Transcriptomic Profiling of Ca2+ Transport Systems during the Formation of the Cerebral Cortex in Mice

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Primary Cultures of Cortical Cells
2.3. Single-Cell Calcium Imaging
2.4. Single-Cell Zinc Imaging
3. Results and Discussion
3.1. Transcriptomic Analysis
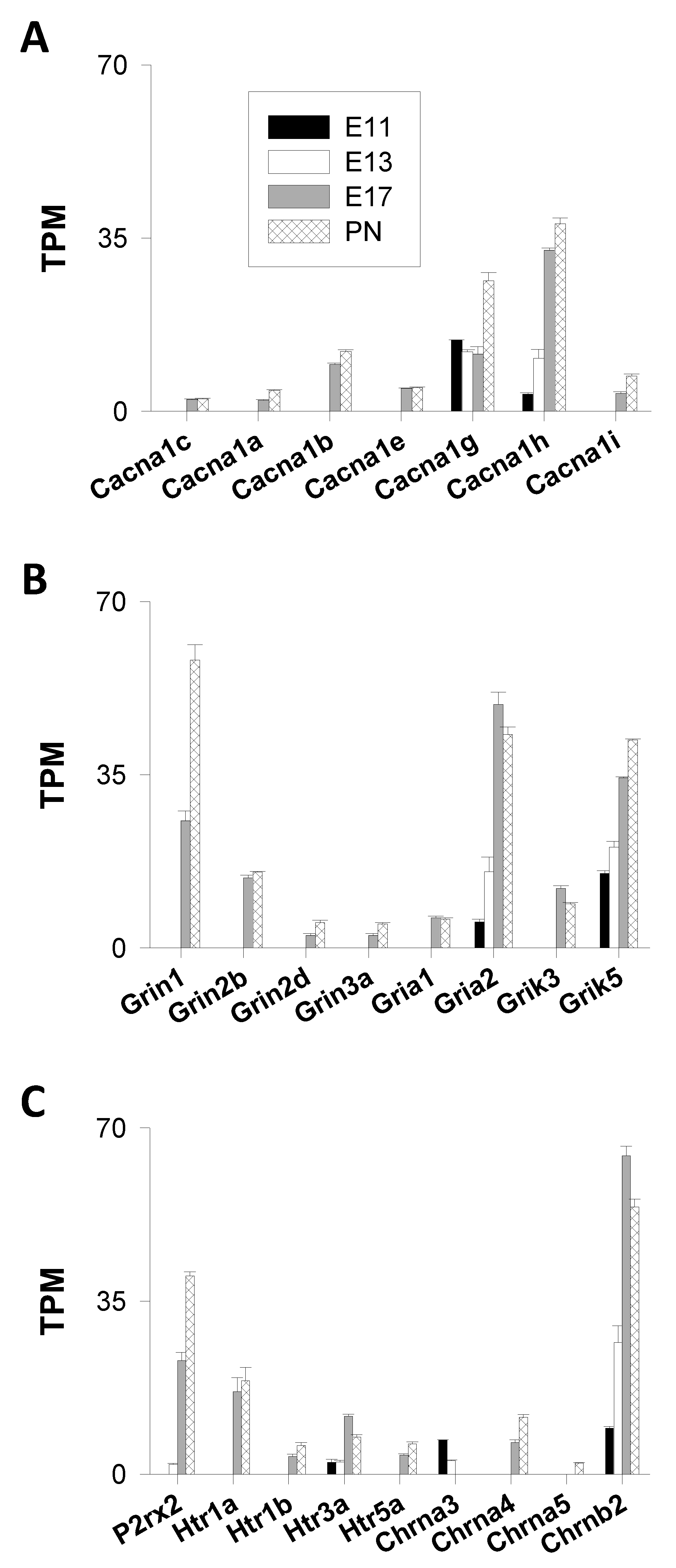
3.1.1. Channels
Voltage-Gated Ca2+ Channels (VGCC)
Ligand-Gated Cation Channels (LGCC)
- Ionotropic Glutamatergic Receptors
- (a)
- AMPA Receptors (GluA1-4)
- (b)
- NMDA Receptors (GluN1–3)
- (c)
- Kainate Receptors (GluK1-5)
- 2.
- Ionotropic Purinergic Receptors
- 3.
- Ionotropic Serotoninergic (5-HT) Receptors
- 4.
- Nicotinic Acetylcholine Receptors
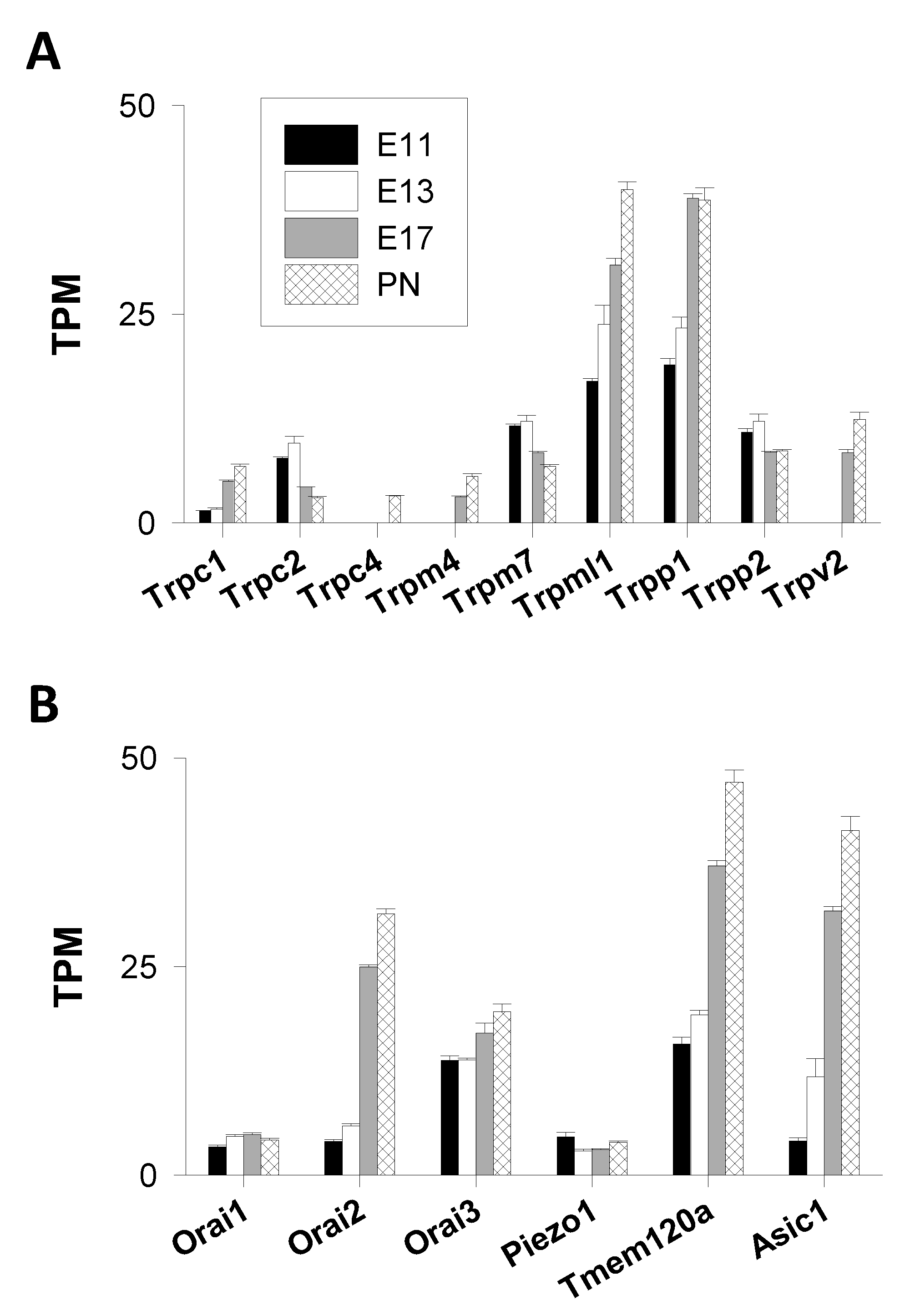
Transient Receptor Potential (TRP) Channels
- TRPA Channels
- 2.
