Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. OPL Dataset
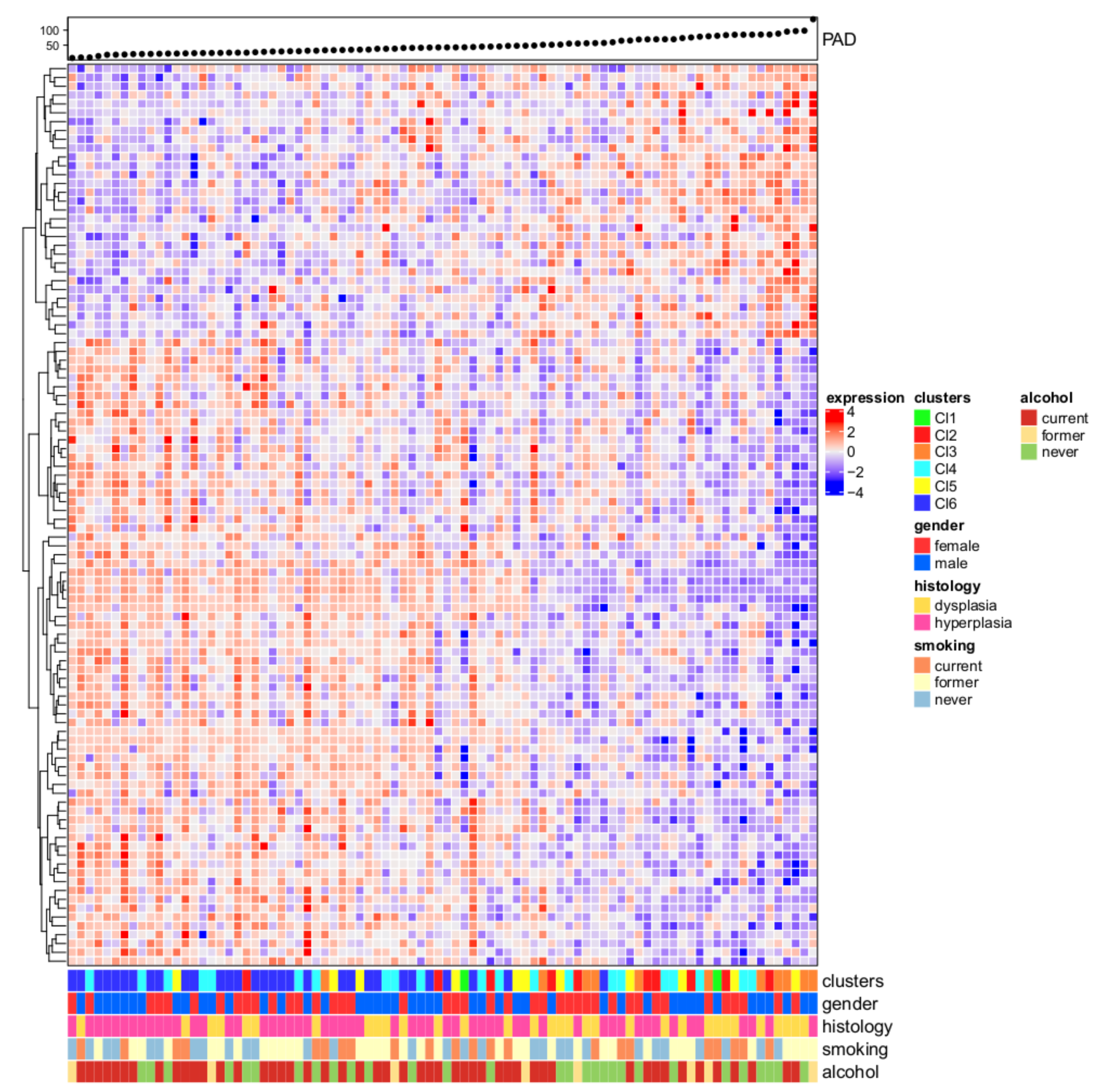
2.2. Gene Expression-Based Molecular Subtyping
2.3. Gene Set Enrichment Analysis (GSEA) Functional Analysis
2.4. Tumor Microenvironment Analysis
2.5. Prognostic Signatures
2.6. Variance Partition Analysis
2.7. Bioinformatics Analysis and Data Visualization
3. Results
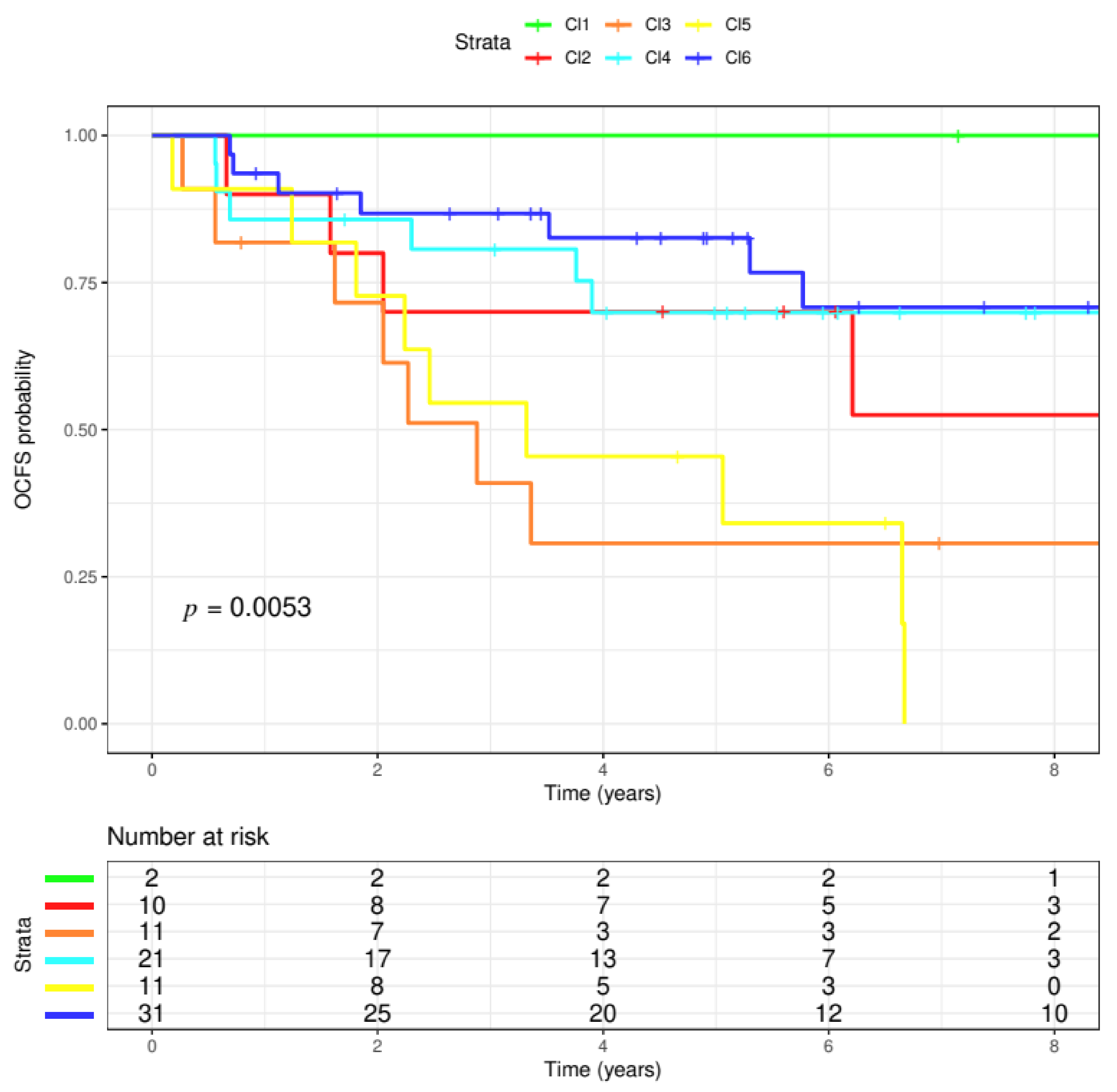
3.1. Six Molecular Subtypes
3.2. Progression Analysis of Disease
3.3. Functional Analysis and Tumor Microenvironment Composition
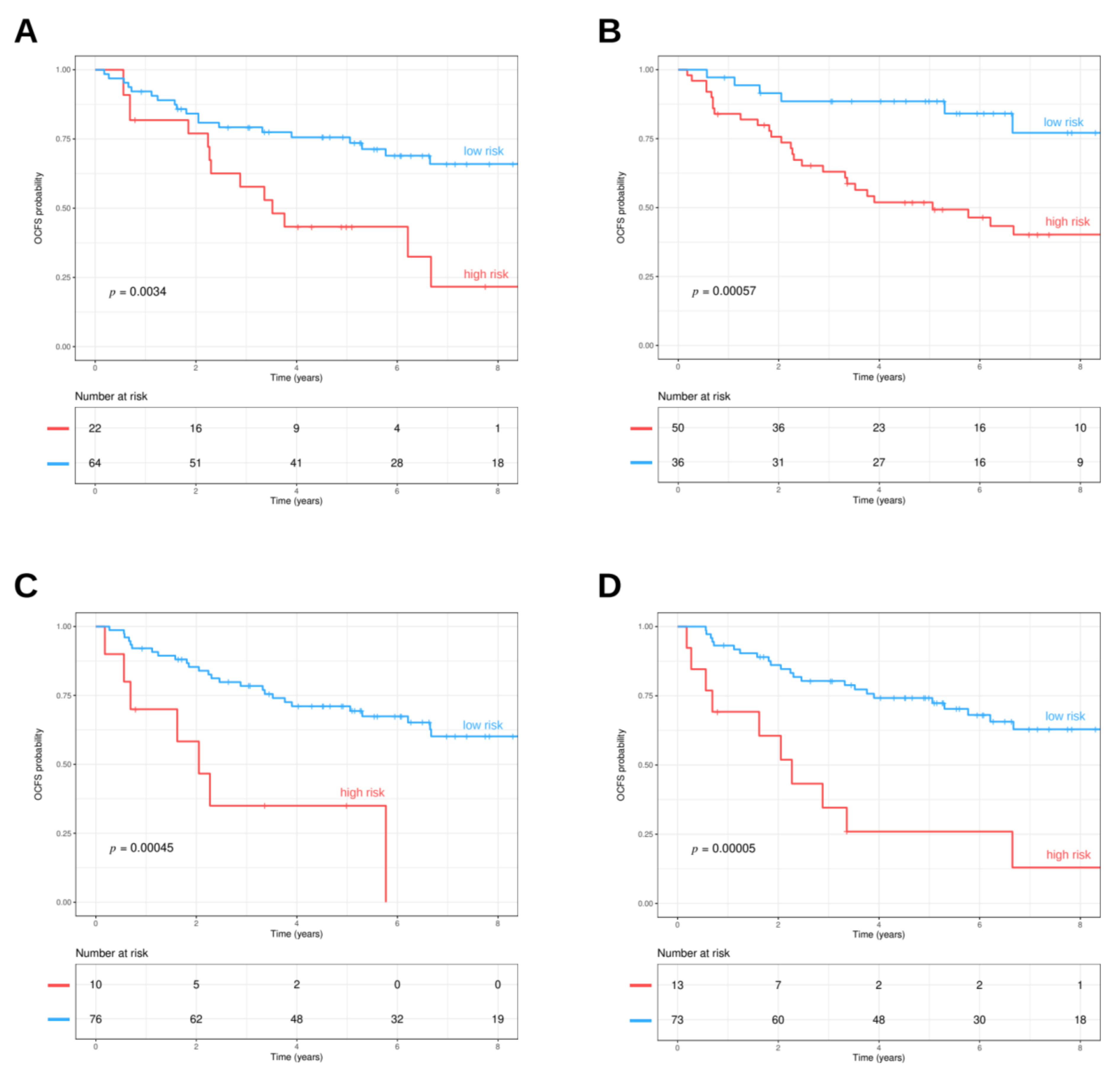
3.4. Prognostic Value of Oral Cavity Signatures
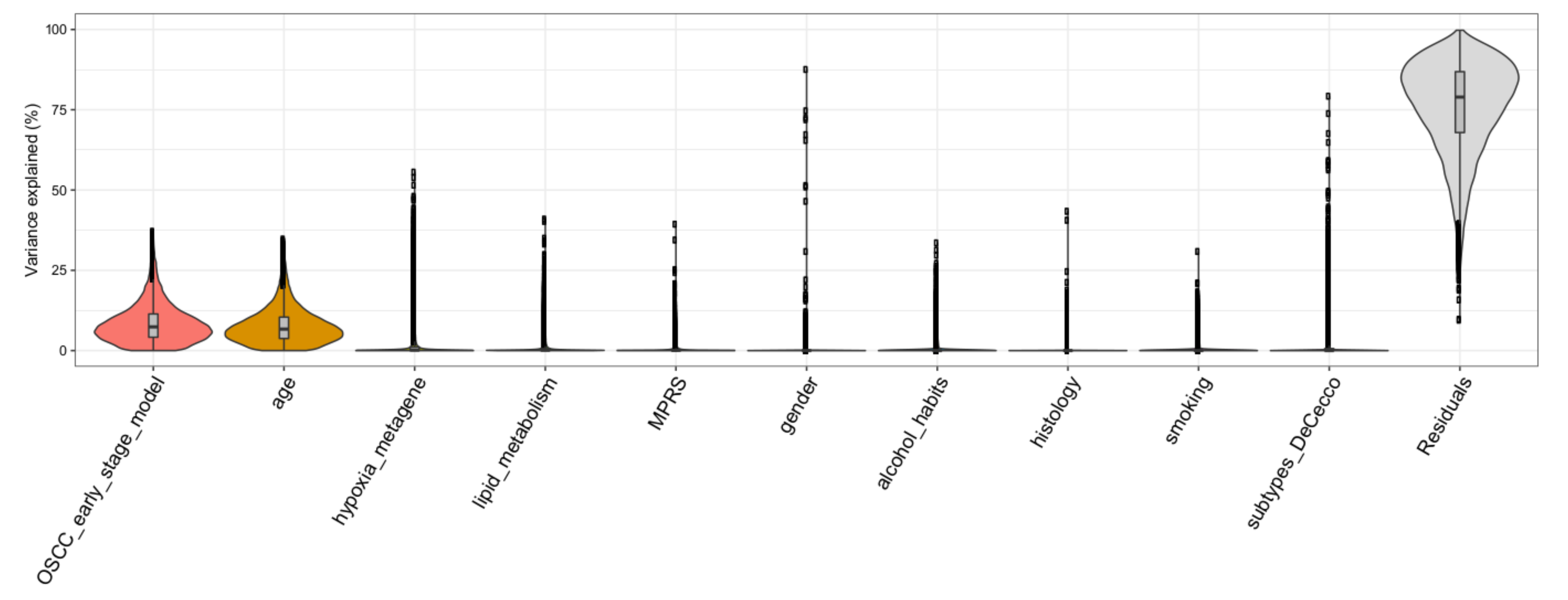
3.5. Variance Partition Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poh, C.F.; Macaulay, C.E.; Laronde, D.M.; Williams, P.M.; Zhang, L.; Rosin, M.P. Squamous cell carcinoma and precursor lesions: Diagnosis and screening in a technical era. Periodontolgy 2000 2011, 57, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2017, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, H.; Baharvand, M.; Mehdipour, M. Oral Potentially Malignant Disorders: An Overview of More than 20 Entities. J. Dent. Res. Dent. Clin. Dent. Prospect. 2014, 8, 6–14. [Google Scholar]
- Warnakulasuriya, S.; Johnson, N.W.; Van Der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 2007, 36, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.C.; Rodrigo, J.P.; Rodríguez-Santamarta, T.; Lequerica-Fernández, P.; Allonca, E.; García-Pedrero, J.M. Podoplanin expression in oral leukoplakia: Tumorigenic role. Oral Oncol. 2013, 49, 598–603. [Google Scholar] [CrossRef]
- El-Sakka, H.; Kujan, O.; Farah, C.S. Assessing miRNAs profile expression as a risk stratification biomarker in oral potentially malignant disorders: A systematic review. Oral Oncol. 2018, 77, 57–82. [Google Scholar] [CrossRef]
