Signals Orchestrating Peripheral Nerve Repair


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Central vs. Peripheral Axonal Regeneration
3. The Injury Response in the PNS: Intrinsic and Extrinsic Controls of Axonal Regeneration
3.1. Local Protein Synthesis in the Axon
3.2. Epigenetic Regulation of Axonal Re-Growth Program
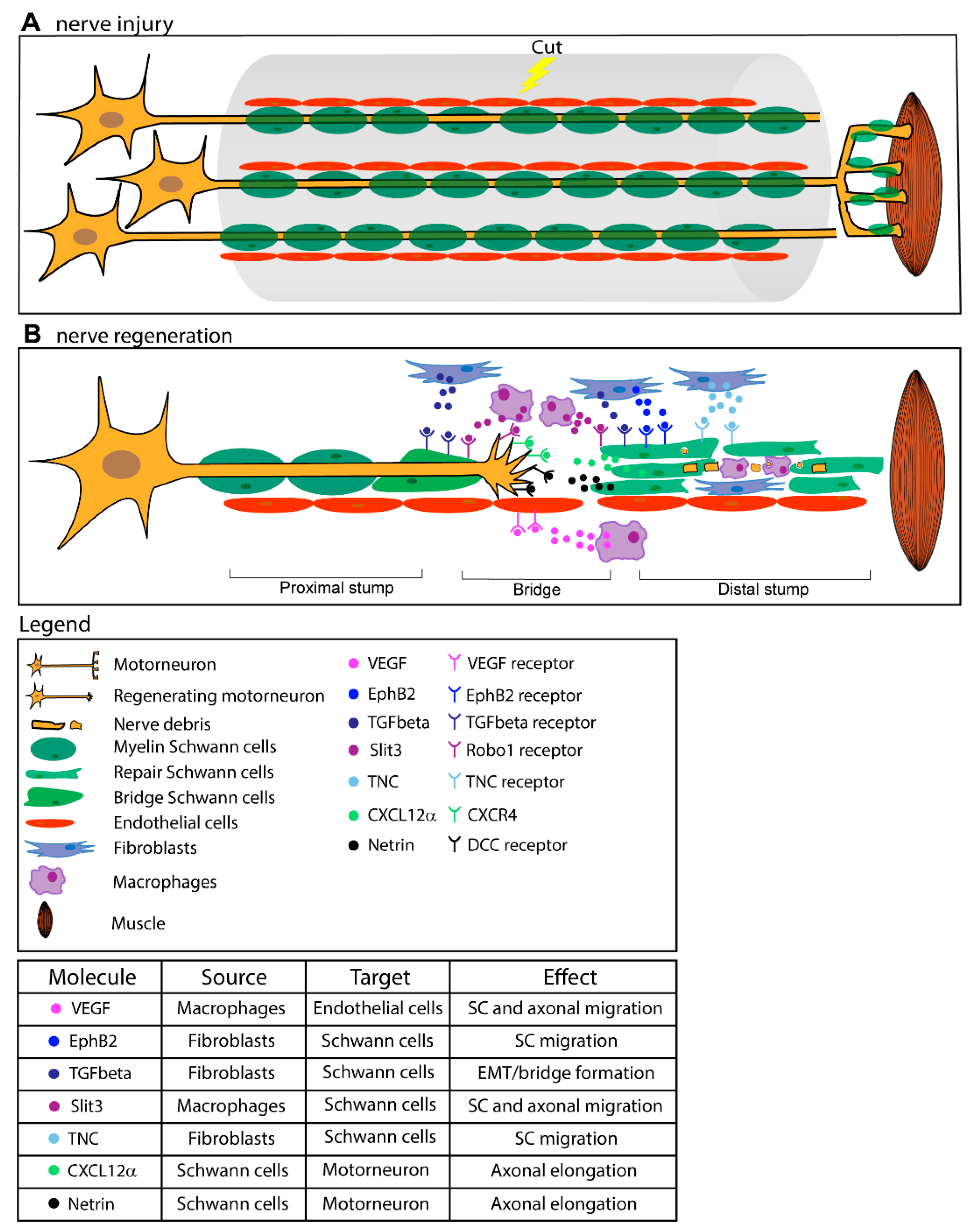
3.3. The Importance of a Pro-Regenerative Environment: Schwann Cells as a Central Hub Coordinating Multiple Partners
3.3.1. The Injury Response of Distal SC
3.3.2. Bridge Formation
3.4. The Mechanobiology of Schwann Cells
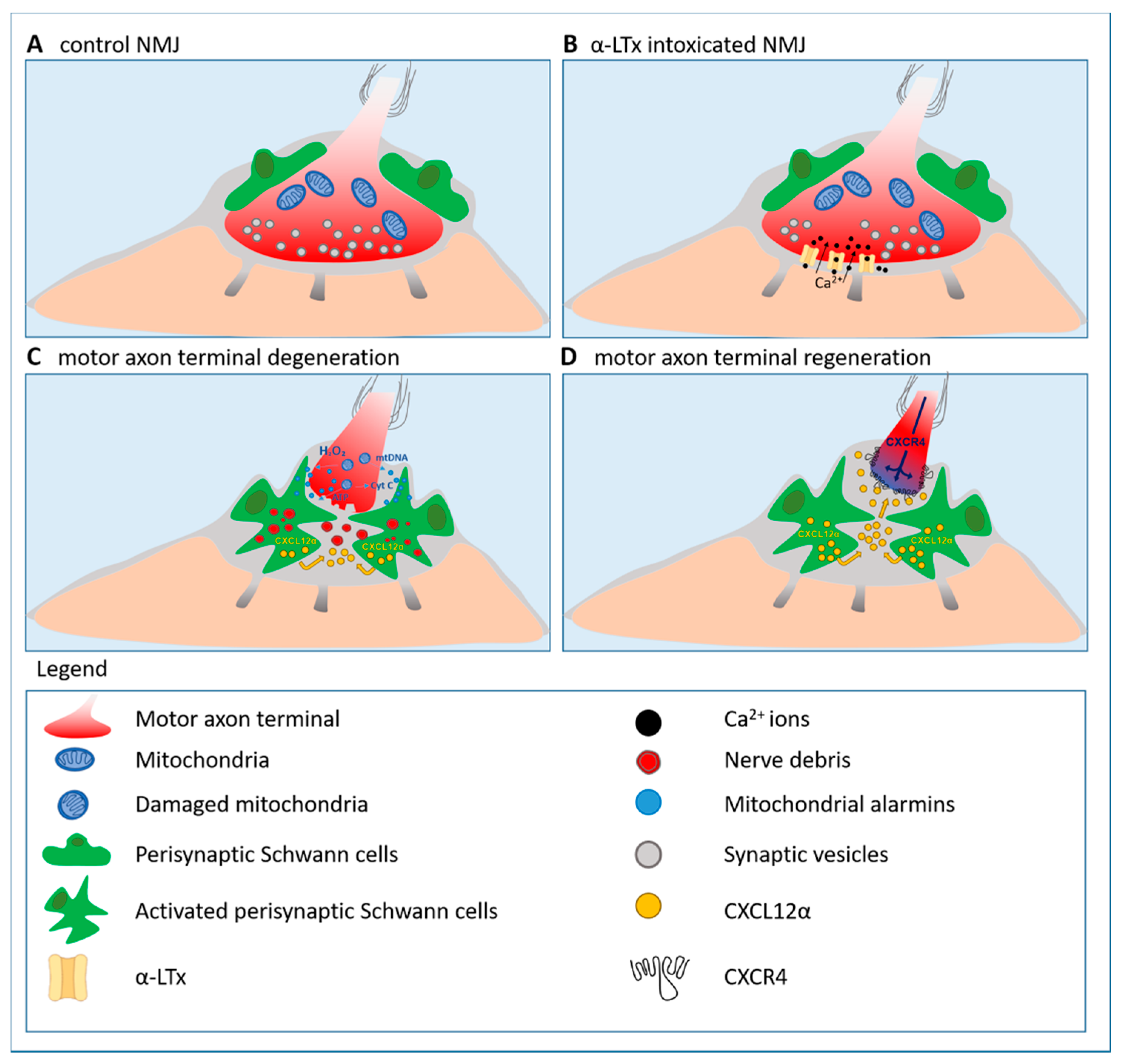
3.5. Presynaptic Neurotoxins: Valuable Tools to Identify Alarm Molecules and Signaling Pathways Driving Motor Axon Regeneration
3.5.1. Hydrogen Peroxide: A Key Alarm Signal Driving Peripheral Nerve Regeneration
3.5.2. Reactivation of the Developmental Axis CXCL12α-CXCR4 Promotes Peripheral Nerve Regeneration
3.6. Regeneration at Sensory Terminals
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BL | basal lamina |
| CNS | central nervous system |
| ECM | extracellular matrix |
| EMT | epithelial to mesenchymal transition |
| HAT | histone acetyl-transferase |
| HDAC | histone deacetylase |
| H2O2 | hydrogen peroxide |
| α-LTx | α-Latrotoxin |
| NMJ | neuromuscular junction |
| OEC | olfactory ensheathing cells |
| PNS | peripheral nervous system |
| PSC | perisynaptic Schwann cells |
| RAG | regeneration associated genes |
| SC | Schwann cells |
References
- Brosius Lutz, A.; Barres, B.A. Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev. Cell 2014, 28, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. Nerve regeneration in the peripheral and central nervous systems. J. Physiol. 2016, 594, 3517–3520. [Google Scholar] [CrossRef] [PubMed]
- Curcio, M.; Bradke, F. Axon Regeneration in the Central Nervous System: Facing the Challenges from the Inside. Annu. Rev. Cell Dev. Biol. 2018, 34, 495–521. [Google Scholar] [CrossRef]
- Tedeschi, A. Tuning the orchestra: Transcriptional pathways controlling axon regeneration. Front. Mol. Neurosci. 2011, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Keynes, R.J. Peripheral nerve regeneration. Annu. Rev. Neurosci. 1990, 13, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, E.A.; Tuszynski, M.H.; Lu, P.; Dulin, J.N. Molecular and Cellular Mechanisms of Axonal Regeneration After Spinal Cord Injury. Mol. Cell Proteom. 2016, 15, 394–408. [Google Scholar] [CrossRef]
- Sanes, J.R.; Lichtman, J.W. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999, 22, 389–442. [Google Scholar] [CrossRef]
- Darabid, H.; Perez-Gonzalez, A.P.; Robitaille, R. Neuromuscular synaptogenesis: Coordinating partners with multiple functions. Nat. Rev. Neurosci. 2014, 15, 703–718. [Google Scholar] [CrossRef]
- Fitzsimonds, R.M.; Poo, M.M. Retrograde signaling in the development and modification of synapses. Physiol. Rev. 1998, 78, 143–170. [Google Scholar] [CrossRef]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of neuromuscular junction in age and dystrophy. Front. Aging Neurosci. 2014, 6, 99. [Google Scholar] [CrossRef]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; Del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: Part II, results and discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76 Pt A, 35–47. [Google Scholar] [CrossRef]
- Son, Y.J.; Thompson, W.J. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 1995, 14, 133–141. [Google Scholar] [CrossRef]
- Son, Y.J.; Thompson, W.J. Schwann cell processes guide regeneration of peripheral axons. Neuron 1995, 14, 125–132. [Google Scholar] [CrossRef]
