Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
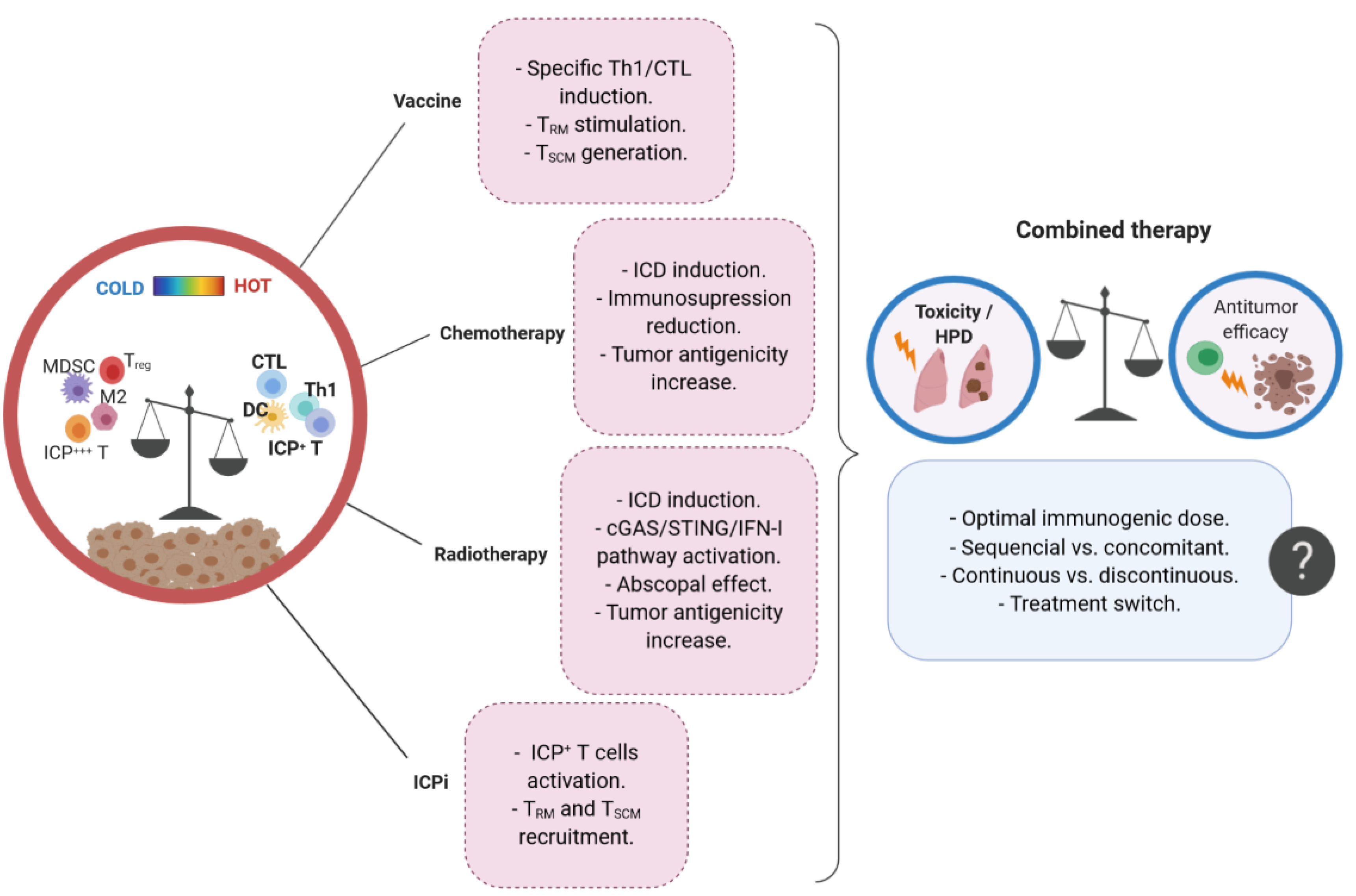
1. Introduction
2. Immunological Parameters Accounting