- Fleskens, S.; Slootweg, P.J. Grading systems in head and neck dysplasia: Their prognostic value, weaknesses and utility. Head Neck Oncol. 2009, 1, 11. [Google Scholar] [CrossRef]
- Pinto, A.C.; Caramês, J.; Francisco, H.; Chen, A.; Azul, A.M.; Marques, D. Malignant transformation rate of oral leukoplakia—systematic review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2020. [Google Scholar] [CrossRef]
- Goodson, M.; Sloan, P.; Robinson, M.; Cocks, K.; Thomson, P. Oral precursor lesions and malignant transformation—Who, where, what, and when? Br. J. Oral Maxillofac. Surg. 2015, 53, 831–835. [Google Scholar] [CrossRef]
- William, W.N.; Papadimitrakopoulou, V.; Lee, J.J.; Mao, L.; Cohen, E.E.W.; Lin, H.Y.; Gillenwater, A.M.; Martin, J.W.; Lingen, M.W.; Boyle, J.O.; et al. Erlotinib and the risk of oral cancer the erlotinib Prevention of Oral Cancer (EPOC) randomized clinical trial. JAMA Oncol. 2016, 2, 209–216. [Google Scholar] [CrossRef]
- William, W.N.; Lee, W.-C.; Lee, J.J.; Lin, H.Y.; Eterovic, A.K.; El-Naggar, A.K.; Gillenwater, A.M.; Pisegna, M.; Tong, P.; Li, L.; et al. Genomic and transcriptomic landscape of oral pre-cancers (OPCs) and risk of oral cancer (OC). J. Clin. Oncol. 2019, 37, 6009. [Google Scholar] [CrossRef]
- Chen, C.; Méndez, E.; Houck, J.; Fan, W.; Lohavanichbutr, P.; Doody, D.; Yueh, B.; Futran, N.D.; Upton, M.; Farwell, D.G.; et al. Gene expression profiling identifies genes predictive of oral squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Mallery, S.R.; Zwick, J.C.; Pei, P.; Tong, M.; Larsen, P.E.; Shumway, B.S.; Lu, B.; Fields, H.W.; Mumper, R.J.; Stoner, G.D. Topical application of a bioadhesive black raspberry gel modulates gene expression and reduces cyclooxygenase 2 protein in human premalignant oral lesions. Cancer Res. 2008, 68, 4945–4957. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.G.; Cristea, S.; Ambatipudi, S.; Desai, R.S.; Kumar, R.; Patil, A.; Kane, S.; Borges, A.M.; Schäffer, A.A.; Beerenwinkel, N.; et al. Chromosomal Alterations and Gene Expression Changes Associated with the Progression of Leukoplakia to Advanced Gingivobuccal Cancer. Transl. Oncol. 2017, 10, 396–409. [Google Scholar] [CrossRef]
