
- Chawanpaiboon, S.; Vogel, J.P.; Moller, A.B.; Lumbiganon, P.; Petzold, M.; Hogan, D.; Landoulsi, S.; Jampathong, N.; Kongwattanakul, K.; Laopaiboon, M.; et al. Global, regional, and national estimates of levels of preterm birth in 2014: A systematic review and modelling analysis. Lancet Glob. Health 2019, 7, e37–e46. [Google Scholar] [CrossRef]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Walsh, M.C.; Carlo, W.A.; Shankaran, S.; Laptook, A.R.; Sanchez, P.J.; Van Meurs, K.P.; Wyckoff, M.; et al. Trends in Care Practices, Morbidity, and Mortality of Extremely Preterm Neonates, 1993–2012. JAMA 2015, 314, 1039–1051. [Google Scholar] [CrossRef]
- Bhatt, A.J.; Pryhuber, G.S.; Huyck, H.; Watkins, R.H.; Metlay, L.A.; Maniscalco, W.M. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, Flt-1, and TIE-2 in human infants dying with bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001, 164, 1971–1980. [Google Scholar] [CrossRef]
- Popova, A.P.; Bentley, J.K.; Cui, T.X.; Richardson, M.N.; Linn, M.J.; Lei, J.; Chen, Q.; Goldsmith, A.M.; Pryhuber, G.S.; Hershenson, M.B. Reduced platelet-derived growth factor receptor expression is a primary feature of human bronchopulmonary dysplasia. Am. J. Physiol. Lung C 2014, 307, L231–L239. [Google Scholar] [CrossRef]
- Speer, C.P. Inflammation and bronchopulmonary dysplasia: A continuing story. Semin. Fetal Neonat. Med. 2006, 11, 354–362. [Google Scholar] [CrossRef]
- Bose, C.; Van Marter, L.J.; Laughon, M.; O’Shea, T.M.; Allred, E.N.; Karna, P.; Ehrenkranz, R.A.; Boggess, K.; Leviton, A.; Extremely Low Gestational Age Newborn Study Investigators. Fetal growth restriction and chronic lung disease among infants born before the 28th week of gestation. Pediatrics 2009, 124, e450–e458. [Google Scholar] [CrossRef]
- Reiss, I.; Landmann, E.; Heckmann, M.; Misselwitz, B.; Gortner, L. Increased risk of bronchopulmonary dysplasia and increased mortality in very preterm infants being small for gestational age. Arch. Gynecol. Obstet. 2003, 269, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Sasi, A.; Abraham, V.; Davies-Tuck, M.; Polglase, G.R.; Jenkin, G.; Miller, S.L.; Malhotra, A. Impact of intrauterine growth restriction on preterm lung disease. Acta Paediatr. 2015, 104, e552–e556. [Google Scholar] [CrossRef] [PubMed]
- Curle, D.C.; Adamson, I.Y. Retarded development of noenatal rat lung by maternal malnutrition. J. Histochem. Cytochem. 1978, 26, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Joss-Moore, L.A.; Wang, Y.; Yu, X.; Campbell, M.S.; Callaway, C.W.; McKnight, R.A.; Wint, A.; Dahl, M.J.; Dull, R.O.; Albertine, K.H.; et al. IUGR decreases elastin mRNA expression in the developing rat lung and alters elastin content and lung compliance in the mature rat lung. Physiol. Genom. 2011, 43, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Dodson, R.B.; Powers, K.N.; Gien, J.; Rozance, P.J.; Seedorf, G.; Astling, D.; Jones, K.; Crombleholme, T.M.; Abman, S.H.; Alvira, C.M. Intrauterine growth restriction decreases NF-kappaB signaling in fetal pulmonary artery endothelial cells of fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L348–L359. [Google Scholar] [CrossRef] [PubMed]
- Morty, R.E. Recent advances in the pathogenesis of BPD. Semin. Perinatol. 2018, 42, 404–412. [Google Scholar] [CrossRef]
- Kalikkot Thekkeveedu, R.; Guaman, M.C.; Shivanna, B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir. Med. 2017, 132, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Jobe, A.H. Mechanisms of Lung Injury and Bronchopulmonary Dysplasia. Am. J. Perinatol. 2016, 33, 1076–1078. [Google Scholar] [CrossRef] [PubMed]
- Isayama, T.; Lee, S.K.; Yang, J.; Lee, D.; Daspal, S.; Dunn, M.; Shah, P.S.; Canadian Neonatal Network and Canadian Neonatal Follow-Up Network Investigators. Revisiting the Definition of Bronchopulmonary Dysplasia: Effect of Changing Panoply of Respiratory Support for Preterm Neonates. JAMA Pediatr. 2017, 171, 271–279. [Google Scholar] [CrossRef]
