Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Behavioural Procedures
2.3. Antidepressant Treatment in FSL Rats
2.4. Neuronal Cultures
2.5. RNA Extraction and cDNA Synthesis
2.6. qPCR
2.7. Transcriptional Analysis
2.8. Statistical Analysis
3. Results
3.1. Antidepressant Treatments Alter Negr1-Fgfr2 Pathway Expression in Hypothalamus of FSL Rats
3.2. Antidepressant Treatments Induce Selective Alterations in Corticolimbic Regions of FSL Rats
3.3. Negr1-Fgfr2 Pathway Expression in the Behavioural Response to Fluoxetine in the Chronic Corticosterone Mice Model
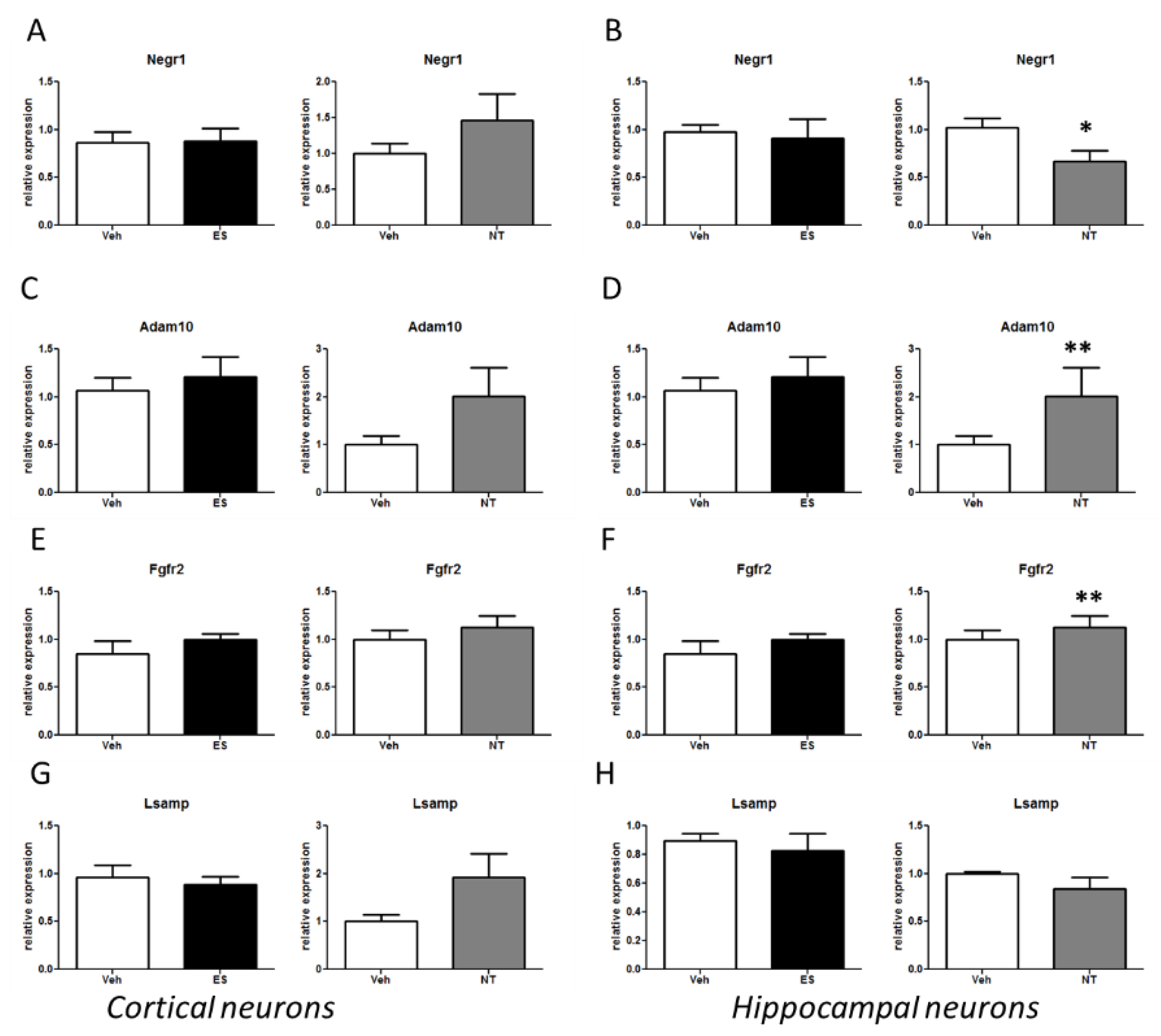
3.4. Alterations of the Negr1-Fgfr2 Pathway in Primary Neuronal Cultures Treated with Antidepressants
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Gerring, Z.F.; Gamazon, E.R.; Derks, E.M. Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet. 2019, 15, e1008245. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cheng, W.; Zhu, J.; Yin, H.; Chang, S.; Yue, W.; Yu, H. Integrating genome-wide association study and expression quantitative trait loci data identifies NEGR1 as a causal risk gene of major depression disorder. J. Affect. Disord. 2020, 265, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Maccarrone, G.; Ditzen, C.; Yassouridis, A.; Rewerts, C.; Uhr, M.; Uhlen, M.; Holsboer, F.; Turck, C.W. Psychiatric patient stratification using biosignatures based on cerebrospinal fluid protein expression clusters. J. Psychiatr. Res. 2013, 47, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Amare, A.T.; Schubert, K.O.; Tekola-Ayele, F.; Hsu, Y.H.; Sangkuhl, K.; Jenkins, G.; Whaley, R.M.; Barman, P.; Batzler, A.; Altman, R.B.; et al. The association of obesity and coronary artery disease genes with response to SSRIs treatment in major depression. J. Neural Transm. 2019, 126, 35–45. [Google Scholar] [CrossRef]
- Raghavan, N.S.; Vardarajan, B.; Mayeux, R. Genomic variation in educational attainment modifies Alzheimer disease risk. Neurol. Genet. 2019, 5. [Google Scholar] [CrossRef]
- Ni, H.; Xu, M.; Zhan, G.-L.; Fan, Y.; Zhou, H.; Jiang, H.-Y.; Lu, W.-H.; Tan, L.; Zhang, D.-F.; Yao, Y.-G.; et al. The GWAS Risk Genes for Depression May Be Actively Involved in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1149–1161. [Google Scholar] [CrossRef]
- Willer, C.; Speliotes, E.; Loos, R.; Li, S.; Lindgren, C.; Heid, I.; Berndt, S.; Elliott, A.; Jackson, A.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar]
- Jarick, I.; Vogel, C.I.G.; Scherag, S.; Schäfer, H.; Hebebrand, J.; Hinney, A.; Scherag, A. Novel common copy number variation for early onset extreme obesity on chromosome 11q11 identified by a genome-wide analysis. Hum. Mol. Genet. 2011, 20, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.S.; Hengstler, H.; Schwald, K.; Berriel-Diaz, M.; Loreth, D.; Kirsch, M.; Kretz, O.; Haas, C.A.; de Angelis, M.H.; Herzig, S.; et al. Functional inactivation of the genome-wide association study obesity gene neuronal growth regulator 1 in mice causes a body mass phenotype. PLoS ONE 2012, 7, e41537. [Google Scholar] [CrossRef] [PubMed]