- TRPC Channels
- 3.
- TRPM Channels
- 4.
- TRPML Channels
- 5.
- TRPP Channels
- 6.
- TRPV Channels
Orai Channels
Stretch-Activated Channels
Acid-Sensing Ion Channels (ASIC)
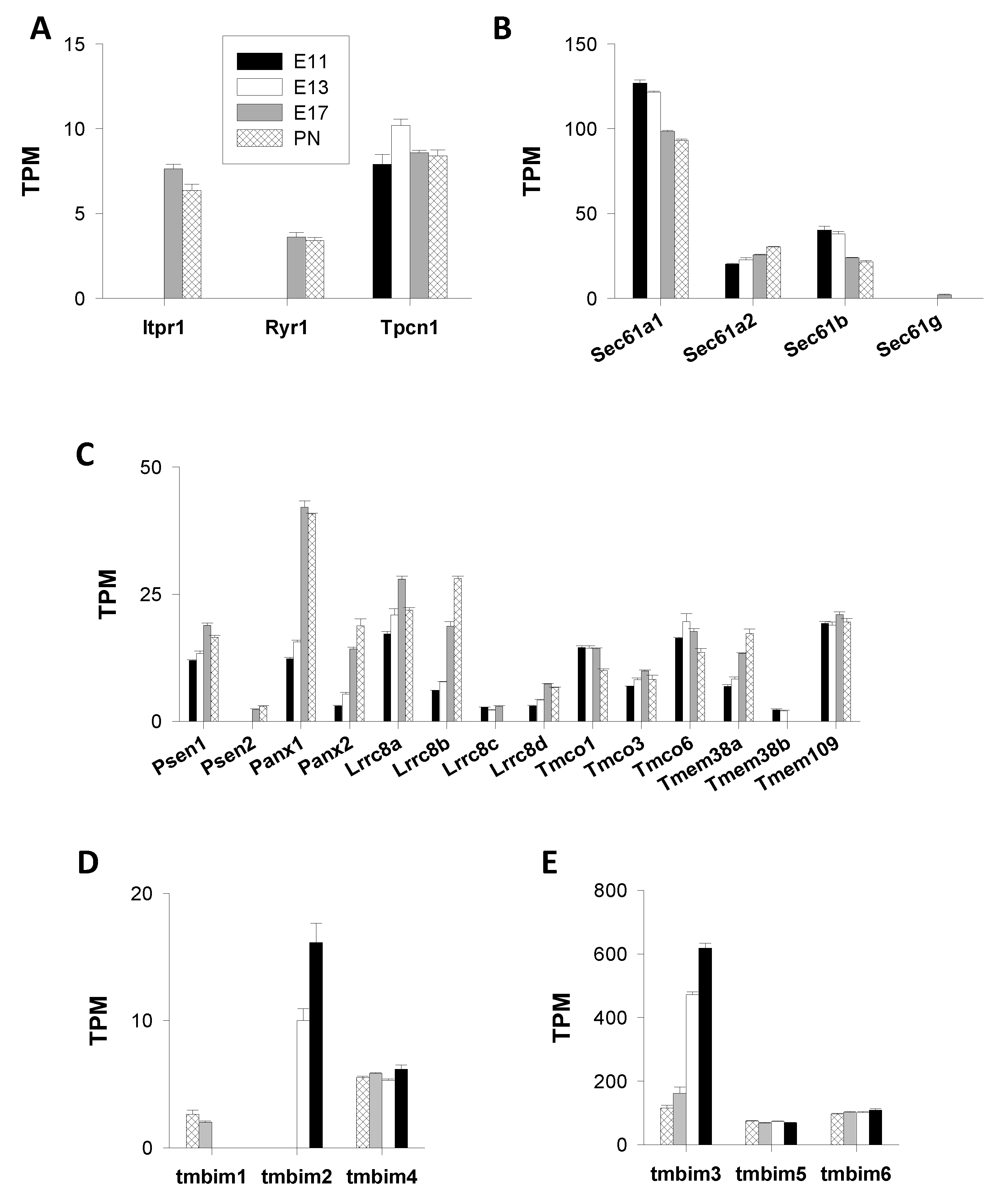
Inositol 1,4,5-Trisphosphate and Ryanodine Receptors (IP3R, RyR), and Two-Pore Channels (TPC)
Intracellular “leak Channels”
- Translocons
- 2.
- Presenilins
- 3.
- Pannexins
- 4.
- LRRC8
- 5.
- TMCO1
- 6.
- TRIC
- 7.
- Mitsugumin23
- 8.