- Saintigny, P.; Zhang, L.; Fan, Y.-H.; El-Naggar, A.K.; A Papadimitrakopoulou, V.; Feng, L.; Lee, J.J.; Kim, E.S.; Hong, W.K.; Mao, L.; et al. Gene expression profiling predicts the development of oral cancer. Cancer Prev. Res. 2011, 4, 218–229. [Google Scholar] [CrossRef]
- Foy, J.-P.; Bertolus, C.; Ortiz-Cuaran, S.; Albaret, M.-A.; Williams, W.N.; Lang, W.; Destandau, S.; De Souza, G.; Sohier, E.; Kielbassa, J.; et al. Immunological and classical subtypes of oral premalignant lesions. OncoImmunology 2018, 7, e1496880. [Google Scholar] [CrossRef]
- De Cecco, L.; Nicolau, M.; Giannoccaro, M.; Daidone, M.G.; Bossi, P.; Locati, L.D.; Licitra, L.; Canevari, S. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget 2015, 6, 9627–9642. [Google Scholar] [CrossRef]
- Nicolau, M.; Levine, A.J.; Carlsson, G. Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proc. Natl. Acad. Sci. USA 2011, 108, 7265–7270. [Google Scholar] [CrossRef]
- Tonella, L.; Giannoccaro, M.; Alfieri, S.; Canevari, S.; De Cecco, L. Gene Expression Signatures for Head and Neck Cancer Patient Stratification: Are Results Ready for Clinical Application? Curr. Treat. Options Oncol. 2017, 18, 2013. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Lee, J.J.; William, W.N.; Martin, J.W.; Thomas, M.; Kim, E.S.; Khuri, F.R.; Shin, D.M.; Feng, L.; Hong, W.K.; et al. Randomized Trial of 13-cis Retinoic Acid Compared With Retinyl Palmitate With or Without Beta-Carotene in Oral Premalignancy. J. Clin. Oncol. 2009, 27, 599–604. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, M.; Tibshirani, R.; Børresen-Dale, A.-L.; Jeffrey, S.S. Disease-specific genomic analysis: Identifying the signature of pathologic biology. Bioinformatics 2007, 23, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Mémoli, F.; Carlsson, G.E. Topological Methods for the Analysis of High Dimensional Data Sets and 3D Object Recognition. SPBG 2007, 91, 100. [Google Scholar]