- Boender, A.J.; van Gestel, M.A.; Garnerv, K.M.; Luijendijk, M.C.M.; Adan, R.A.H. The Obesity-Associated gene NEGR1 regulates aspects of energy balance in rat hypothalamic areas. Physiol. Rep. 2014, 2, e12083. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Loreth, D.; Pöttker, B.; Hefti, K.; Innos, J.; Schwald, K.; Hengstler, H.; Menzel, L.; Sommer, C.J.; Radyushkin, K.; et al. Neuronal growth and behavioral alterations in mice deficient for the psychiatric disease-associated negr1 gene. Front. Mol. Neurosci. 2018, 11, 30. [Google Scholar] [CrossRef]
- Singh, K.; Jayaram, M.; Kaare, M.; Leidmaa, E.; Jagomäe, T.; Heinla, I.; Hickey, M.A.; Kaasik, A.; Schäfer, M.K.; Innos, J.; et al. Neural cell adhesion molecule Negr1 deficiency in mouse results in structural brain endophenotypes and behavioral deviations related to psychiatric disorders. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Funatsu, N.; Miyata, S.; Kumanogoh, H.; Shigeta, M.; Hamada, K.; Endo, Y.; Sokawa, Y.; Maekawa, S. Characterization of a novel rat brain glycosylphosphatidylinositol- anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 1999, 274, 8224–8230. [Google Scholar] [CrossRef]
- Ramani, S.R.; Tom, I.; Lewin-Koh, N.; Wranik, B.; Depalatis, L.; Zhang, J.; Eaton, D.; Gonzalez, L.C. A secreted protein microarray platform for extracellular protein interaction discovery. Anal. Biochem. 2012, 420, 127–138. [Google Scholar] [CrossRef]
- Pourhaghighi, R.; Ash, P.E.A.; Phanse, S.; Goebels, F.; Hu, L.Z.M.; Chen, S.; Zhang, Y.; Wierbowski, S.D.; Boudeau, S.; Moutaoufik, M.T.; et al. BraInMap Elucidates the Macromolecular Connectivity Landscape of Mammalian Brain. Cell Syst. 2020, 10, 333–350. [Google Scholar] [CrossRef]
- Innos, J.; Philips, M.A.; Leidmaa, E.; Heinla, I.; Raud, S.; Reemann, P.; Plaas, M.; Nurk, K.; Kurrikoff, K.; Matto, V.; et al. Lower anxiety and a decrease in agonistic behaviour in Lsamp-deficient mice. Behav. Brain Res. 2011, 217, 21–31. [Google Scholar] [CrossRef]
- Innos, J.; Leidmaa, E.; Philips, M.A.; Sütt, S.; Alttoa, A.; Harro, J.; Kõks, S.; Vasar, E. Lsamp-/- mice display lower sensitivity to amphetamine and have elevated 5-HT turnover. Biochem. Biophys. Res. Commun. 2013, 430, 413–418. [Google Scholar] [CrossRef]
- Koido, K.; Janno, S.; Traks, T.; Parksepp, M.; Ljubajev, Ü.; Veiksaar, P.; Must, A.; Shlik, J.; Vasar, V.; Vasar, E. Associations between polymorphisms of LSAMP gene and schizophrenia. Psychiatry Res. 2014, 215, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Traks, T.; Balõtšev, R.; Eller, T.; Must, A.; Koks, S.; Maron, E.; Tõru, I.; Shlik, J.; Vasar, V.; et al. Associations between LSAMP gene polymorphisms and major depressive disorder and panic disorder. Transl. Psychiatry 2012, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.L.; Jahanshad, N.; Braskie, M.N.; Warstadt, N.M.; Hibar, D.P.; Kohannim, O.; Nir, T.M.; McMahon, K.L.; de