- Bax Inhibitor-1 Motif-Containing (TMBIM) Proteins
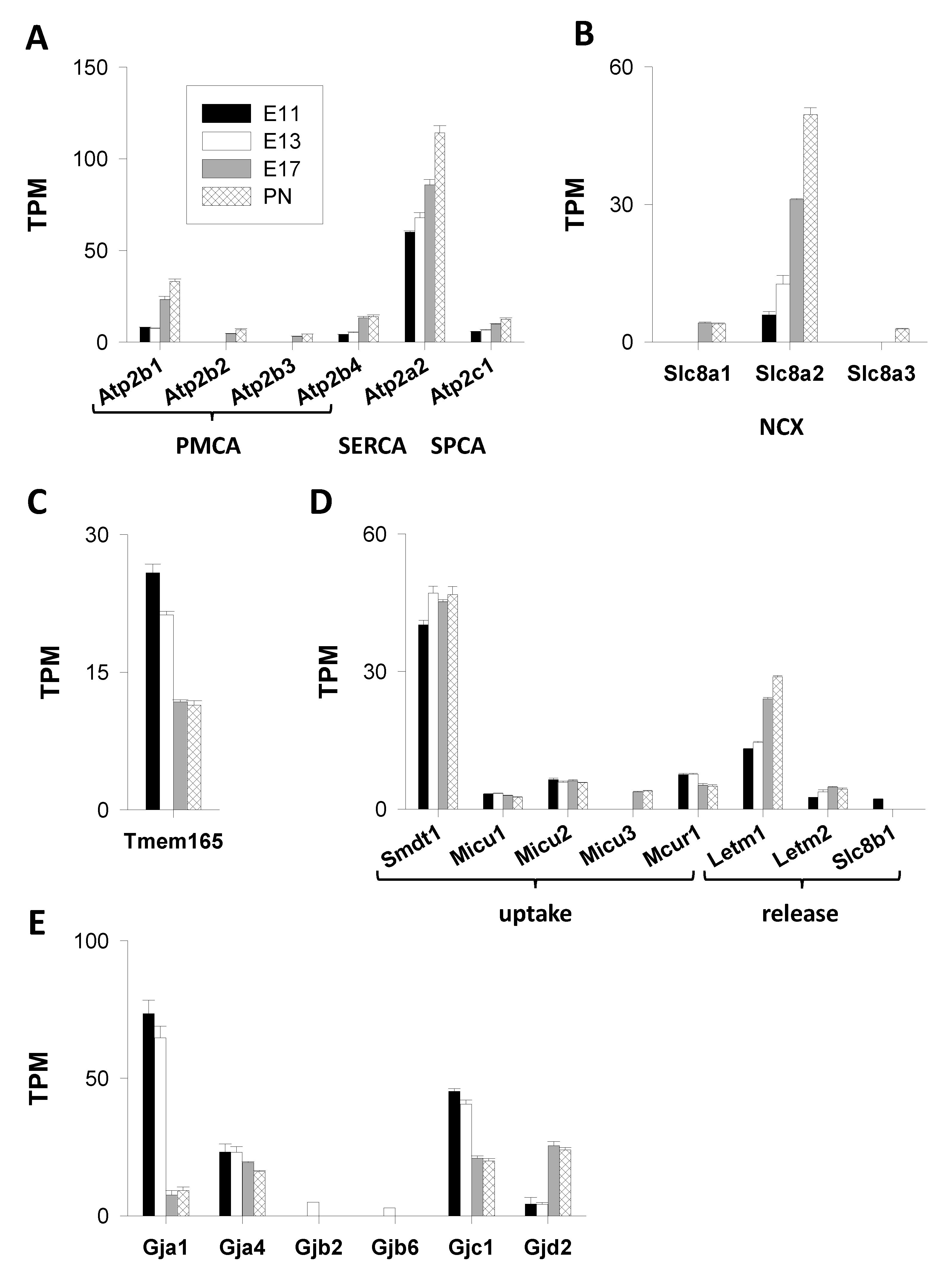
3.1.2. Ca2+ ATPases
3.1.3. Exchangers
3.1.4. Cation Antiporter
3.1.5. Mitochondrial Actors (Figure 4D)
Mitochondrial Ca2+ Uptake
Mitochondrial Ca2+ Release
3.1.6. Connexins
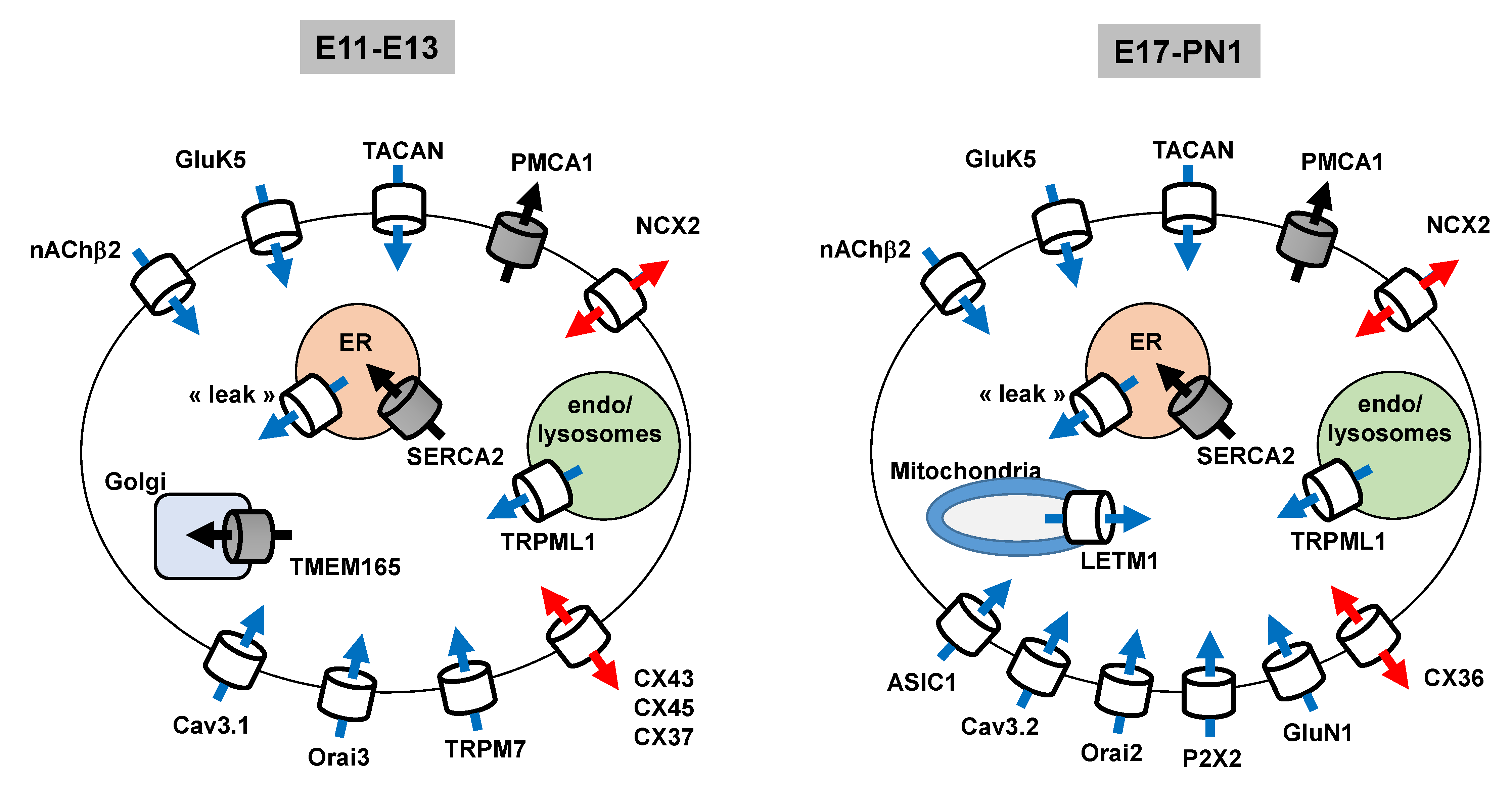
3.1.7. Expression Profile of The Main Ca2+ Transport Systems Throughout Corticogenesis
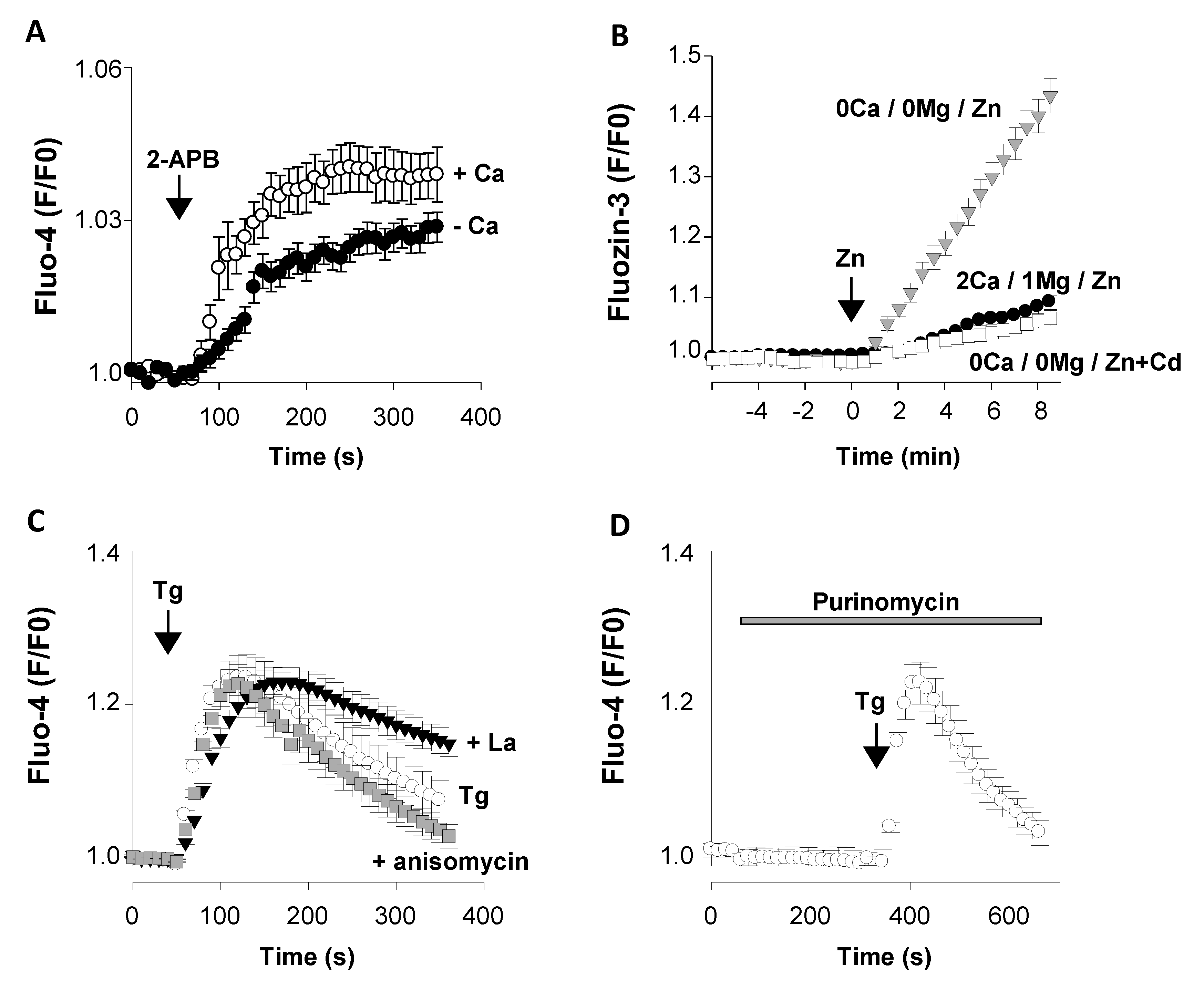
3.2. Presence of Functional Ca2+ Transport Systems at E13
3.2.1. Functional Ca2+ Transport Systems of The Plasma Membrane
Acetylcholine Nicotinic Receptors AChR β2
Orai3
TRPM7
3.2.2. Functional Intracellular Ca2+ Transport Systems
The Ca2+ Pump SERCA2
The Leak Channel Translocon
3.3. Summary
4. Conclusions
Supplementary Materials
Supplementary File 1Funding
Acknowledgments
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Komuro, H.; Rakic, P. Intracellular Ca2+ Fluctuations Modulate the Rate of Neuronal Migration. Neuron 1996, 17, 275–285. [Google Scholar] [CrossRef]
- Owens, D.F.; Kriegstein, A.R. Patterns of Intracellular Calcium Fluctuation in Precursor Cells of the Neocortical Ventricular Zone. J. Neurosci. 1998, 18, 5374–5388. [Google Scholar] [CrossRef] [PubMed]
- Gomez, T.M.; Spitzer, N.C. Regulation of Growth Cone Behavior by Calcium: New Dynamics to Earlier Perspectives. J. Neurobiol. 2000, 44, 174–183. [Google Scholar] [CrossRef]
- Kriegstein, A.R.; Noctor, S.C. Patterns of Neuronal Migration in the Embryonic Cortex. Trends Neurosci. 2004, 27, 392–399. [Google Scholar] [CrossRef]
- Jabaudon, D. Fate and Freedom in Developing Neocortical Circuits. Nat. Commun. 2017, 8, 16042. [Google Scholar] [CrossRef]
- Finlay, B.L.; Uchiyama, R. Developmental Mechanisms Channeling Cortical Evolution. Trends Neurosci. 2015, 38, 69–76. [Google Scholar] [CrossRef]
- Sun, T.; Hevner, R.F. Growth and Folding of the Mammalian Cerebral Cortex: From Molecules to Malformations. Nat. Rev. Neurosci. 2014, 15, 217–232. [Google Scholar] [CrossRef]
- Kriegstein, A.; Parnavelas, J.G. Changing Concepts of Cortical Development. Cereb. Cortex 2003, 13, 541. [Google Scholar] [CrossRef]
- Barkovich, A.J.; Guerrini, R.; Kuzniecky, R.I.; Jackson, G.D.; Dobyns, W.B. A Developmental and Genetic Classification for Malformations of Cortical Development: Update 2012. Brain A J. Neurol. 2012, 135, 1348–1369. [Google Scholar] [CrossRef]
- Behar, T.N.; Schaffner, A.E.; Catherine, A.S.; Casey, O.C.; Jeffery, L.B. Differential Response of Cortical Plate and Ventricular Zone Cells to Gaba as a Migration Stimulus. J. Neurosci. 1998, 18, 6378–6387. [Google Scholar] [CrossRef] [PubMed]
- Hasna, J.; Bohic, S.; Lemoine, S.; Blugeon, C.; Bouron, A. Zinc Uptake and Storage during the Formation of the Cerebral Cortex in Mice. Mol. Neurobiol. 2019, 56, 6928–6940. [Google Scholar] [CrossRef] [PubMed]
- Gibon, J.; Deloulme, J.C.; Chevallier, T.; Ladeveze, E.; Abrous, D.N.; Bouron, A. The Antidepressant Hyperforin Increases the Phosphorylation of Creb and the Expression of Trkb in a Tissue—Specific Manner. Int. J. Neuropsychopharmacol. 2013, 16, 189–198. [Google Scholar] [CrossRef]
- Chauvet, S.; Louis, J.; Mireille, C.; Niroj, S.; Klaus, G.; Alexandre, B. Pharmacological Characterization of the Native Store-Operated Calcium Channels of Cortical Neurons from Embryonic Mouse Brain. Front. Pharmacol. 2016, 7, 486. [Google Scholar] [CrossRef]
- Bouron, A. Phyto and Endocannabinoids Exert Complex Actions on Calcium and Zinc Signaling in Mouse Cortical Neurons. Biochem. Pharm. 2018, 152, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Bouron, A.C.; Mbebi, C.; Loeffler, J.P.; Waard, M.D. The Beta-Amyloid Precursor Protein Controls a Store-Operated Ca2+ Entry in Cortical Neurons. Eur. J. Neurosci. 2004, 20, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Deveci, A.; Hasna, J.; Bouron, A. Inhibition of Store-Operated Calcium Channels by N-Arachidonoyl Glycine (Nagly): No Evidence for the Involvement of Lipid-Sensing G Protein Coupled Receptors. Sci. Rep. 2020, 10, 2649. [Google Scholar] [CrossRef]