- Son, Y.J.; Trachtenberg, J.T.; Thompson, W.J. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996, 19, 280–285. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Jin, Y. Intrinsic Control of Axon Regeneration. Neuron 2016, 90, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W.; Schwab, M.E.; Montani, L.; Brazda, N.; Müller, H.W. Defeating inhibition of regeneration by scar and myelin components. Handb. Clin. Neurol. 2012, 109, 503–522. [Google Scholar]
- Liu, K.; Tedeschi, A.; Park, K.K.; He, Z. Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 2011, 34, 131–152. [Google Scholar] [CrossRef]
- Richardson, P.M.; Verge, V.M. Axonal regeneration in dorsal spinal roots is accelerated by peripheral axonal transection. Brain Res. 1987, 411, 406–408. [Google Scholar] [CrossRef]
- Neumann, S.; Woolf, C.J. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 1999, 23, 83–91. [Google Scholar] [CrossRef]
- Richardson, P.M.; Issa, V.M. Peripheral injury enhances central regeneration of primary sensory neurones. Nature 1984, 309, 791–793. [Google Scholar] [CrossRef]
- Tedeschi, A.; Dupraz, S.; Laskowski, C.J.; Xue, J.; Ulas, T.; Beyer, M.; Schultze, J.L.; Bradke, F. The Calcium Channel Subunit Alpha2delta2 Suppresses Axon Regeneration in the Adult CNS. Neuron 2016, 92, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Enes, J.; Langwieser, N.; Ruschel, J.; Carballosa-Gonzalez, M.M.; Klug, A.; Traut, M.H.; Ylera, B.; Tahirovic, S.; Hofmann, F.; Stein, V.; et al. Electrical activity suppresses axon growth through Ca(v)1.2 channels in adult primary sensory neurons. Curr. Biol. 2010, 20, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, P.N. A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp. Neurol. 2010, 223, 11–18. [Google Scholar] [CrossRef]
- Duraikannu, A.; Krishnan, A.; Chandrasekhar, A.; Zochodne, D.W. Beyond Trophic Factors: Exploiting the Intrinsic Regenerative Properties of Adult Neurons. Front. Cell Neurosci. 2019, 13, 128. [Google Scholar] [CrossRef]
- Chen, Z.L.; Yu, W.M.; Strickland, S. Peripheral regeneration. Annu. Rev. Neurosci. 2007, 30, 209–233. [Google Scholar] [CrossRef]
- Hellal, F.; Hurtado, A.; Ruschel, J.; Flynn, K.C.; Laskowski, C.J.; Umlauf, M.; Kapitein, L.C.; Strikis, D.; Lemmon, V.; Bixby, J.; et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science 2011, 331, 928–931. [Google Scholar] [CrossRef]
- Bradke, F.; Fawcett, J.W.; Spira, M.E. Assembly of a new growth cone after axotomy: The precursor to axon regeneration. Nat. Rev. Neurosci. 2012, 13, 183–193. [Google Scholar] [CrossRef]
- Tom, V.J.; Steinmetz, M.P.; Miller, J.H.; Doller, C.M.; Silver, J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J. Neurosci. 2004, 24, 6531–6539. [Google Scholar] [CrossRef]
- Ertürk, A.; Hellal, F.; Enes, J.; Bradke, F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 2007, 27, 9169–9180. [Google Scholar] [CrossRef] [PubMed]
- Ziv, N.E.; Spira, M.E. Axotomy induces a transient and localized elevation of the free intracellular calcium concentration to the millimolar range. J. Neurophysiol. 1995, 74, 2625–2637. [Google Scholar] [CrossRef] [PubMed]
- Ziv, N.E.; Spira, M.E. Localized and transient elevations of intracellular Ca2+ induce the dedifferentiation of axonal segments into growth cones. J. Neurosci. 1997, 17, 3568–3579. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Sloutsky, R.; Naegle, K.M.; Cavalli, V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell 2013, 155, 894–908. [Google Scholar] [CrossRef]
- Rishal, I.; Fainzilber, M. Axon-soma communication in neuronal injury. Nat. Rev. Neurosci. 2014, 15, 32–42. [Google Scholar] [CrossRef]
- Czogalla, A.; Sikorski, A.F. Spectrin and calpain: A ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol. Life Sci. 2005, 62, 1913–1924. [Google Scholar] [CrossRef]