Zubicaray, G.I.; Montgomery, G.W.; et al. Obesity gene NEGR1 associated with white matter integrity in healthy young adults. Neuroimage 2014, 102, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Szczurkowska, J.; Cirnaru, M.D.; Giesert, F.; Vezzoli, E.; Ueffing, M.; Sala, C.; Francolini, M.; Hauck, S.M.; Cancedda, L.; et al. A cell surface biotinylation assay to reveal membrane-associated neuronal cues: Negr1 regulates dendritic arborization. Mol. Cell. Proteom. 2014, 13, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Piccoli, G. The IgLON Family Member Negr1 Promotes Neuronal Arborization Acting as Soluble Factor via FGFR2. Front. Mol. Neurosci. 2016, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.J.; Choudary, P.V.; Neal, C.R.; Li, J.Z.; Vawter, M.P.; Tomita, H.; Lopez, J.F.; Thompson, R.C.; Meng, F.; Stead, J.D.; et al. Dysregulation of the fibroblast growth factor system in major depression. Proc. Natl. Acad. Sci. USA 2004, 101, 15506–15511. [Google Scholar] [CrossRef]
- Wittchen, H.U.; Jacobi, F.; Rehm, J.; Gustavsson, A.; Svensson, M.; Jönsson, B.; Olesen, J.; Allgulander, C.; Alonso, J.; Faravelli, C.; et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur. Neuropsychopharmacol. 2011, 21, 655–679. [Google Scholar] [CrossRef]
- Rehm, J.; Shield, K.D. Global Burden of Disease and the Impact of Mental and Addictive Disorders. Curr. Psychiatry Rep. 2019, 21, 10. [Google Scholar] [CrossRef]
- Catalá-López, F.; Gènova-Maleras, R.; Vieta, E.; Tabarés-Seisdedos, R. The increasing burden of mental and neurological disorders. Eur. Neuropsychopharmacol. 2013, 23, 1337–1339. [Google Scholar] [CrossRef]
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 16065. [Google Scholar] [CrossRef]
- Park, L.T.; Zarate, C.A. Depression in the Primary Care Setting. N. Engl. J. Med. 2019, 380, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Tundo, A.; de Filippis, R.; Proietti, L. Pharmacologic approaches to treatment resistant depression: Evidences and personal experience. Worldj. Psychiatry 2015, 5, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, D.H.; Friedman, E.; Mathé, A.A.; Yadid, G. The Flinders Sensitive Line rat: A selectively bred putative animal model of depression. Neurosci. Biobehav. Rev. 2005, 29, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, D.H.; Wegener, G. The flinders sensitive line rat model of depression--25 years and still producing. Pharmacol. Rev. 2013, 65, 143–155. [Google Scholar] [CrossRef]
- Nelson, J.; Klumparendt, A.; Doebler, P.; Ehring, T. Childhood maltreatment and characteristics of adult depression: Meta-analysis. Br. J. Psychiatry 2017, 210, 96–104. [Google Scholar] [CrossRef]
- Piubelli, C.; Carboni, L.; Becchi, S.; Mathé, A.A.; Domenici, E. Regulation of cytoskeleton machinery, neurogenesis and energy metabolism pathways in a rat gene-environment model of depression revealed by proteomic analysis. Neuroscience 2011, 176, 349–380. [Google Scholar] [CrossRef]