- Tu, P.; Gibon, J.; Bouron, A. The Trpc6 Channel Activator Hyperforin Induces the Release of Zinc and Calcium from Mitochondria. J. Neurochem. 2010, 112, 204–213. [Google Scholar] [CrossRef]
- Yunker, A.M.; Sharp, A.H.; Sundarraj, S.; Ranganathan, V.; Copeland, T.D.; McEnery, M.W. Immunological Characterization of T-Type Voltage-Dependent Calcium Channel Cav3.1 (Alpha 1g) and Cav3.3 (Alpha 1i) Isoforms Reveal Differences in Their Localization, Expression, and Neural Development. Neuroscience 2003, 117, 321–335. [Google Scholar] [CrossRef]
- Frischknecht, F.; Randall, A.D. Voltage- and Ligand-Gated Ion Channels in Floor Plate Neuroepithelia of the Rat. Neuroscience 1998, 85, 1135–1149. [Google Scholar] [CrossRef]
- Bouron, A.S.; Boisseau, A.; Waard, M.D.; Peris, L. Differential Down-Regulation of Voltage-Gated Calcium Channel Currents by Glutamate and Bdnf in Embryonic Cortical Neurons. Eur. J. Neurosci. 2006, 24, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, D.B.; Lund, T.M.; Belhage, B.; Schousboe, A. Localization and Pharmacological Characterization of Voltage Dependent Calcium Channels in Cultured Neocortical Neurons. Int. J. Dev. Neurosci. 2001, 19, 1–10. [Google Scholar] [CrossRef]
- Bahrey, H.L.; Moody, W.J. Voltage-Gated Currents, Dye and Electrical Coupling in the Embryonic Mouse Neocortex. Cereb. Cortex 2003, 13, 239–251. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hadzic, M.; Jack, A.; Wahle, P. Ionotropic Glutamate Receptors: Which Ones, When, and Where in the Mammalian Neocortex. J. Comp. Neurol. 2017, 525, 976–1033. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and Regional Expression in the Rat Brain and Functional Properties of Four Nmda Receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Gallyas, F., Jr.; Ball, S.M.; Molnar, E. Assembly and Cell Surface Expression of Ka-2 Subunit-Containing Kainate Receptors. J. Neurochem. 2003, 86, 1414–1427. [Google Scholar] [CrossRef]
- Zoli, M.; Le, N.; Novere, J.A.; Hill, J.; Changeux, J.P. Developmental Regulation of Nicotinic Ach Receptor Subunit Mrnas in the Rat Central and Peripheral Nervous Systems. J. Neurosci. 1995, 15, 1912–1939. [Google Scholar] [CrossRef]
- Tanaka, H.; Grooms, S.Y.; Bennett, M.V.; Zukin, R.S. The Ampar Subunit Glur2: Still Front and Center-Stage. Brain Res. 2000, 886, 190–207. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic Ampa Receptor Composition in Development, Plasticity and Disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef]
- Whitney, N.P.; Peng, H.; Erdmann, N.B.; Tian, C.; Monaghan, D.T.; Zheng, J.C. Calcium-Permeable Ampa Receptors Containing Q/R-Unedited Glur2 Direct Human Neural Progenitor Cell Differentiation to Neurons. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 2888–2900. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. Transient Receptor Potential Channelopathies. Pflug. Arch. 2010, 460, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.J.; Sweet, T.B.; Clapham, D.E. International Union of Basic and Clinical Pharmacology. Lxxvi. Current Progress in the Mammalian Trp Ion Channel Family. Pharm. Rev. 2010, 62, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Owsianik, G.; Talavera, K.; Voets, T.; Nilius, B. Permeation and Selectivity of Trp Channels. Annu. Rev. Physiol. 2006, 68, 685–717. [Google Scholar] [CrossRef] [PubMed]
- Fliniaux, I.; Germain, E.; Farfariello, V.; Prevarskaya, N. Trps and Ca(2+) in Cell Death and Survival. Cell Calcium. 2018, 69, 4–18. [Google Scholar] [CrossRef]
- Busch, T.; Kottgen, M.; Hofherr, A. Trpp2 Ion Channels: Critical Regulators of Organ Morphogenesis in Health and Disease. Cell Calcium 2017, 66, 25–32. [Google Scholar] [CrossRef]
- Nilius, B.; Szallasi, A. Transient Receptor Potential Channels as Drug Targets: From the Science of Basic Research to the Art of Medicine. Pharm. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef]