- Alvira, C.M.; Morty, R.E. Can We Understand the Pathobiology of Bronchopulmonary Dysplasia? J. Pediatr. 2017, 190, 27–37. [Google Scholar] [CrossRef]
- Jakkula, M.; Le Cras, T.D.; Gebb, S.; Hirth, K.P.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L600–L607. [Google Scholar] [CrossRef]
- Thebaud, B.; Ladha, F.; Michelakis, E.D.; Sawicka, M.; Thurston, G.; Eaton, F.; Hashimoto, K.; Harry, G.; Haromy, A.; Korbutt, G.; et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 2005, 112, 2477–2486. [Google Scholar] [CrossRef]
- Galambos, C.; Ng, Y.S.; Ali, A.; Noguchi, A.; Lovejoy, S.; D’Amore, P.A.; DeMello, D.E. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am. J. Respir. Cell Mol. Biol. 2002, 27, 194–203. [Google Scholar] [CrossRef]
- Gerber, H.P.; Hillan, K.J.; Ryan, A.M.; Kowalski, J.; Keller, G.A.; Rangell, L.; Wright, B.D.; Radtke, F.; Aguet, M.; Ferrara, N. VEGF is required for growth and survival in neonatal mice. Development 1999, 126, 1149–1159. [Google Scholar] [PubMed]
- Lazarus, A.; Del-Moral, P.M.; Ilovich, O.; Mishani, E.; Warburton, D.; Keshet, E. A perfusion-independent role of blood vessels in determining branching stereotypy of lung airways. Development 2011, 138, 2359–2368. [Google Scholar] [CrossRef]
- Asikainen, T.M.; Waleh, N.S.; Schneider, B.K.; Clyman, R.I.; White, C.W. Enhancement of angiogenic effectors through hypoxia-inducible factor in preterm primate lung in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L588–L595. [Google Scholar] [CrossRef] [PubMed]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Seedorf, G.; Gien, J.; Abman, S.H. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1068–L1078. [Google Scholar] [CrossRef] [PubMed]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Morgan, D.; Montgomery, G.; Grover, T.R.; Abman, S.H. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L529–L535. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.J.; Amin, S.B.; Chess, P.R.; Watkins, R.H.; Maniscalco, W.M. Expression of vascular endothelial growth factor and Flk-1 in developing and glucocorticoid-treated mouse lung. Pediatr. Res. 2000, 47, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Lassus, P.; Ristimaki, A.; Ylikorkala, O.; Viinikka, L.; Andersson, S. Vascular endothelial growth factor in human preterm lung. Am. J. Respir. Crit. Care Med. 1999, 159, 1429–1433. [Google Scholar] [CrossRef]
- Lassus, P.; Turanlahti, M.; Heikkila, P.; Andersson, L.C.; Nupponen, I.; Sarnesto, A.; Andersson, S. Pulmonary vascular endothelial growth factor and Flt-1 in fetuses, in acute and chronic lung disease, and in persistent pulmonary hypertension of the newborn. Am. J. Respir. Crit. Care Med. 2001, 164, 1981–1987. [Google Scholar] [CrossRef]
- Maniscalco, W.M.; Watkins, R.H.; D’Angio, C.T.; Ryan, R.M. Hyperoxic injury decreases alveolar epithelial cell expression of vascular endothelial growth factor (VEGF) in neonatal rabbit lung. Am. J. Respir. Cell Mol. Biol. 1997, 16, 557–567. [Google Scholar] [CrossRef]
- Maniscalco, W.M.; Watkins, R.H.; Pryhuber, G.S.; Bhatt, A.; Shea, C.; Huyck, H. Angiogenic factors and alveolar vasculature: Development and alterations by injury in very premature baboons. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L811–L823. [Google Scholar] [CrossRef]
- Roberts, R.J.; Weesner, K.M.; Bucher, J.R. Oxygen-Induced alterations in lung vascular development in the newborn rat. Pediatr. Res. 1983, 17, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghanem, G.; Shah, P.; Thomas, S.; Banfield, L.; El Helou, S.; Fusch, C.; Mukerji, A. Bronchopulmonary dysplasia and pulmonary hypertension: A meta-analysis. J. Perinatol. 2017, 37, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, S.; Zwart, E.A.H.; Ploegstra, M.J.; Bos, A.F.; Kooi, E.M.W.; Hillege, H.L.; Berger, R.M.F. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 2018, 32, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Lighthouse, J.K.; Greif, D.M. Recapitulation of developing artery muscularization in pulmonary hypertension. Cell Rep. 2014, 6, 809–817. [Google Scholar] [CrossRef]