- Yoo, S.; Nguyen, M.P.; Fukuda, M.; Bittner, G.D.; Fishman, H.M. Plasmalemmal sealing of transected mammalian neurites is a gradual process mediated by Ca(2+)-regulated proteins. J. Neurosci. Res. 2003, 74, 541–551. [Google Scholar] [CrossRef]
- Gitler, D.; Spira, M.E. Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron 1998, 20, 1123–1135. [Google Scholar] [CrossRef]
- Gitler, D.; Spira, M.E. Short window of opportunity for calpain induced growth cone formation after axotomy of Aplysia neurons. J. Neurobiol. 2002, 52, 267–279. [Google Scholar] [CrossRef]
- Shin, J.E.; Cho, Y.; Beirowski, B.; Milbrandt, J.; Cavalli, V.; DiAntonio, A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 2012, 74, 1015–1022. [Google Scholar] [CrossRef]
- Saito, A.; Cavalli, V. Signaling Over Distances. Mol. Cell Proteom. 2016, 15, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; van Niekerk, E.A.; Merianda, T.T.; Twiss, J.L. Dynamics of axonal mRNA transport and implications for peripheral nerve regeneration. Exp. Neurol. 2010, 223, 19–27. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Court, F.A.; Hendriks, W.T.; MacGillavry, H.D.; Alvarez, J.; van Minnen, J. Schwann cell to axon transfer of ribosomes: Toward a novel understanding of the role of glia in the nervous system. J. Neurosci. 2008, 28, 11024–11029. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, P.K.; Smith, D.S.; Perrone-Bizzozero, N.; Twiss, J.L. Axonal mRNA transport and translation at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Terenzio, M.; Schiavo, G.; Fainzilber, M. Compartmentalized Signaling in Neurons: From Cell Biology to Neuroscience. Neuron 2017, 96, 667–679. [Google Scholar] [CrossRef]
- Terenzio, M.; Koley, S.; Samra, N.; Rishal, I.; Zhao, Q.; Sahoo, P.K.; Urisman, A.; Marvaldi, L.; Oses-Prieto, J.A.; Forester, C.; et al. Locally translated mTOR controls axonal local translation in nerve injury. Science 2018, 359, 1416–1421. [Google Scholar] [CrossRef]
- Smith, D.S.; Skene, J.H. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 1997, 17, 646–658. [Google Scholar] [CrossRef]
- Ma, K.H.; Svaren, J. Epigenomic reprogramming in peripheral nerve injury. Neural Regen. Res. 2016, 11, 1930–1931. [Google Scholar]
- Palmisano, I.; Di Giovanni, S. Advances and Limitations of Current Epigenetic Studies Investigating Mammalian Axonal Regeneration. Neurotherapeutics 2018, 15, 529–540. [Google Scholar] [CrossRef]
- Verdú, E.; Ceballos, D.; Vilches, J.J.; Navarro, X. Influence of aging on peripheral nerve function and regeneration. J. Peripher Nerv. Syst. 2000, 5, 191–208. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; Zhou, L.; Palmisano, I.; McLachlan, E.; Kong, G.; Hutson, T.H.; Danzi, M.C.; Lemmon, V.P.; Bixby, J.L.; Matamoros-Angles, A.; et al. PP4-dependent HDAC3 dephosphorylation discriminates between axonal regeneration and regenerative failure. EMBO J. 2019, 38, e101032. [Google Scholar] [CrossRef] [PubMed]
- Puttagunta, R.; Tedeschi, A.; Sória, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat. Commun. 2014, 5, 3527. [Google Scholar] [CrossRef]
- Shin, J.E.; Cho, Y. Epigenetic Regulation of Axon Regeneration after Neural Injury. Mol. Cells 2017, 40, 10–16. [Google Scholar] [CrossRef]
- Palmisano, I.; Danzi, M.C.; Hutson, T.H.; Zhou, L.; McLachlan, E.; Serger, E.; Shkura, K.; Srivastava, P.K.; Hervera, A.; Neill, N.O.; et al. Epigenomic signatures underpin the axonal regenerative ability of dorsal root ganglia sensory neurons. Nat. Neurosci. 2019, 22, 1913–1924. [Google Scholar] [CrossRef]
- Zhou, S.; Ding, F.; Gu, X. Non-coding RNAs as Emerging Regulators of Neural Injury Responses and Regeneration. Neurosci. Bull. 2016, 32, 253–264. [Google Scholar] [CrossRef]