- Jang, Y.; Lee, Y.; Kim, S.M.; Yang, Y.D.; Jung, J.; Oh, U. Quantitative Analysis of Trp Channel Genes in Mouse Organs. Arch. Pharm. Res. 2012, 35, 1823–1830. [Google Scholar] [CrossRef]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-Specific Expression of Trp Channel Genes in the Mouse and Its Variation in Three Different Mouse Strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular Trpml1-3 Channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Wong, C.O.; Zhu, M.X. The Role of Trpmls in Endolysosomal Trafficking and Function. Cell Calcium 2015, 58, 48–56. [Google Scholar] [CrossRef]
- Tai, Y.; Feng, S.; Du, W.; Wang, Y. Functional Roles of Trpc Channels in the Developing Brain. Pflug. Arch. 2009, 458, 283–289. [Google Scholar] [CrossRef]
- Lucas, P.; Ukhanov, K.; Leinders-Zufall, T.; Zufall, F.A. Diacylglycerol-Gated Cation Channel in Vomeronasal Neuron Dendrites Is Impaired in Trpc2 Mutant Mice: Mechanism of Pheromone Transduction. Neuron 2003, 40, 551–561. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. The Neglected Crac Proteins: Orai2, Orai3, and Stim2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R. Stim and Orai Proteins in the Nervous System. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Meuth, G.S.; Nieswandt, B. Stim2 Regulates Capacitive Ca2+ Entry in Neurons and Plays a Key Role in Hypoxic Neuronal Cell Death. Sci. Signal. 2009, 2, 67. [Google Scholar] [CrossRef]
- Honoré, E.; Martins, J.R.; Penton, D.; Patel, A.; Demolombe, S. The Piezo Mechanosensitive Ion Channels: May the Force Be with You! Rev. Physiol. Biochem. Pharm. 2015, 169, 25–41. [Google Scholar] [CrossRef]
- Koser, D.E.; Thompson, A.J.; Foster, S.K.; Dwivedy, A.; Pillai, E.K.; Sheridan, G.K.; Svoboda, H.; Viana, M.; Costa, L.D.; Guck, C.E.; et al. Mechanosensing Is Critical for Axon Growth in the Developing Brain. Nat. Neurosci. 2016, 19, 1592–1598. [Google Scholar] [CrossRef]
- Beaulieu-Laroche, L.; Christin, M.; Donoghue, A.; Agosti, F.; Yousefpour, N.; Petitjean, H.; Davidova, A.; Petitjean, H.; Faure, E.; Stanton, C.; et al. Tacan Is an Ion Channel Involved in Sensing Mechanical Pain. Cell 2020, 180, 956–967. [Google Scholar] [CrossRef]
- Grunder, S.; Pusch, M. Biophysical Properties of Acid-Sensing Ion Channels (Asics). Neuropharmacology 2015, 94, 9–18. [Google Scholar] [CrossRef]
- Deval, E.; Lingueglia, E. Acid-Sensing Ion Channels and Nociception in the Peripheral and Central Nervous Systems. Neuropharmacology 2015, 94, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kratzer, E.; Inoue, K.; Simon, R.P.; Xiong, Z.G. Developmental Change in the Electrophysiological and Pharmacological Properties of Acid-Sensing Ion Channels in Cns Neurons. J. Physiol. 2010, 588, 3883–3900. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Askwith, C.C.; Lamani, E.; Cassell, M.D.; Freeman, J.H.; Welsh, M.J. Acid-Sensing Ion Channel 1 Is Localized in Brain Regions with High Synaptic Density and Contributes to Fear Conditioning. J. Neurosci. 2003, 23, 5496–5502. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Chen, J.; Askwith, C.C.; Hruska-Hageman, A.M.; Price, M.P.; Nolan, B.C.; Welsh, M.J. The Acid-Activated Ion Channel Asic Contributes to Synaptic Plasticity, Learning, and Memory. Neuron 2002, 34, 463–477. [Google Scholar] [CrossRef]
- Ruas, M.; Galione, A.; Parrington, J. Two-Pore Channels: Lessons from Mutant Mouse Models. Messenger 2015, 4, 4–22. [Google Scholar] [CrossRef]
- Szlufcik, K.; Missiaen, L.; Parys, J.B.; Callewaert, G.; De Smedt, H. Uncoupled Ip3 Receptor Can Function as a Ca2+-Leak Channel: Cell Biological and Pathological Consequences. Biol. Cell. 2006, 98, 1–14. [Google Scholar] [CrossRef]
- Takeshima, H.; Venturi, E.; Sitsapesan, R. New and Notable Ion-Channels in the Sarcoplasmic/Endoplasmic Reticulum: Do They Support the Process of Intracellular Ca(2)(+) Release? J. Physiol. 2015, 593, 3241–3251. [Google Scholar] [CrossRef]
- Berbey, C.; Weiss, N.; Legrand, C.; Allard, B. Transient Receptor Potential Canonical Type 1 (Trpc1) Operates as a Sarcoplasmic Reticulum Calcium Leak Channel in Skeletal Muscle. J. Biol. Chem. 2009, 284, 36387–36394. [Google Scholar] [CrossRef]