- Shimoda, L.A.; Laurie, S.S. Vascular remodeling in pulmonary hypertension. J. Mol. Med. Berl. 2013, 91, 297–309. [Google Scholar] [CrossRef]
- Tan, W.; Madhavan, K.; Hunter, K.S.; Park, D.; Stenmark, K.R. Vascular stiffening in pulmonary hypertension: Cause or consequence? (2013 Grover Conference series). Pulm. Circ. 2014, 4, 560–580. [Google Scholar] [CrossRef]
- Frank, D.B.; Crystal, M.A.; Morales, D.L.; Gerald, K.; Hanna, B.D.; Mallory, G.B., Jr.; Rossano, J.W. Trends in pediatric pulmonary hypertension-related hospitalizations in the United States from 2000–2009. Pulm. Circ. 2015, 5, 339–348. [Google Scholar] [CrossRef]
- Haworth, S.G.; Hislop, A.A. Treatment and survival in children with pulmonary arterial hypertension: The UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009, 95, 312–317. [Google Scholar] [CrossRef]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric Pulmonary Hypertension: Guidelines From the American Heart Association and American Thoracic Society. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef]
- Krishnan, U.; Feinstein, J.A.; Adatia, I.; Austin, E.D.; Mullen, M.P.; Hopper, R.K.; Hanna, B.; Romer, L.; Keller, R.L.; Fineman, J.; et al. Evaluation and Management of Pulmonary Hypertension in Children with Bronchopulmonary Dysplasia. J. Pediatr. 2017, 188, 24–34. [Google Scholar] [CrossRef]
- Hansmann, G.; Apitz, C.; Abdul-Khaliq, H.; Alastalo, T.P.; Beerbaum, P.; Bonnet, D.; Dubowy, K.O.; Gorenflo, M.; Hager, A.; Hilgendorff, A.; et al. Executive summary. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102 (Suppl. S2), ii86–ii100. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Ezekian, J.E.; Hill, K.D. Management of Pulmonary Arterial Hypertension in the Pediatric Patient. Curr. Cardiol. Rep. 2019, 21, 162. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.R.; Lawrie, A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol. Med. 2017, 23, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T. Vascular Niche in Lung Alveolar Development, Homeostasis, and Regeneration. Front. Bioeng. Biotechnol. 2019, 7, 318. [Google Scholar] [CrossRef]
- Chao, C.M.; El Agha, E.; Tiozzo, C.; Minoo, P.; Bellusci, S. A breath of fresh air on the mesenchyme: Impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front. Med. Lausanne 2015, 2, 27. [Google Scholar] [CrossRef]
- Gebb, S.A.; Shannon, J.M. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev. Dyn. 2000, 217, 159–169. [Google Scholar] [CrossRef]
- Schittny, J.C. Development of the lung. Cell Tissue Res. 2017, 367, 427–444. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Abman, S.H. Lung vascular development: Implications for the pathogenesis of bronchopulmonary dysplasia. Annu. Rev. Physiol. 2005, 67, 623–661. [Google Scholar] [CrossRef]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Schittny, J.C.; Mund, S.I.; Stampanoni, M. Evidence and structural mechanism for late lung alveolarization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L246–L254. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.M.; Thompson, F.Y.; Brooks, S.K.; Shannon, J.M.; McCormick-Shannon, K.; Cameron, J.E.; Mallory, B.P.; Akeson, A.L. Mesenchymal expression of vascular endothelial growth factors D and A defines vascular patterning in developing lung. Dev. Dyn. 2002, 224, 144–153. [Google Scholar] [CrossRef] [PubMed]