- Efron, B.; Hastie, T.; Johnstone, I.; Tibshirani, R.; Ishwaran, H.; Knight, K.; Loubes, J.M.; Massart, P.; Madigan, D.; Ridgeway, G.; et al. Least angle regression. Ann. Stat. 2004, 32, 407–499. [Google Scholar]
- Zhao, Y.; Simon, R. Development and Validation of Predictive Indices for a Continuous Outcome Using Gene Expression Profiles. Cancer Inform. 2010, 9, 105–114. [Google Scholar] [CrossRef]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef]
- Dudoit, S.; Fridlyand, J.; Speed, T. Comparison of Discrimination Methods for the Classification of Tumors Using Gene Expression Data. J. Am. Stat. Assoc. 2002, 97, 77–87. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. Improved shrunken centroid classifiers for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 64. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Boil. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lohavanichbutr, P.; Zhang, Y.; Houck, J.R.; Upton, M.P.; Abedi-Ardekani, B.; Agudo, A.; Ahrens, W.; Alemany, L.; Anantharaman, D.; et al. Prediction of survival of HPV16-negative, p16-negative oral cavity cancer patients using a 13-gene signature: A multicenter study using FFPE samples. Oral Oncol. 2020, 100, 104487. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Guo, M.; Zhang, X.; Liu, F. A transcriptional metabolic gene-set based prognostic signature is associated with clinical and mutational features in head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 621–630. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a Hypoxia Metagene Derived from Head and Neck Cancer to Prognosis of Multiple Cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef]