- Leon-Aparicio, D.; Pacheco, J.; Chavez-Reyes, J.; Galindo, J.M.; Valdes, J.; Vaca, L.; Guerrero-Hernandez, A. Orai3 Channel Is the 2-Apb-Induced Endoplasmic Reticulum Calcium Leak. Cell Calcium 2017, 65, 91–101. [Google Scholar] [CrossRef]
- Lisak, D.A.; Schacht, T.; Enders, V.; Habicht, J.; Kiviluoto, S.; Schneider, J.; Methner, A. The Transmembrane Bax Inhibitor Motif (Tmbim) Containing Protein Family: Tissue Expression, Intracellular Localization and Effects on the Er Ca(2)(+)-Filling State. Biochim. Biophys. Acta. 2015, 1853, 2104–2114. [Google Scholar] [CrossRef]
- Johnson, A.E.; Van Waes, M.A. The Translocon: A Dynamic Gateway at the Er Membrane. Annu. Rev. Cell. Dev. Biol. 1999, 15, 799–842. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Van Coppenolle, F.; Lehen’Kyi, V.Y.; Beck, B.; Skryma, R.; Prevarskaya, N.; Skryma, R. Passive Calcium Leak Via Translocon Is a First Step for Ipla2-Pathway Regulated Store Operated Channels Activation. FASEB J. 2006, 20, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Erdmann, F.; Jung, M.; Wagner, R.; Cavalie, A.; Zimmermann, R. Sec61 Complexes Form Ubiquitous Er Ca2+ Leak Channels. Channels 2011, 5, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Bezprozvanny, I. Presenilins Form Er Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Bezprozvanny, I. Lack of Evidence for Presenilins as Endoplasmic Reticulum Ca2+ Leak Channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef]
- Bond, S.R.; Naus, C.C. The Pannexins: Past and Present. Front. Physiol. 2014, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Penuela, S.; Gehi, R.; Laird, D.W. The Biochemistry and Function of Pannexin Channels. Biochim. Biophys. Acta 2013, 1828, 15–22. [Google Scholar] [CrossRef]
- Ghosh, A.; Khandelwal, N.; Kumar, A.; Bera, A.K. Leucine-Rich Repeat-Containing 8b Protein Is Associated with the Endoplasmic Reticulum Ca(2+) Leak in Hek293 Cells. J. Cell Sci. 2017, 130, 3818–3828. [Google Scholar] [CrossRef]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. Tmco1 Is an Er Ca(2+) Load-Activated Ca(2+) Channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef]
- Zhou, X.; Lin, P.; Yamazaki, D.; Park, K.H.; Komazaki, S.; Chen, S.W.; Ma, J. Trimeric Intracellular Cation Channels and Sarcoplasmic/Endoplasmic Reticulum Calcium Homeostasis. Circ. Res. 2014, 114, 706–716. [Google Scholar] [CrossRef]
- Shrestha, N.; Bacsa, B.; Ong, H.L.; Scheruebel, S.; Bischof, H.; Malli, R.; Groschner, K. Tric-a Shapes Oscillatory Ca2+ Signals by Interaction with Stim1/Orai1 Complexes. PLoS Biol. 2020, 18, e3000700. [Google Scholar] [CrossRef] [PubMed]
- Venturi, E.; Mio, K.; Nishi, M.; Ogura, T.; Moriya, T.; Pitt, S.J.; Okuda, K.; Kakizawa, S.; Sitsapesan, R.; Sato, C.; et al. Mitsugumin 23 Forms a Massive Bowl-Shaped Assembly and Cation-Conducting Channel. Biochemistry 2011, 50, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q. Tmbim-Mediated Ca(2+) Homeostasis and Cell Death. Biochim. Et Biophys. Acta. Mol. Cell Res. 2017, 1864, 850–857. [Google Scholar] [CrossRef]
- Brini, M. Plasma Membrane Ca(2+)-Atpase: From a Housekeeping Function to a Versatile Signaling Role. Pflug. Arch. 2009, 457, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Maric, D.; Maric, I.; Barker, J.L. Developmental Changes in Cell Calcium Homeostasis during Neurogenesis of the Embryonic Rat Cerebral Cortex. Cereb. Cortex 2000, 10, 561–573. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stribny, J.; Thines, L.; Deschamps, A.; Goffin, P.; Morsomme, P. The Human Golgi Protein Tmem165 Transports Calcium and Manganese in Yeast and Bacterial Cells. J. Biol. Chem. 2020, 295, 3865–3874. [Google Scholar] [CrossRef]