- El Khoury, A.; Gruber, S.H.M.; Mørk, A.; Mathé, A.A. Adult life behavioral consequences of early maternal separation are alleviated by escitalopram treatment in a rat model of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 535–540. [Google Scholar] [CrossRef]
- Marchetti, L.; Lauria, M.; Caberlotto, L.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E.; Carboni, L. Gene expression signature of antidepressant treatment response/non-response in Flinders Sensitive Line rats subjected to maternal separation. Eur. Neuropsychopharmacol. 2020, 31, 69–85. [Google Scholar] [CrossRef]
- Musazzi, L.; Mallei, A.; Tardito, D.; Gruber, S.H.M.; El Khoury, A.; Racagni, G.; Mathé, A.A.; Popoli, M. Early-life stress and antidepressant treatment involve synaptic signaling and Erk kinases in a gene-environment model of depression. J. Psychiatr. Res. 2010, 44, 511–520. [Google Scholar] [CrossRef]
- Piubelli, C.; Gruber, S.; El Khoury, A.; Mathé, A.A.; Domenici, E.; Carboni, L. Nortriptyline influences protein pathways involved in carbohydrate metabolism and actin-related processes in a rat gene–environment model of depression. Eur. Neuropsychopharmacol. 2011, 21, 545–562. [Google Scholar] [CrossRef]
- Piubelli, C.; Vighini, M.; Mathé, A.A.; Domenici, E.; Carboni, L. Escitalopram affects cytoskeleton and synaptic plasticity pathways in a rat gene–environment interaction model of depression as revealed by proteomics. Part II: Environmental challenge. Int. J. Neuropsychopharmacol. 2011, 14, 834–855. [Google Scholar] [CrossRef] [PubMed]
- Piubelli, C.; Vighini, M.; Mathé, A.A.; Domenici, E.; Carboni, L. Escitalopram modulates neuron-remodelling proteins in a rat gene–environment interaction model of depression as revealed by proteomics. Part I: Genetic background. Int. J. Neuropsychopharmacol. 2011, 14, 796–833. [Google Scholar] [CrossRef] [PubMed]
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.; et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009, 62, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Berton, O.; Nestler, E.J. New approaches to antidepressant drug discovery: Beyond monoamines. Nat. Rev. Neurosci. 2006, 7, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Disner, S.G.; Beevers, C.G.; Haigh, E.A.P.; Beck, A.T. Neural mechanisms of the cognitive model of depression. Nat. Rev. Neurosci. 2011, 12, 467–477. [Google Scholar] [CrossRef]
- Mallei, A.; Giambelli, R.; Gass, P.; Racagni, G.; Math??, A.A.; Vollmayr, B.; Popoli, M. Synaptoproteomics of learned helpless rats involve energy metabolism and cellular remodeling pathways in depressive-like behavior and antidepressant response. Neuropharmacology 2011, 60, 1243–1253. [Google Scholar] [CrossRef]