- Hu, Q.; Peng, J.; Chen, X.; Li, H.; Song, M.; Cheng, B.; Wu, T. Obesity and genes related to lipid metabolism predict poor survival in oral squamous cell carcinoma. Oral Oncol. 2019, 89, 14–22. [Google Scholar] [CrossRef]
- Maglott, D.; Ostell, J.; Pruitt, K.D.; Tatusova, T. Entrez Gene: Gene-centered information at NCBI. Nucleic Acids Res. 2011, 39, D52–D57. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Schadt, E.E. variancePartition: Interpreting drivers of variation in complex gene expression studies. BMC Bioinform. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; European Environment Agency: Vienna, Austria, 2019. [Google Scholar]
- Simon, R.; Lam, A.; Li, M.-C.; Ngan, M.; Menenzes, S.; Zhao, Y. Analysis of Gene Expression Data Using BRB-Array Tools. Cancer Inform. 2007, 3, 11–17. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A Comprehensive and Straightforward Web Application Enabling Rapid Biomarker Cutoff Optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 0387987843. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Lohavanichbutr, P.; Méndez, E.; Holsinger, F.C.; Rue, T.C.; Zhang, Y.; Houck, J.; Upton, M.P.; Futran, N.; Schwartz, S.M.; Wang, P.; et al. A 13-gene signature prognostic of HPV-negative OSCC: Discovery and external validation. Clin. Cancer Res. 2013, 19, 1197–1203. [Google Scholar] [CrossRef]
- EI-Naggar, A.K.; Chan, J.K.C.; Grandis, J.R.; Takata T, S.P.J. (Eds.) WHO Classification of Head and Neck Tumors, 4th ed.; The International Agency for Research on Cancer: Lyon, France, 2017; ISBN 978-92-832-2438-9. [Google Scholar]
- Mehanna, H.; Rattay, T.; Smith, J.; McConkey, C.C. Treatment and follow-up of oral dysplasia—A systematic review and meta-analysis. Head Neck 2009, 31, 1600–1609. [Google Scholar] [CrossRef]
- Van der Waal, I. Potentially malignant disorders of the oral and oropharyngeal mucosa; terminology, classification and present concepts of management. Oral Oncol. 2009, 45, 317–323. [Google Scholar] [CrossRef]