- White, A.C.; Lavine, K.J.; Ornitz, D.M. FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonary capillary network. Development 2007, 134, 3743–3752. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, F.; Rossant, J.; Yamaguchi, T.P.; Gertsenstein, M.; Wu, X.F.; Breitman, M.L.; Schuh, A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yun, E.J.; Gerber, H.P.; Ferrara, N.; Whitsett, J.A.; Vu, T.H. Epithelial-Vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev. Biol. 2007, 308, 44–53. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, K.; Ferrara, N.; Vu, T.H. Vascular endothelial growth factor co-ordinates proper development of lung epithelium and vasculature. Mech. Dev. 2005, 122, 877–886. [Google Scholar] [CrossRef]
- Yang, J.; Hernandez, B.J.; Martinez Alanis, D.; Narvaez del Pilar, O.; Vila-Ellis, L.; Akiyama, H.; Evans, S.E.; Ostrin, E.J.; Chen, J. The development and plasticity of alveolar type 1 cells. Development 2016, 143, 54–65. [Google Scholar] [CrossRef]
- Greif, D.M.; Kumar, M.; Lighthouse, J.K.; Hum, J.; An, A.; Ding, L.; Red-Horse, K.; Espinoza, F.H.; Olson, L.; Offermanns, S.; et al. Radial construction of an arterial wall. Dev. Cell. 2012, 23, 482–493. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.W.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef]
- Paiva, A.E.; Lousado, L.; Guerra, D.A.P.; Azevedo, P.O.; Sena, I.F.G.; Andreotti, J.P.; Santos, G.S.P.; Goncalves, R.; Mintz, A.; Birbrair, A. Pericytes in the Premetastatic Niche. Cancer Res. 2018, 78, 2779–2786. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Dieguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.W.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1574. [Google Scholar] [CrossRef]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Jarad, M.; Kuczynski, E.A.; Morrison, J.; Viloria-Petit, A.M.; Coomber, B.L. Release of endothelial cell associated VEGFR2 during TGF-beta modulated angiogenesis in vitro. BMC Cell Biol. 2017, 18, 10. [Google Scholar] [CrossRef]
- Wakui, S.; Yokoo, K.; Muto, T.; Suzuki, Y.; Takahashi, H.; Furusato, M.; Hano, H.; Endou, H.; Kanai, Y. Localization of Ang-1, -2, Tie-2, and VEGF expression at endothelial-pericyte interdigitation in rat angiogenesis. Lab. Investig. 2006, 86, 1172–1184. [Google Scholar] [CrossRef]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/Pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef]
- Kato, K.; Dieguez-Hurtado, R.; Park, D.Y.; Hong, S.P.; Kato-Azuma, S.; Adams, S.; Stehling, M.; Trappmann, B.; Wrana, J.L.; Koh, G.Y.; et al. Pulmonary pericytes regulate lung morphogenesis. Nat. Commun. 2018, 9, 2448. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Shamskhou, E.A.; Orcholski, M.E.; Nathan, A.; Reddy, S.; Honda, H.; Mani, V.; Zeng, Y.; Ozen, M.O.; Wang, L.; et al. Loss of Endothelium-Derived Wnt5a Is Associated With Reduced Pericyte Recruitment and Small Vessel Loss in Pulmonary Arterial Hypertension. Circulation 2019, 139, 1710–1724. [Google Scholar] [CrossRef] [PubMed]
- Shammout, B.; Johnson, J.R. Pericytes in Chronic Lung Disease. Adv. Exp. Med. Biol. 2019, 1147, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, T.; Raj, J.U. Endothelial and Smooth Muscle Cell Interactions in the Pathobiology of Pulmonary Hypertension. Am. J. Respir. Cell. Mol. Biol. 2016, 54, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Hadchouel, A.; Durrmeyer, X.; Bouzigon, E.; Incitti, R.; Huusko, J.; Jarreau, P.H.; Lenclen, R.; Demenais, F.; Franco-Montoya, M.L.; Layouni, I.; et al. Identification of SPOCK2 as a susceptibility gene for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2011, 184, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Tiozzo, C.; Carraro, G.; Al Alam, D.; Baptista, S.; Danopoulos, S.; Li, A.; Lavarreda-Pearce, M.; Li, C.; De Langhe, S.; Chan, B.; et al. Mesodermal Pten inactivation leads to alveolar capillary dysplasia- like phenotype. J. Clin. Investig. 2012, 122, 3862–3872. [Google Scholar] [CrossRef]
- Abman, S.H. Bronchopulmonary dysplasia: A vascular hypothesis. Am. J. Respir. Crit. Care Med. 2001, 164, 1755–1756. [Google Scholar] [CrossRef]