- Zochodne, D.W. The challenges and beauty of peripheral nerve regrowth. J. Peripher Nerv. Syst. 2012, 17, 1–18. [Google Scholar] [CrossRef]
- Cattin, A.L.; Lloyd, A.C. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol. 2016, 39, 38–46. [Google Scholar] [CrossRef]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Stierli, S.; Imperatore, V.; Lloyd, A.C. Schwann cell plasticity-roles in tissue homeostasis, regeneration, and disease. Glia 2019, 67, 2203–2215. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Clements, M.P.; Byrne, E.; Camarillo Guerrero, L.F.; Cattin, A.L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114.e7. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Pilch, K.S.; van der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef]
- Fontana, X.; Hristova, M.; Da Costa, C.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.R.; et al. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef]
- Harrisingh, M.C.; Perez-Nadales, E.; Parkinson, D.B.; Malcolm, D.S.; Mudge, A.W.; Lloyd, A.C. The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J. 2004, 23, 3061–3071. [Google Scholar] [CrossRef]
- Napoli, I.; Noon, L.A.; Ribeiro, S.; Kerai, A.P.; Parrinello, S.; Rosenberg, L.H.; Collins, M.J.; Harrisingh, M.C.; White, I.J.; Woodhoo, A.; et al. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 2012, 73, 729–742. [Google Scholar] [CrossRef]
- Mogha, A.; Harty, B.L.; Carlin, D.; Joseph, J.; Sanchez, N.E.; Suter, U.; Piao, X.; Cavalli, V.; Monk, K.R. Gpr126/Adgrg6 Has Schwann Cell Autonomous and Nonautonomous Functions in Peripheral Nerve Injury and Repair. J. Neurosci. 2016, 36, 12351–12367. [Google Scholar] [CrossRef] [PubMed]
- Martini, R.; Fischer, S.; López-Vales, R.; David, S. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 2008, 56, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. NeuroInflamm. 2011, 8, 109. [Google Scholar] [CrossRef]
- Duman, M.; Martinez-Moreno, M.; Jacob, C.; Tapinos, N. Functions of histone modifications and histone modifiers in Schwann cells. Glia 2020, 68, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.; Moyon, S. DNA methylation in Schwann cells and in oligodendrocytes. Glia 2020, 68, 1568–1583. [Google Scholar] [CrossRef]
- Arthur-Farraj, P.J.; Morgan, C.C.; Adamowicz, M.; Gomez-Sanchez, J.A.; Fazal, S.V.; Beucher, A.; Razzaghi, B.; Mirsky, R.; Jessen, K.R.; Aitman, T.J. Changes in the Coding and Non-coding Transcriptome and DNA Methylome that Define the Schwann Cell Repair Phenotype after Nerve Injury. Cell Rep. 2017, 20, 2719–2734. [Google Scholar] [CrossRef]
- Cattin, A.L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; Hoving, J.J.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef]
- Dun, X.P.; Carr, L.; Woodley, P.K.; Barry, R.W.; Drake, L.K.; Mindos, T.; Roberts, S.L.; Lloyd, A.C.; Parkinson, D.B. Macrophage-Derived Slit3 Controls Cell Migration and Axon Pathfinding in the Peripheral Nerve Bridge. Cell Rep. 2019, 26, 1458–1472.e4. [Google Scholar] [CrossRef]
- Dun, X.P.; Parkinson, D.B. Classic axon guidance molecules control correct nerve bridge tissue formation and precise axon regeneration. Neural. Regen. Res. 2020, 15, 6–9. [Google Scholar] [CrossRef]
- Chen, B.; Carr, L.; Dun, X.P. Dynamic expression of Slit1-3 and Robo1-2 in the mouse peripheral nervous system after injury. Neural. Regen. Res. 2020, 15, 948–958. [Google Scholar] [CrossRef]