- Demaegd, D.; Foulquier, F.; Colinet, A.S.; Gremillon, L.; Legrand, D.; Mariot, P.; Morsomme, P. Newly Characterized Golgi-Localized Family of Proteins Is Involved in Calcium and Ph Homeostasis in Yeast and Human Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6859–6864. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. Nclx Protein, but Not Letm1, Mediates Mitochondrial Ca2+ Extrusion, Thereby Limiting Ca2+-Induced Nad(P)H Production and Modulating Matrix Redox State. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-Wide Rnai Screen Identifies Letm1 as a Mitochondrial Ca2+/H+ Antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef]
- Tsai, M.F.; Jiang, D.; Zhao, L.; Clapham, D.; Miller, C. Functional Reconstitution of the Mitochondrial Ca2+/H+ Antiporter Letm1. J. Gen. Physiol. 2014, 143, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Nowikovsky, K. Letm1: Essential for Mitochondrial Biology and Cation Homeostasis? Trends Biochem. Sci. 2019, 44, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Turco, J.L.; Kriegstein, A.R. Clusters of Coupled Neuroblasts in Embryonic Neocortex. Science 1991, 252, 563–566. [Google Scholar] [CrossRef]
- Uhlen, P.; Fritz, N.; Smedler, E.; Malmersjö, S.; Kanatani, S. Calcium Signaling in Neocortical Development. Dev. Neurobiol. 2015, 75, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Mathie, A.; Peters, J.A.; Veale, E.L.; Striessnig, J.; Kelly, E.; Armstron, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J. The Concise Guide to Pharmacology 2019/20: Ion Channels. Br. J. Pharmacol. 2019, 176, S142–S228. [Google Scholar] [CrossRef]
- Elias, L.A.; Kriegstein, A.R. Gap Junctions: Multifaceted Regulators of Embryonic Cortical Development. Trends Neurosci. 2008, 31, 243–250. [Google Scholar] [CrossRef]
- Platel, J.C.; Boisseau, S.; Dupuis, A.; Brocard, J.; Poupard, A.; Savasta, M.; Villaz, M.; Albrieux, M. Na+ Channel-Mediated Ca2+ Entry Leads to Glutamate Secretion in Mouse Neocortical Preplate. Proc. Natl. Acad. Sci. USA 2005, 102, 19174–19179. [Google Scholar] [CrossRef]
- Zhang, S.L.; Kozak, J.A.; Jiang, W.; Yeromin, A.V.; Chen, J.; Yu, Y.; Penna, A.; Shen, W.; Chi, V.; Cahalan, M.D. Store-Dependent and -Independent Modes Regulating Ca2+ Release-Activated Ca2+ Channel Activity of Human Orai1 and Orai3. J. Biol. Chem. 2008, 283, 17662–17671. [Google Scholar] [CrossRef]
- Peinelt, C.; Lis, A.; Beck, A.; Fleig, A.; Penner, R. 2-Aminoethoxydiphenyl Borate Directly Facilitates and Indirectly Inhibits Stim1-Dependent Gating of Crac Channels. J. Physiol. 2008, 586, 3061–3073. [Google Scholar] [CrossRef]
- Gibon, J.; Tu, P.; Bouron, A. Store-Depletion and Hyperforin Activate Distinct Types of Ca(2+)-Conducting Channels in Cortical Neurons. Cell Calcium 2010, 47, 538–543. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Mochida, S.; Krapivinsky, L.; Cibulsky, S.M.; Clapham, D.E. The Trpm7 Ion Channel Functions in Cholinergic Synaptic Vesicles and Affects Transmitter Release. Neuron 2006, 52, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.L.; Sun, H.S.; Olah, M.E.; Sun, X.; Czerwinska, E.; Czerwinski, W.; Mori, Y.; Orser, B.A.; Xiong, Z.-G.; Jackson, M.; et al. Trpm7 Channels in Hippocampal Neurons Detect Levels of Extracellular Divalent Cations. Proc. Natl. Acad. Sci. USA 2007, 104, 16323–16328. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Branigan, D.; Xiong, Z.G. Zinc-Induced Neurotoxicity Mediated by Transient Receptor Potential Melastatin 7 Channels. J. Biol. Chem. 2010, 285, 7430–7439. [Google Scholar] [CrossRef] [PubMed]
- Fleig, A.; Chubanov, V. Trpm7. Handb Exp. Pharm. 2014, 222, 521–546. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Liu, Y.; Li, W.; Wang, Z.; Sun, Q.; Zhou, H.; Chen, X.; Yu, Y.; Wang, Y.; et al. Trpm7 Is Required for Normal Synapse Density, Learning, and Memory at Different Developmental Stages. Cell Rep. 2018, 23, 3480–3491. [Google Scholar] [CrossRef]
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a Tumor Promoter, Discharges Intracellular Ca2+ Stores by Specific Inhibition of the Endoplasmic Reticulum Ca2(+)-Atpase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef]
- Di Capite, J.; Ng, S.W.; Parekh, A.B. Decoding of Cytoplasmic Ca(2+) Oscillations through the Spatial Signature Drives Gene Expression. Curr. Biol. 2009, 19, 853–858. [Google Scholar] [CrossRef]