- Holmstrup, P.; Vedtofte, P.; Reibel, J.; Stoltze, K. Oral premalignant lesions: Is a biopsy reliable? J. Oral Pathol. Med. 2007, 36, 262–266. [Google Scholar] [CrossRef]
- Graveland, A.P.; Bremmer, J.F.; De Maaker, M.; Brink, A.; Cobussen, P.; Zwart, M.; Braakhuis, B.J.; Bloemena, E.; Van Der Waal, I.; Leemans, C.R.; et al. Molecular screening of oral precancer. Oral Oncol. 2013, 49, 1129–1135. [Google Scholar] [CrossRef]
- Saba, N.F.; Haigentz, M.; Vermorken, J.B.; Strojan, P.; Bossi, P.; Rinaldo, A.; Takes, R.P.; Ferlito, A. Prevention of head and neck squamous cell carcinoma: Removing the “chemo” from “chemoprevention”. Oral Oncol. 2015, 51, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Serafini, M.S.; Lopez-Perez, L.; Fico, G.; Licitra, L.; De Cecco, L.; Resteghini, C. Transcriptomics and Epigenomics in head and neck cancer: Available repositories and molecular signatures. Cancers Head Neck 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Ribatti, D. The concept of immune surveillance against tumors: The first theories. Oncotarget 2016, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Yagyuu, T.; Hatakeyama, K.; Imada, M.; Kurihara, M.; Matsusue, Y.; Yamamoto, K.; Obayashi, C.; Kirita, T. Programmed death ligand 1 (PD-L1) expression and tumor microenvironment: Implications for patients with oral precancerous lesions. Oral Oncol. 2017, 68, 36–43. [Google Scholar] [CrossRef]
- Young, M. Immunological Phenotypes of Premalignant Oral Lesions and the Immune Shifts with the Development of Head and Neck Cancer. Austin J. Otolaryngol. 2014, 1, 7. [Google Scholar]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.H.-F.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef]