- Dominici, C.; Moreno-Bravo, J.A.; Puiggros, S.R.; Rappeneau, Q.; Rama, N.; Vieugue, P.; Bernet, A.; Mehlen, P.; Chédotal, A. Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 2017, 545, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Dun, X.P.; Parkinson, D.B. Role of Netrin-1 Signaling in Nerve Regeneration. Int. J. Mol. Sci. 2017, 18, 491. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S.; Napoli, I.; Ribeiro, S.; Wingfield Digby, P.; Fedorova, M.; Parkinson, D.B.; Doddrell, R.D.; Nakayama, M.; Adams, R.H.; Lloyd, A.C. EphB signaling directs peripheral nerve regeneration through Sox2-dependent Schwann cell sorting. Cell 2010, 143, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, B.; Gu, Y.; Zhou, S.; Qian, T.; Wang, Y.; Ding, G.; Ding, F.; Gu, X. Fibroblast-derived tenascin-C promotes Schwann cell migration through β1-integrin dependent pathway during peripheral nerve regeneration. Glia 2016, 64, 374–385. [Google Scholar] [CrossRef]
- Taveggia, C.; Zanazzi, G.; Petrylak, A.; Yano, H.; Rosenbluth, J.; Einheber, S.; Xu, X.; Esper, R.M.; Loeb, J.A.; Shrager, P.; et al. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 2005, 47, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Bogni, S.; Matafora, V.; Della-Flora Nunes, G.; Hurley, E.; Ghidinelli, M.; Katzenellenbogen, B.S.; Taveggia, C.; Silvestri, N.; Bachi, A.; et al. Spatial mapping of juxtacrine axo-glial interactions identifies novel molecules in peripheral myelination. Nat. Commun. 2015, 6, 8303. [Google Scholar] [CrossRef]
- Belin, S.; Zuloaga, K.L.; Poitelon, Y. Influence of Mechanical Stimuli on Schwann Cell Biology. Front. Cell Neurosci. 2017, 11, 347. [Google Scholar] [CrossRef]
- Poitelon, Y.; Lopez-Anido, C.; Catignas, K.; Berti, C.; Palmisano, M.; Williamson, C.; Ameroso, D.; Abiko, K.; Hwang, Y.; Gregorieff, A.; et al. YAP and TAZ control peripheral myelination and the expression of laminin receptors in Schwann cells. Nat. Neurosci. 2016, 19, 879–887. [Google Scholar] [CrossRef]
- Shimizu, T.; Osanai, Y.; Tanaka, K.F.; Abe, M.; Natsume, R.; Sakimura, K.; Ikenaka, K. YAP functions as a mechanotransducer in oligodendrocyte morphogenesis and maturation. Glia 2017, 65, 360–374. [Google Scholar] [CrossRef]
- Grove, M.; Kim, H.; Santerre, M.; Krupka, A.J.; Han, S.B.; Zhai, J.; Cho, J.Y.; Park, R.; Harris, M.; Kim, S.; et al. YAP/TAZ initiate and maintain Schwann cell myelination. Elife 2017, 6. [Google Scholar] [CrossRef]
- Deng, Y.; Wu, L.M.N.; Bai, S.; Zhao, C.; Wang, H.; Wang, J.; Xu, L.; Sakabe, M.; Zhou, W.; Xin, M.; et al. A reciprocal regulatory loop between TAZ/YAP and G-protein Gαs regulates Schwann cell proliferation and myelination. Nat. Commun. 2017, 8, 15161. [Google Scholar] [CrossRef] [PubMed]
- Grove, M.; Lee, H.; Zhao, H.; Son, Y.J. Axon-dependent expression of YAP/TAZ mediates Schwann cell remyelination but not proliferation after nerve injury. Elife 2020, 9. [Google Scholar]
- Song, Y.; Li, D.; Farrelly, O.; Miles, L.; Li, F.; Kim, S.E.; Lo, T.Y.; Wang, F.; Li, T.; Thompson-Peer, K.L.; et al. The Mechanosensitive Ion Channel Piezo Inhibits Axon Regeneration. Neuron 2019, 102, 373–389.e6. [Google Scholar] [CrossRef] [PubMed]
- Mokalled, M.H.; Poss, K.D. A Regeneration Toolkit. Dev. Cell 2018, 47, 267–280. [Google Scholar] [CrossRef]
- Rasmussen, J.P.; Sagasti, A. Learning to swim, again: Axon regeneration in fish. Exp. Neurol. 2017, 287 Pt 3, 318–330. [Google Scholar] [CrossRef]
- Rigoni, M.; Montecucco, C. Animal models for studying motor axon terminal paralysis and recovery. J. Neurochem. 2017. [Google Scholar] [CrossRef]
- Li, L.; Xiong, W.C.; Mei, L. Neuromuscular Junction Formation, Aging, and Disorders. Annu. Rev. Physiol. 2018, 80, 159–188. [Google Scholar] [CrossRef]
- Plomp, J.J.; Willison, H.J. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction. J. Physiol. 2009, 587 (Pt 16), 3979–3999. [Google Scholar] [CrossRef]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef]
- Goulet, B.B.; Kothary, R.; Parks, R.J. At the “junction” of spinal muscular atrophy pathogenesis: The role of neuromuscular junction dysfunction in SMA disease progression. Curr. Mol. Med. 2013, 13, 1160–1174. [Google Scholar] [CrossRef]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef] [PubMed]