- Pischedda, F.; Montani, C.; Obergasteiger, J.; Frapporti, G.; Corti, C.; Rosato Siri, M.; Volta, M.; Piccoli, G. Cryopreservation of Primary Mouse Neurons: The Benefit of Neurostore Cryoprotective Medium. Front. Cell. Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Samuels, B.A.; Leonardo, E.D.; Dranovsky, A.; Williams, A.; Wong, E.; Nesbitt, A.M.I.; McCurdy, R.D.; Hen, R.; Alter, M. Global state measures of the dentate gyrus gene expression system predict antidepressant-sensitive behaviors. PLoS ONE 2014, 9, e85136. [Google Scholar] [CrossRef]
- Bate, S.T.; Clark, R.A. The Design and Statistical Analysis of Animal Experiments; Cambridge University Press: Cambridge, UK, 2014; ISBN 978-1-107-69094-3. [Google Scholar]
- Clark, R.A.; Shoaib, M.; Hewitt, K.N.; Stanford, S.C.; Bate, S.T. A comparison of InVivoStat with other statistical software packages for analysis of data generated from animal experiments. J. Psychopharmacol. 2012, 26, 1136–1142. [Google Scholar] [CrossRef]
- Milaneschi, Y.; Simmons, W.K.; van Rossum, E.F.C.; Penninx, B.W. Depression and obesity: Evidence of shared biological mechanisms. Mol. Psychiatry 2019, 24, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.C.; Clark, D.L.; Hassel, S.; MacQueen, G.; Ramasubbu, R. Thalamocortical connectivity in major depressive disorder. J. Affect. Disord. 2017, 217, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mulders, P.C.; van Eijndhoven, P.F.; Schene, A.H.; Beckmann, C.F.; Tendolkar, I. Resting-state functional connectivity in major depressive disorder: A review. Neurosci. Biobehav. Rev. 2015, 56, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhu, Y.; Zhu, Y.; Wu, S.; Liu, H.; Zhang, W.; Xu, C.; Zhang, H.; Hayashi, T.; Tian, M. Molecular, Functional, and Structural Imaging of Major Depressive Disorder. Neurosci. Bull. 2016, 32, 273–285. [Google Scholar] [CrossRef]
- Helm, K.; Viol, K.; Weiger, T.M.; Tass, P.A.; Grefkes, C.; Del Monte, D.; Schiepek, G. Neuronal connectivity in major depressive disorder: A systematic review. Neuropsychiatr. Dis. Treat. 2018, 14, 2715–2737. [Google Scholar] [CrossRef] [PubMed]
- Mayberg, H.S.; Liotti, M.; Brannan, S.K.; McGinnis, S.; Mahurin, R.K.; Jerabek, P.A.; Silva, J.A.; Tekell, J.L.; Martin, C.C.; Lancaster, J.L.; et al. Reciprocal limbic-cortical function and negative mood: Converging PET findings in depression and normal sadness. Am. J. Psychiatry 1999, 156, 675–682. [Google Scholar]
- Brakowski, J.; Spinelli, S.; Dörig, N.; Bosch, O.G.; Manoliu, A.; Holtforth, M.G.; Seifritz, E. Resting state brain network function in major depression–Depression symptomatology, antidepressant treatment effects, future research. J. Psychiatr. Res. 2017, 92, 147–159. [Google Scholar] [CrossRef]
- Dichter, G.S.; Gibbs, D.; Smoski, M.J. A systematic review of relations between resting-state functional-MRI and treatment response in major depressive disorder. J. Affect. Disord. 2015, 172, 8–17. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef]
- Harmer, C.J.; Duman, R.S.; Cowen, P.J. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet. Psychiatry 2017, 4, 409–418. [Google Scholar] [CrossRef]