- Kwan, C.Y.; Takemura, H.; Obie, J.F.; Thastrup, O.; Putney, J.W., Jr. Effects of Mech, Thapsigargin, and La3+ on Plasmalemmal and Intracellular Ca2+ Transport in Lacrimal Acinar Cells. Am. J. Physiol. 1990, 258, C1006–C1015. [Google Scholar] [CrossRef]
- Amer, M.S.; Li, J.; O’Regan, D.J.; Steele, D.S.; Porter, K.E.; Sivaprasadarao, A.; Beech, D.J. Translocon Closure to Ca2+ Leak in Proliferating Vascular Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H910–H916. [Google Scholar] [CrossRef]
- Zima, A.V.; Bovo, E.; Bers, D.M.; Blatter, L.A. Ca(2)+ Spark-Dependent and-Independent Sarcoplasmic Reticulum Ca(2)+ Leak in Normal and Failing Rabbit Ventricular Myocytes. J. Physiol. 2010, 588, 4743–4757. [Google Scholar] [CrossRef]
- Du, H.; Ye, C.; Wu, D.; Zang, Y.Y.; Zhang, L.; Chen, C.; He, X.Y.; Yang, J.J.; Hu, P.; Xu, G.; et al. The Cation Channel Tmem63b Is an Osmosensor Required for Hearing. Cell Rep. 2020, 31, 107596. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, Z.; Li, J.; Hulse, R.E.; Santa-Cruz, A.; Valinsky, W.C.; Krapivinsky, G.; Zhang, J.; Clapham, D.E. Structure of the Mammalian Trpm7, a Magnesium Channel Required During Embryonic Development. Proc. Natl. Acad. Sci. USA 2018, 115, E8201–E8210. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca Transport System | Gene | Protein | Rank Number | |||
|---|---|---|---|---|---|---|
| E11 | E13 | E17 | PN | |||
| Connexins | Gja1 | CX43 | 1 | 1 | / | / |
| Connexins | Gjc1 | CX45 | 2 | 2 | / | / |
| Connexins | Gja4 | CX37 | 3 | 4 | / | / |
| Voltage-gated channels | Cacna1g | Cav3.1 | 6 | 10 | / | / |
| Other channels | Trpm7 | TRPM7 | 8 | 9 | / | / |
| Store-operated channels | Orai3 | Orai3 | 7 | 7 | / | / |
| Exchanger | Slc8a2 | NCX2 | / | 8 | 6 | 3 |
| Connexins | Gjd2 | CX36 | / | / | 8 | / |
| Voltage-gated channels | Cacna1h | Cav3.2 | / | / | 4 | 8 |
| Ligand-gated channels | Grin1 | GluN1 | / | / | 7 | 1 |
| Ligand-gated channels | P2rx2 | P2X2 | / | / | / | 7 |
| Store-operated channels | Orai2 | Orai2 | / | / | 9 | 10 |
| Acid-sensing channels | Asic1 | ASIC1 | / | / | 5 | 6 |
| Ligand-gated channels | Chrnb2 | nAChR β2 | 10 | 3 | 1 | 2 |
| Ligand-gated channels | Grik5 | GluK5 | 5 | 5 | 3 | 5 |
| Mechanosensitive channels | Tmem120A | TACAN | 4 | 6 | 2 | 4 |
| Pump | Atp2b1 | PMCA1 | 9 | / | 10 | 9 |
| Ca Transport System | Gene | Protein | Rank Number | |||
|---|---|---|---|---|---|---|
| E11 | E13 | E17 | PN1 | |||
| « Leak » pathways | Tmem109 | TMEM109 | 9 | / | / | / |
| « Leak » pathways | Tmco6 | TMCO6 | / | 10 | / | / |
| Transporter | Tmem165 | TMEM165 | 7 | 9 | / | / |
| « Leak » pathways | Sec61b | SEC61B | 6 | 6 | 10 | / |
| « Leak » pathways | Panx1 | PANX1 | / | / | 6 | 6 |
| Mitochondrial actor | Letm1 | LETM1 | / | / | 9 | 9 |
| « Leak » pathways | Lrrc8b | LRRC8B | / | / | / | 10 |
| « Leak » pathways | Tmbim3 | TMBIM3 | 2 | 1 | 1 | 1 |
| « Leak » pathways | Tmbim5 | TMBIM5 | 4 | 4 | 5 | 5 |
| « Leak » pathways | Sec61a1 | SEC61A1 | 1 | 2 | 3 | 4 |
| « Leak » pathways | Sec61a2 | SEC61A2 | 8 | 8 | 8 | 8 |
| « Leak » pathways | Tmbim6 | TMBIM6 | 3 | 3 | 2 | 3 |
| Channels | Trpml1 | TRPML1 | 10 | 7 | 7 | 7 |
| Pump | Atp2a2 | SERCA2 | 5 | 5 | 4 | 2 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouron, A. Transcriptomic Profiling of Ca2+ Transport Systems during the Formation of the Cerebral Cortex in Mice. Cells 2020, 9, 1800. https://doi.org/10.3390/cells9081800
Bouron A. Transcriptomic Profiling of Ca2+ Transport Systems during the Formation of the Cerebral Cortex in Mice. Cells. 2020; 9(8):1800. https://doi.org/10.3390/cells9081800
Chicago/Turabian StyleBouron, Alexandre. 2020. "Transcriptomic Profiling of Ca2+ Transport Systems during the Formation of the Cerebral Cortex in Mice" Cells 9, no. 8: 1800. https://doi.org/10.3390/cells9081800
APA StyleBouron, A. (2020). Transcriptomic Profiling of Ca2+ Transport Systems during the Formation of the Cerebral Cortex in Mice. Cells, 9(8), 1800. https://doi.org/10.3390/cells9081800