- Negro, S.; Lessi, F.; Duregotti, E.; Aretini, P.; La Ferla, M.; Franceschi, S.; Menicagli, M.; Bergamin, E.; Radice, E.; Thelen, M.; et al. CXCL12α/SDF-1 from perisynaptic Schwann cells promotes regeneration of injured motor axon terminals. Embo Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hurlbut, W.P.; Ceccarelli, B. Use of black widow spider venom to study the release of neurotransmitters. Adv. Cytopharmacol. 1979, 3, 87–115. [Google Scholar]
- Ceccarelli, B.; Hurlbut, W.P. Vesicle hypothesis of the release of quanta of acetylcholine. Physiol. Rev. 1980, 60, 396–441. [Google Scholar] [CrossRef]
- Duchen, L.W.; Gomez, S.; Queiroz, L.S. The neuromuscular junction of the mouse after black widow spider venom. J. Physiol. 1981, 316, 279–291. [Google Scholar] [CrossRef]
- Rosenthal, L.; Zacchetti, D.; Madeddu, L.; Meldolesi, J. Mode of action of alpha-latrotoxin: Role of divalent cations in Ca2(+)-dependent and Ca2(+)-independent effects mediated by the toxin. Mol. Pharm. 1990, 38, 917–923. [Google Scholar]
- Südhof, T.C. alpha-Latrotoxin and its receptors: Neurexins and CIRL/latrophilins. Annu. Rev. Neurosci. 2001, 24, 933–962. [Google Scholar] [CrossRef]
- Ushkaryov, Y.A.; Rohou, A.; Sugita, S. alpha-Latrotoxin and its receptors. Handb. Exp. Pharm. 2008, 184, 171–206. [Google Scholar]
- Tedesco, E.; Rigoni, M.; Caccin, P.; Grishin, E.; Rossetto, O.; Montecucco, C. Calcium overload in nerve terminals of cultured neurons intoxicated by alpha-latrotoxin and snake PLA2 neurotoxins. Toxicon 2009, 54, 138–144. [Google Scholar] [CrossRef]
- Negro, S.; Bergamin, E.; Rodella, U.; Duregotti, E.; Scorzeto, M.; Jalink, K.; Montecucco, C.; Rigoni, M. ATP Released by Injured Neurons Activates Schwann Cells. Front. Cell Neurosci. 2016, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Rieger, S.; Sagasti, A. Hydrogen peroxide promotes injury-induced peripheral sensory axon regeneration in the zebrafish skin. PLoS ONE Biol. 2011, 9, e1000621. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Gauron, C.; Meda, F.; Dupont, E.; Albadri, S.; Quenech’Du, N.; Ipendey, E.; Volovitch, M.; Del Bene, F.; Joliot, A.; Rampon, C.; et al. Hydrogen peroxide (H2O2) controls axon pathfinding during zebrafish development. Dev. Biol. 2016, 414, 133–141. [Google Scholar] [CrossRef]
- Meda, F.; Gauron, C.; Rampon, C.; Teillon, J.; Volovitch, M.; Vriz, S. Nerves Control Redox Levels in Mature Tissues Through Schwann Cells and Hedgehog Signaling. Antioxid. Redox Signal. 2016, 24, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.M.G.; McCathie, G.; Jankun, P.; Roehl, H.H. Damage-induced reactive oxygen species enable zebrafish tail regeneration by repositioning of Hedgehog expressing cells. Nat. Commun. 2018, 9, 4010. [Google Scholar] [CrossRef] [PubMed]
- Love, N.R.; Chen, Y.; Ishibashi, S.; Kritsiligkou, P.; Lea, R.; Koh, Y.; Gallop, J.L.; Dorey, K.; Amaya, E. Amputation-induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nat. Cell Biol. 2013, 15, 222–228. [Google Scholar] [CrossRef]
- Klyubin, I.V.; Kirpichnikova, K.M.; Gamaley, I.A. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur. J. Cell Biol. 1996, 70, 347–351. [Google Scholar] [PubMed]
- Li, L.; Yan, B.; Shi, Y.Q.; Zhang, W.Q.; Wen, Z.L. Live imaging reveals differing roles of macrophages and neutrophils during zebrafish tail fin regeneration. J. Biol. Chem. 2012, 287, 25353–25360. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.D.; Armstrong, J.J.; Kupchak, B.R. Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochem. Biophys. Res. Commun. 2004, 315, 568–574. [Google Scholar] [CrossRef]