- Baker, C.D.; Abman, S.H. Impaired pulmonary vascular development in bronchopulmonary dysplasia. Neonatology 2015, 107, 344–351. [Google Scholar] [CrossRef]
- Chao, C.M.; Moiseenko, A.; Kosanovic, D.; Rivetti, S.; El Agha, E.; Wilhelm, J.; Kampschulte, M.; Yahya, F.; Ehrhardt, H.; Zimmer, K.P.; et al. Impact of Fgf10 deficiency on pulmonary vasculature formation in a mouse model of bronchopulmonary dysplasia. Hum. Mol. Genet. 2019, 28, 1429–1444. [Google Scholar] [CrossRef]
- Yee, M.; White, R.J.; Awad, H.A.; Bates, W.A.; McGrath-Morrow, S.A.; O’Reilly, M.A. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am. J. Pathol. 2011, 178, 2601–2610. [Google Scholar] [CrossRef]
- Greco, F.; Wiegert, S.; Baumann, P.; Wellmann, S.; Pellegrini, G.; Cannizzaro, V. Hyperoxia-Induced lung structure-function relation, vessel rarefaction, and cardiac hypertrophy in an infant rat model. J. Transl. Med. 2019, 17, 91. [Google Scholar] [CrossRef] [PubMed]
- Crnkovic, S.; Marsh, L.M.; El Agha, E.; Voswinckel, R.; Ghanim, B.; Klepetko, W.; Stacher-Priehse, E.; Olschewski, H.; Bloch, W.; Bellusci, S.; et al. Resident cell lineages are preserved in pulmonary vascular remodeling. J. Pathol. 2018, 244, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-To-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH. Circulation 2016, 133, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Assaad, A.M.; Kawut, S.M.; Arcasoy, S.M.; Rosenzweig, E.B.; Wilt, J.S.; Sonett, J.R.; Borczuk, A.C. Platelet-Derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest 2007, 131, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef]
- Rozance, P.J.; Seedorf, G.J.; Brown, A.; Roe, G.; O’Meara, M.C.; Gien, J.; Tang, J.R.; Abman, S.H. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L860–L871. [Google Scholar] [CrossRef]
- Wallace, B.; Peisl, A.; Seedorf, G.; Nowlin, T.; Kim, C.; Bosco, J.; Kenniston, J.; Keefe, D.; Abman, S.H. Anti-sFlt-1 Therapy Preserves Lung Alveolar and Vascular Growth in Antenatal Models of Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2018, 197, 776–787. [Google Scholar] [CrossRef]
- DeLisser, H.M.; Helmke, B.P.; Cao, G.; Egan, P.M.; Taichman, D.; Fehrenbach, M.; Zaman, A.; Cui, Z.; Mohan, G.S.; Baldwin, H.S.; et al. Loss of PECAM-1 function impairs alveolarization. J. Biol. Chem. 2006, 281, 8724–8731. [Google Scholar] [CrossRef]
- Bordenave, J.; Tu, L.; Berrebeh, N.; Thuillet, R.; Cumont, A.; Le Vely, B.; Fadel, E.; Nadaud, S.; Savale, L.; Humbert, M.; et al. Lineage Tracing Reveals the Dynamic Contribution of Pericytes to the Blood Vessel Remodeling in Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 766–782. [Google Scholar] [CrossRef]
- Lin, S.L.; Chang, F.C.; Schrimpf, C.; Chen, Y.T.; Wu, C.F.; Wu, V.C.; Chiang, W.C.; Kuhnert, F.; Kuo, C.J.; Chen, Y.M.; et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am. J. Pathol. 2011, 178, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Chen, Q.; Luo, J.M.; Nie, J.; Meng, Q.H.; Shuai, W.; Xie, H.; Xia, J.M.; Wang, H. Notch1 promotes the pericyte-myofibroblast transition in idiopathic pulmonary fibrosis through the PDGFR/ROCK1 signal pathway. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lee, M.K.; Gao, F.; Webster, S.; Di, H.; Duan, J.; Yang, C.Y.; Bhopal, N.; Peinado, N.; Pryhuber, G.; et al. Secondary crest myofibroblast PDGFRalpha controls the elastogenesis pathway via a secondary tier of signaling networks during alveologenesis. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Oak, P.; Pritzke, T.; Thiel, I.; Koschlig, M.; Mous, D.S.; Windhorst, A.; Jain, N.; Eickelberg, O.; Foerster, K.; Schulze, A.; et al. Attenuated PDGF signaling drives alveolar and microvascular defects in neonatal chronic lung disease. EMBO Mol. Med. 2017, 9, 1504–1520. [Google Scholar] [CrossRef]
- Xing, Y.; Fu, J.; Yang, H.; Yao, L.; Qiao, L.; Du, Y.; Xue, X. MicroRNA expression profiles and target prediction in neonatal Wistar rat lungs during the development of bronchopulmonary dysplasia. Int. J. Mol. Med. 2015, 36, 1253–1263. [Google Scholar] [CrossRef][Green Version]