- Rosin, M.P.; Cheng, X.; Poh, C.; Lam, W.L.; Huang, Y.; Lovas, J.; Berean, K.; Epstein, J.B.; Priddy, R.; Le, N.D.; et al. Use of allelic loss to predict malignant risk for low-grade oral epithelial dysplasia. Clin. Cancer Res. 2000, 6, 357–362. [Google Scholar]
- Zhang, L.; Poh, C.F.; Williams, M.; Laronde, D.; Berean, K.; Gardner, P.J.; Jiang, H.; Wu, L.; Lee, J.J.; Rosin, M.P. Loss of heterozygosity (LOH) profiles--validated risk predictors for progression to oral cancer. Cancer Prev. Res. 2012, 5, 1081–1089. [Google Scholar] [CrossRef]
- Lodi, G.; Franchini, R.; Warnakulasuriya, S.; Varoni, E.M.; Sardella, A.; Kerr, A.R.; Carrassi, A.; MacDonald, L.C.I.; Worthington, H. Interventions for treating oral leukoplakia to prevent oral cancer. Cochrane Database Syst. Rev. 2016, 2016, CD001829. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carenzo, A.; Serafini, M.S.; Roca, E.; Paderno, A.; Mattavelli, D.; Romani, C.; Saintigny, P.; Koljenović, S.; Licitra, L.; De Cecco, L.; et al. Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions. Cells 2020, 9, 1828. https://doi.org/10.3390/cells9081828
Carenzo A, Serafini MS, Roca E, Paderno A, Mattavelli D, Romani C, Saintigny P, Koljenović S, Licitra L, De Cecco L, et al. Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions. Cells. 2020; 9(8):1828. https://doi.org/10.3390/cells9081828
Chicago/Turabian StyleCarenzo, Andrea, Mara S. Serafini, Elisa Roca, Alberto Paderno, Davide Mattavelli, Chiara Romani, Pierre Saintigny, Senada Koljenović, Lisa Licitra, Loris De Cecco, and et al. 2020. "Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions" Cells 9, no. 8: 1828. https://doi.org/10.3390/cells9081828
APA StyleCarenzo, A., Serafini, M. S., Roca, E., Paderno, A., Mattavelli, D., Romani, C., Saintigny, P., Koljenović, S., Licitra, L., De Cecco, L., & Bossi, P. (2020). Gene Expression Clustering and Selected Head and Neck Cancer Gene Signatures Highlight Risk Probability Differences in Oral Premalignant Lesions. Cells, 9(8), 1828. https://doi.org/10.3390/cells9081828