- Sato, K.; Ogawa, K.; Tokmakov, A.A.; Iwasaki, T.; Fukami, Y. Hydrogen peroxide induces Src family tyrosine kinase-dependent activation of Xenopus eggs. Dev. Growth Differ. 2001, 43, 55–72. [Google Scholar] [CrossRef]
- Yoo, S.K.; Freisinger, C.M.; LeBert, D.C.; Huttenlocher, A. Early redox, Src family kinase, and calcium signaling integrate wound responses and tissue regeneration in zebrafish. J. Cell Biol. 2012, 199, 225–234. [Google Scholar] [CrossRef]
- Wilson, C.; González-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef]
- Xu, S.; Chisholm, A.D. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 2014, 31, 48–60. [Google Scholar] [CrossRef]
- Negro, S.; Stazi, M.; Marchioretto, M.; Tebaldi, T.; Rodella, U.; Duregotti, E.; Gerke, V.; Quattrone, A.; Montecucco, C.; Rigoni, M.; et al. Hydrogen peroxide is a neuronal alarmin that triggers specific RNAs, local translation of Annexin A2, and cytoskeletal remodeling in Schwann cells. Rna 2018, 24, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Hervera, A.; Santos, C.X.; De Virgiliis, F.; Shah, A.M.; Di Giovanni, S. Paracrine Mechanisms of Redox Signalling for Postmitotic Cell and Tissue Regeneration. Trends Cell Biol. 2019, 29, 514–530. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Srikun, D.; Chang, C.J. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr. Opin Chem. Biol. 2010, 14, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Rodella, U.; Scorzeto, M.; Duregotti, E.; Negro, S.; Dickinson, B.C.; Chang, C.J.; Yuki, N.; Rigoni, M.; Montecucco, C. An animal model of Miller Fisher syndrome: Mitochondrial hydrogen peroxide is produced by the autoimmune attack of nerve terminals and activates Schwann cells. Neurobiol. Dis. 2016, 96, 95–104. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Subcell Biochem. 2007, 45, 481–506. [Google Scholar]
- Hervera, A.; De Virgiliis, F.; Palmisano, I.; Zhou, L.; Tantardini, E.; Kong, G.; Hutson, T.; Danzi, M.C.; Perry, R.B.; Santos, C.X.C.; et al. Publisher Correction: Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat. Cell Biol. 2018, 20, 1098. [Google Scholar] [CrossRef]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef]
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204. [Google Scholar] [CrossRef]
- Tashiro, K.; Tada, H.; Heilker, R.; Shirozu, M.; Nakano, T.; Honjo, T. Signal sequence trap: A cloning strategy for secreted proteins and type I membrane proteins. Science 1993, 261, 600–603. [Google Scholar] [CrossRef]
- Nagasawa, T.; Kikutani, H.; Kishimoto, T. Molecular cloning and structure of a pre-B-cell growth-stimulating factor. Proc. Natl. Acad. Sci. USA 1994, 91, 2305–2309. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, S.H.; Sabelko, K.A.; Sunshine, M.J.; Littman, D.R.; Raper, J.A. A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon pathfinding. J. Neurosci. 2003, 23, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Stumm, R.K.; Zhou, C.; Ara, T.; Lazarini, F.; Dubois-Dalcq, M.; Nagasawa, T.; Höllt, V.; Schulz, S. CXCR4 regulates interneuron migration in the developing neocortex. J. Neurosci. 2003, 23, 5123–5130. [Google Scholar] [CrossRef]
- Lu, M.; Grove, E.A.; Miller, R.J. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 7090–7095. [Google Scholar] [CrossRef]
- Zhu, Y.; Matsumoto, T.; Mikami, S.; Nagasawa, T.; Murakami, F. SDF1/CXCR4 signalling regulates two distinct processes of precerebellar neuronal migration and its depletion leads to abnormal pontine nuclei formation. Development 2009, 136, 1919–1928. [Google Scholar] [CrossRef]
- Lieberam, I.; Agalliu, D.; Nagasawa, T.; Ericson, J.; Jessell, T.M. A Cxcl12-CXCR4 chemokine signaling pathway defines the initial trajectory of mammalian motor axons. Neuron 2005, 47, 667–679. [Google Scholar] [CrossRef]