- Yang, Y.; Qiu, J.; Kan, Q.; Zhou, X.G.; Zhou, X.Y. MicroRNA expression profiling studies on bronchopulmonary dysplasia: A systematic review and meta-analysis. Genet. Mol. Res. 2013, 12, 5195–5206. [Google Scholar] [CrossRef]
- Chen, J.; Cui, X.; Li, L.; Qu, J.; Raj, J.U.; Gou, D. MiR-339 inhibits proliferation of pulmonary artery smooth muscle cell by targeting FGF signaling. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef]
- Yuan, H.S.; Xiong, D.Q.; Huang, F.; Cui, J.; Luo, H. MicroRNA-421 inhibition alleviates bronchopulmonary dysplasia in a mouse model via targeting Fgf10. J. Cell. Biochem. 2019, 120, 16876–16887. [Google Scholar] [CrossRef]
- Yang, S.; Banerjee, S.; Freitas, A.; Cui, H.; Xie, N.; Abraham, E.; Liu, G. miR-21 regulates chronic hypoxia-induced pulmonary vascular remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L521–L529. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Xi, D.; Wang, Y.; Huang, C.; Narasaraju, T.; Shu, W.; Zhao, C.; Xiao, X.; More, S.; Breshears, M.; et al. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol. Genom. 2012, 44, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Talati, M.; Fessel, J.P.; Hemnes, A.R.; Gladson, S.; French, J.; Shay, S.; Trammell, A.; Phillips, J.A.; Hamid, R.; et al. Estrogen Metabolite 16alpha-Hydroxyestrone Exacerbates Bone Morphogenetic Protein Receptor Type II-Associated Pulmonary Arterial Hypertension Through MicroRNA-29-Mediated Modulation of Cellular Metabolism. Circulation 2016, 133, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xie, L.; Yu, J.; Fu, H.; Zhou, D.; Liu, H. Inhibition of microRNA-29a alleviates hyperoxia-induced bronchopulmonary dysplasia in neonatal mice via upregulation of GAB1. Mol. Med. 2019, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Carey, W.A.; Abel, S.; Collura, C.; Jiang, G.; Tomaszek, S.; Sutor, S.; Roden, A.C.; Asmann, Y.W.; Prakash, Y.S.; et al. MicroRNA-mRNA interactions in a murine model of hyperoxia-induced bronchopulmonary dysplasia. BMC Genom. 2012, 13, 204. [Google Scholar] [CrossRef] [PubMed]
- Syed, M.; Das, P.; Pawar, A.; Aghai, Z.H.; Kaskinen, A.; Zhuang, Z.W.; Ambalavanan, N.; Pryhuber, G.; Andersson, S.; Bhandari, V. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 2017, 8, 1173. [Google Scholar] [CrossRef]
- Ruiz-Camp, J.; Quantius, J.; Lignelli, E.; Arndt, P.F.; Palumbo, F.; Nardiello, C.; Surate Solaligue, D.E.; Sakkas, E.; Mizikova, I.; Rodriguez-Castillo, J.A.; et al. Targeting miR-34a/Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef]
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, E.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943. [Google Scholar] [CrossRef]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef]
- Chao, C.M.; Carraro, G.; Rako, Z.A.; Kolck, J.; Sedighi, J.; Zimmermann, V.; Moiseenko, A.; Wilhelm, J.; Young, B.M.; Chong, L.; et al. Failure to Down-Regulate miR-154 Expression in Early Postnatal Mouse Lung Epithelium Suppresses Alveologenesis, with Changes in Tgf-beta Signaling Similar to those Induced by Exposure to Hyperoxia. Cells 2020, 9, 859. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.M.; Tham, Y.K.; Ooi, J.Y.; Patterson, N.L.; Kiriazis, H.; Su, Y.; Thomas, C.J.; Lin, R.C.; et al. Inhibition of miR-154 Protects Against Cardiac Dysfunction and Fibrosis in a Mouse Model of Pressure Overload. Sci. Rep. 2016, 6, 22442. [Google Scholar] [CrossRef]