- Castrén, E.; Hen, R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013, 36, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Kavalali, E.T.; Monteggia, L.M. Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 2020, 106, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Sanz, R.; Ferraro, G.B.; Fournier, A.E. IgLON Cell Adhesion Molecules Are Shed from the Cell Surface of Cortical Neurons to Promote Neuronal Growth. J. Biol. Chem. 2015, 290, 4330–4342. [Google Scholar] [CrossRef] [PubMed]
- Sanz, R.L.; Ferraro, G.B.; Girouard, M.P.; Fournier, A.E. Ectodomain shedding of Limbic System-Associated Membrane Protein (LSAMP) by ADAM Metallopeptidases promotes neurite outgrowth in DRG neurons. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maekawa, S.; Miyata, S. IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem. Funct. 2009, 27, 496–498. [Google Scholar] [CrossRef]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012, 18, 1413–1417. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K. Synaptic dysfunction in depression: Potential therapeutic targets. Science 2012, 338, 68–72. [Google Scholar] [CrossRef]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2012, 13, 22–37. [Google Scholar] [CrossRef]
- Szczurkowska, J.; Pischedda, F.; Pinto, B.; Managò, F.; Haas, C.A.; Summa, M.; Bertorelli, R.; Papaleo, F.; Schäfer, M.K.; Piccoli, G.; et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 2018, 141, 2772–2794. [Google Scholar] [CrossRef]
- Bianco, O.A.F.M.; Manzine, P.R.; Nascimento, C.M.C.; Vale, F.A.C.; Pavarini, S.C.I.; Cominetti, M.R. Serotoninergic antidepressants positively affect platelet ADAM10 expression in patients with Alzheimer’s disease. Int. Psychogeriatr. 2016, 28, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Tamási, V.; Petschner, P.; Adori, C.; Kirilly, E.; Ando, R.D.; Tothfalusi, L.; Juhasz, G.; Bagdy, G. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also glutatmatergic- and insulin-mediated neuronal processes. PLoS ONE 2014, 9, e113662. [Google Scholar] [CrossRef]
- Carboni, L.; Becchi, S.; Piubelli, C.; Mallei, A.; Giambelli, R.; Razzoli, M.; Mathé, A.A.; Popoli, M.; Domenici, E. Early-life stress and antidepressants modulate peripheral biomarkers in a gene-environment rat model of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, B.A.; Angelucci, F.; Husum, H.; Mathé, A.A. Gene-environment interactions in a rat model of depression. Maternal separation affects neurotensin in selected brain regions. Neuropeptides 2016, 59, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Ellenbroek, B.A.; El Khoury, A.; Mathé, A.A. CGRP in a gene–environment interaction model for depression: Effects of antidepressant treatment. Acta Neuropsychiatr. 