- Wang, J.; Knaut, H. Chemokine signaling in development and disease. Development 2014, 141, 4199–4205. [Google Scholar] [CrossRef]
- Nagasawa, T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. (Berl) 2014, 92, 433–439. [Google Scholar] [CrossRef]
- Guyon, A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front. Cell Neurosci. 2014, 8, 65. [Google Scholar] [CrossRef]
- Bagri, A.; Gurney, T.; He, X.; Zou, Y.R.; Littman, D.R.; Tessier-Lavigne, M.; Pleasure, S.J. The chemokine SDF1 regulates migration of dentate granule cells. Development 2002, 129, 4249–4260. [Google Scholar] [PubMed]
- Negro, S.; Zanetti, G.; Mattarei, A.; Valentini, A.; Megighian, A.; Tombesi, G.; Zugno, A.; Dianin, V.; Pirazzini, M.; Fillo, S.; et al. Agonist of the CXCR4 Receptor Strongly Promotes Regeneration of Degenerated Motor Axon Terminals. Cells 2019, 8, 1183. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Negro, S.; Megighian, A.; Mattarei, A.; Lista, F.; Fillo, S.; Rigoni, M.; Pirazzini, M.; Montecucco, C. A CXCR4 receptor agonist strongly stimulates axonal regeneration after damage. Ann. Clin. Transl. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.I.; Mar, F.M.; Sousa, M.M. The intriguing nature of dorsal root ganglion neurons: Linking structure with polarity and function. Prog. Neurobiol. 2018, 168, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Toth, C.C.; Willis, D.; Twiss, J.L.; Walsh, S.; Martinez, J.A.; Liu, W.Q.; Midha, R.; Zochodne, D.W. Locally synthesized calcitonin gene-related Peptide has a critical role in peripheral nerve regeneration. J. Neuropathol. Exp. Neurol. 2009, 68, 326–337. [Google Scholar] [CrossRef]
- Höke, A.; Redett, R.; Hameed, H.; Jari, R.; Zhou, C.; Li, Z.B.; Griffin, J.W.; Brushart, T.M. Schwann cells express motor and sensory phenotypes that regulate axon regeneration. J. Neurosci. 2006, 26, 9646–9655. [Google Scholar] [CrossRef]
- Avraham, O.; Deng, P.Y.; Jones, S.; Kuruvilla, R.; Semenkovich, C.F.; Klyachko, V.A.; Cavalli, V. Satellite glial cells promote regenerative growth in sensory neurons. bioRxiv 2019. [Google Scholar] [CrossRef]
- Barton, M.J.; John, J.S.; Clarke, M.; Wright, A.; Ekberg, J. The Glia Response after Peripheral Nerve Injury: A Comparison between Schwann Cells and Olfactory Ensheathing Cells and Their Uses for Neural Regenerative Therapies. Int. J. Mol. Sci. 2017, 18, 287. [Google Scholar] [CrossRef]
- Abdo, H.; Calvo-Enrique, L.; Lopez, J.M.; Song, J.; Zhang, M.; Usoskin, D.; El Manira, A.; Adameyko, I.; Hjerling-Leffler, J.; Ernfors, P. Specialized cutaneous Schwann cells initiate pain sensation. Science 2019, 365, 695–699. [Google Scholar] [CrossRef]
- Guo, X.; Colon, A.; kanda, N.A.; Spradling, S.; Stancescu, M.; Martin, C.; Hickman, J.J. Tissue engineering the mechanosensory circuit of the stretch reflex arc with human stem cells: Sensory neuron innervation of intrafusal muscle fibers. Biomaterials 2017, 122, 179–187. [Google Scholar] [CrossRef]
- Copray, J.C.; Brouwer, N. Neurotrophin-3 mRNA expression in rat intrafusal muscle fibres after denervation and reinnervation. Neurosci. Lett. 1997, 236, 41–44. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigoni, M.; Negro, S. Signals Orchestrating Peripheral Nerve Repair. Cells 2020, 9, 1768. https://doi.org/10.3390/cells9081768
Rigoni M, Negro S. Signals Orchestrating Peripheral Nerve Repair. Cells. 2020; 9(8):1768. https://doi.org/10.3390/cells9081768
Chicago/Turabian StyleRigoni, Michela, and Samuele Negro. 2020. "Signals Orchestrating Peripheral Nerve Repair" Cells 9, no. 8: 1768. https://doi.org/10.3390/cells9081768
APA StyleRigoni, M., & Negro, S. (2020). Signals Orchestrating Peripheral Nerve Repair. Cells, 9(8), 1768. https://doi.org/10.3390/cells9081768