- Milosevic, J.; Pandit, K.; Magister, M.; Rabinovich, E.; Ellwanger, D.C.; Yu, G.; Vuga, L.J.; Weksler, B.; Benos, P.V.; Gibson, K.F.; et al. Profibrotic role of miR-154 in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Lingampally, A.; Dilai, S.; Shrestha, A.; Stripp, B.; Helmbacher, F.; Chen, C.; Chao, C.M.; Bellusci, S. Characterization of Tg(Etv4-GFP) and Etv5 (RFP) Reporter Lines in the Context of Fibroblast Growth Factor 10 Signaling During Mouse Embryonic Lung Development. Front. Genet. 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- Mailleux, A.A.; Kelly, R.; Veltmaat, J.M.; De Langhe, S.P.; Zaffran, S.; Thiery, J.P.; Bellusci, S. Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development 2005, 132, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, H.; Hori, Y.; Yamasaki, M.; Harada, H.; Sekine, K.; Kato, S.; Itoh, N. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem. Biophys. Res. Commun. 2000, 277, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Gupte, V.V.; Ramasamy, S.K.; Reddy, R.; Lee, J.; Weinreb, P.H.; Violette, S.M.; Guenther, A.; Warburton, D.; Driscoll, B.; Minoo, P.; et al. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care. Med. 2009, 180, 424–436. [Google Scholar] [CrossRef]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273. [Google Scholar] [CrossRef]
- Chao, C.M.; Yahya, F.; Moiseenko, A.; Tiozzo, C.; Shrestha, A.; Ahmadvand, N.; El Agha, E.; Quantius, J.; Dilai, S.; Kheirollahi, V.; et al. Fgf10 deficiency is causative for lethality in a mouse model of bronchopulmonary dysplasia. J. Pathol. 2017, 241, 91–103. [Google Scholar] [CrossRef]
- Scott, C.L.; Walker, D.J.; Cwiklinski, E.; Tait, C.; Tee, A.R.; Land, S.C. Control of HIF-1{alpha} and vascular signaling in fetal lung involves cross talk between mTORC1 and the FGF-10/FGFR2b/Spry2 airway branching periodicity clock. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L455–L471. [Google Scholar] [CrossRef][Green Version]
- Del Moral, P.M.; Sala, F.G.; Tefft, D.; Shi, W.; Keshet, E.; Bellusci, S.; Warburton, D. VEGF-A signaling through Flk-1 is a critical facilitator of early embryonic lung epithelial to endothelial crosstalk and branching morphogenesis. Dev. Biol. 2006, 290, 177–188. [Google Scholar] [CrossRef]
- Ramasamy, S.K.; Mailleux, A.A.; Gupte, V.V.; Mata, F.; Sala, F.G.; Veltmaat, J.M.; Del Moral, P.M.; De Langhe, S.; Parsa, S.; Kelly, L.K.; et al. Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Dev. Biol. 2007, 307, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.T.; Smith, R.J.; Halloran, B.A.; Day, T.J.; Kelly, D.R.; Prince, L.S. FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by Toll-like receptor activation. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2007, 292, L550–L558. [Google Scholar] [CrossRef] [PubMed]
- Seghezzi, G.; Patel, S.; Ren, C.J.; Gualandris, A.; Pintucci, G.; Robbins, E.S.; Shapiro, R.L.; Galloway, A.C.; Rifkin, D.B.; Mignatti, P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: An autocrine mechanism contributing to angiogenesis. J. Cell Biol. 1998, 141, 1659–1673. [Google Scholar] [CrossRef]
- Gospodarowicz, D.; Brown, K.D.; Birdwell, C.R.; Zetter, B.R. Control of proliferation of human vascular endothelial cells. Characterization of the response of human umbilical vein endothelial cells to fibroblast growth factor, epidermal growth factor, and thrombin. J. Cell Biol. 1978, 77, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Claus, P.; Grothe, C. Molecular cloning and developmental expression of rat fibroblast growth factor receptor 3. Histochem. Cell Biol. 2001, 115, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Min, M.; Larrieu-Lahargue, F.; Canron, X.; Kunstfeld, R.; Nguyen, L.; Henderson, J.E.; Bikfalvi, A.; Detmar, M.; Hong, Y.K. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: A role for FGF signaling in lymphangiogenesis. Mol. Biol. Cell 2006, 17, 576–584. [Google Scholar] [CrossRef]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef]
- Murakami, M.; Elfenbein, A.; Simons, M. Non-Canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc. Res. 2008, 78, 223–231. [Google Scholar] [CrossRef]