2019, 31, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.S.; Pine, D.S.; Luckenbaugh, D.A.; Varnäs, K.; Henter, I.D.; Innis, R.B.; Mathé, A.A.; Svenningsson, P. Antidepressant effects on serotonin 1A/1B receptors in the rat brain using a gene x environment model. Neurosci. Lett. 2014, 559, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Tansey, K.E.; Guipponi, M.; Perroud, N.; Bondolfi, G.; Domenici, E.; Evans, D.; Hall, S.K.; Hauser, J.; Henigsberg, N.; Hu, X.; et al. Genetic Predictors of Response to Serotonergic and Noradrenergic Antidepressants in Major Depressive Disorder: A Genome-Wide Analysis of Individual-Level Data and a Meta-Analysis. PLoS Med. 2012, 9, e1001326. [Google Scholar] [CrossRef]
- Hagena, H.; Hansen, N.; Manahan-Vaughan, D. β-Adrenergic Control of Hippocampal Function: Subserving the Choreography of Synaptic Information Storage and Memory. Cereb. Cortex 2016, 26, 1349–1364. [Google Scholar] [CrossRef]
- Ramos, B.P.; Arnsten, A.F.T. Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol. Ther. 2007, 113, 523–536. [Google Scholar] [CrossRef]
- Celada, P.; Puig, M.V.; Artigas, F. Serotonin modulation of cortical neurons and networks. Front. Integr. Neurosci. 2013, 7, 25. [Google Scholar] [CrossRef]
- Berumen, L.C.; Rodríguez, A.; Miledi, R.; García-Alcocer, G. Serotonin Receptors in Hippocampus. Sci. Worldj. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hisaoka, K.; Tsuchioka, M.; Yano, R.; Maeda, N.; Kajitani, N.; Morioka, N.; Nakata, Y.; Takebayashi, M. Tricyclic Antidepressant Amitriptyline Activates Fibroblast Growth Factor Receptor Signaling in Glial Cells. J. Biol. Chem. 2011, 286, 21118–21128. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Flinders Sensitive Rats (ES, NT) | Cort-Treated Mice (FLX) | Primary Neurons (ES, NT) | ||||
|---|---|---|---|---|---|---|---|
| Hippocampus | Hypothalamus | Cortex | Hip DG (dorsal) | Hip DG (ventral) | Hippocampus | Cortex | |
| Negr1 | - | ↓↓ nMS ES | - | ↓↓↓sens ↓↓↓res | ↓↓↓sens ↓↓res | ↓ NT | - |
| Adam10 | ↑ MS ES | ↑ MS NT | - | ↑↑ sens | - | ↑↑ NT | - |
| Fgfr2 | - | ↑ MS NT | - | - | - | ↑↑ NT | - |
| Lsamp | - | - | - | ↑ sens | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carboni, L.; Pischedda, F.; Piccoli, G.; Lauria, M.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E. Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells 2020, 9, 1818. https://doi.org/10.3390/cells9081818
Carboni L, Pischedda F, Piccoli G, Lauria M, Musazzi L, Popoli M, Mathé AA, Domenici E. Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells. 2020; 9(8):1818. https://doi.org/10.3390/cells9081818
Chicago/Turabian StyleCarboni, Lucia, Francesca Pischedda, Giovanni Piccoli, Mario Lauria, Laura Musazzi, Maurizio Popoli, Aleksander A. Mathé, and Enrico Domenici. 2020. "Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment" Cells 9, no. 8: 1818. https://doi.org/10.3390/cells9081818
APA StyleCarboni, L., Pischedda, F., Piccoli, G., Lauria, M., Musazzi, L., Popoli, M., Mathé, A. A., & Domenici, E. (2020). Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells, 9(8), 1818. https://doi.org/10.3390/cells9081818