- Murakami, M.; Nguyen, L.T.; Hatanaka, K.; Schachterle, W.; Chen, P.Y.; Zhuang, Z.W.; Black, B.L.; Simons, M. FGF-Dependent regulation of VEGF receptor 2 expression in mice. J. Clin. Investig. 2011, 121, 2668–2678. [Google Scholar] [CrossRef]
- Hatanaka, K.; Lanahan, A.A.; Murakami, M.; Simons, M. Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS ONE 2012, 7, e37600. [Google Scholar] [CrossRef]
- Oladipupo, S.S.; Smith, C.; Santeford, A.; Park, C.; Sene, A.; Wiley, L.A.; Osei-Owusu, P.; Hsu, J.; Zapata, N.; Liu, F.; et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 13379–13384. [Google Scholar] [CrossRef] [PubMed]
- House, S.L.; Castro, A.M.; Lupu, T.S.; Weinheimer, C.; Smith, C.; Kovacs, A.; Ornitz, D.M. Endothelial fibroblast growth factor receptor signaling is required for vascular remodeling following cardiac ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H559–H571. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Herold, S.; Al Alam, D.; Quantius, J.; MacKenzie, B.; Carraro, G.; Moiseenko, A.; Chao, C.M.; Minoo, P.; Seeger, W.; et al. Fgf10-Positive cells represent a progenitor cell population during lung development and postnatally. Development 2014, 141, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, A.R.; Willems-Widyastuti, A.; Mooi, W.J.; Saxena, P.R.; Sterk, P.J.; de Boer, W.I.; Sharma, H.S. Chronic obstructive pulmonary disease is associated with enhanced bronchial expression of FGF-1, FGF-2, and FGFR-1. J. Pathol. 2005, 206, 28–38. [Google Scholar] [CrossRef] [PubMed]
- McAuley, D.F.; Cross, L.M.; Hamid, U.; Gardner, E.; Elborn, J.S.; Cullen, K.M.; Dushianthan, A.; Grocott, M.P.; Matthay, M.A.; O’Kane, C.M. Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Respir. Med. 2017, 5, 484–491. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| miRs Shown to be Involved | Changes in PH and BPD | Potential Roles in BPD-PH | References |
|---|---|---|---|
| miR-21 | increased | Increases PASMCs proliferation through inhibiting Bmpr2 activation | [100,101] |
| miR-29 | increased | Impairs PASMCs and ECs function through down-regulating PPARγ | [102,103,104] |
| miR-34a | increased | increases lung epithelial cell apoptosis through down-regulating ANG1-TIE2 signaling; impairs alveologenesis through increasing PDGFR alpha-expressing myofibroblasts | [105,106] |
| miR-126 | decreased | Decreases microvessel density through up-regulating SPRED-1 | [101,107] |
| miR-130 | increased | Promotes ECM remodeling, increases PASMCs proliferation and crosstalk through down-regulating PPARγ-APOE-LRP8 axis | [96,108] |
| miR-154 | increased | Impairs alveologenesis through increasing TGFβ. | [109,110,111] |
| miR-421 | increased | Impairs alveologenesis through down-regulation of FGF10 signaling | [99] |
| miR-503 | decreased | Impairs ECs function and induces PASMCs proliferation through upregulating FGF2 and FGFR1 | [96,98] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, C.-M.; Chong, L.; Chu, X.; Shrestha, A.; Behnke, J.; Ehrhardt, H.; Zhang, J.; Chen, C.; Bellusci, S. Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System. Cells 2020, 9, 1875. https://doi.org/10.3390/cells9081875
Chao C-M, Chong L, Chu X, Shrestha A, Behnke J, Ehrhardt H, Zhang J, Chen C, Bellusci S. Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System. Cells. 2020; 9(8):1875. https://doi.org/10.3390/cells9081875
Chicago/Turabian StyleChao, Cho-Ming, Lei Chong, Xuran Chu, Amit Shrestha, Judith Behnke, Harald Ehrhardt, Jinsan Zhang, Chengshui Chen, and Saverio Bellusci. 2020. "Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System" Cells 9, no. 8: 1875. https://doi.org/10.3390/cells9081875
APA StyleChao, C.-M., Chong, L., Chu, X., Shrestha, A., Behnke, J., Ehrhardt, H., Zhang, J., Chen, C., & Bellusci, S. (2020). Targeting Bronchopulmonary Dysplasia-Associated Pulmonary Hypertension (BPD-PH): Potential Role of the FGF Signaling Pathway in the Development of the Pulmonary Vascular System. Cells, 9(8), 1875. https://doi.org/10.3390/cells9081875