for Improved Efficacy of Combined Immunotherapies
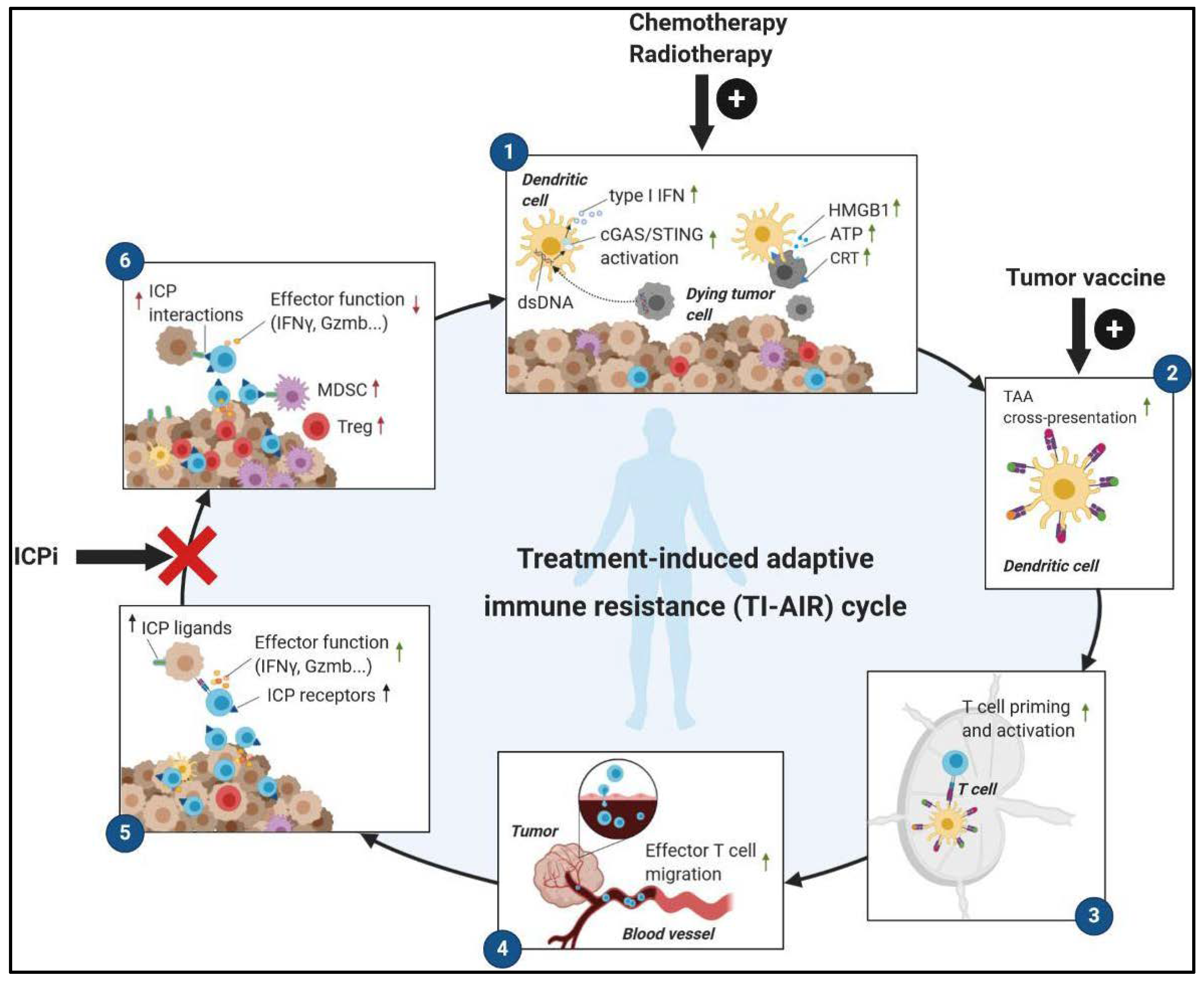
2.1. Immunogenic Anticancer Therapies Convert ‘Cold’ Tumors into ‘Hot’ Tumors
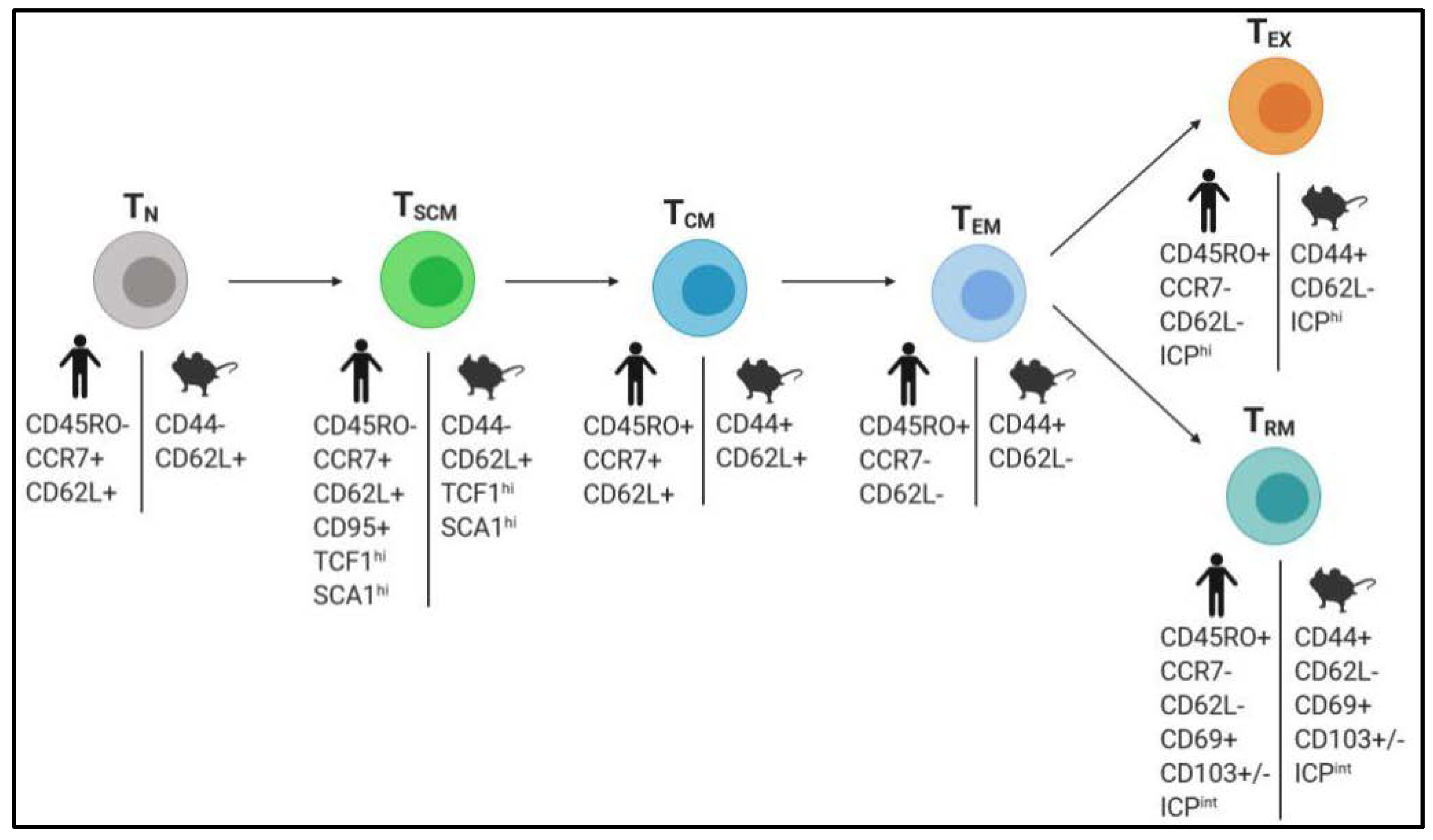
2.2. Boosting the Generation of Memory T Cells with Superior Antitumor Properties
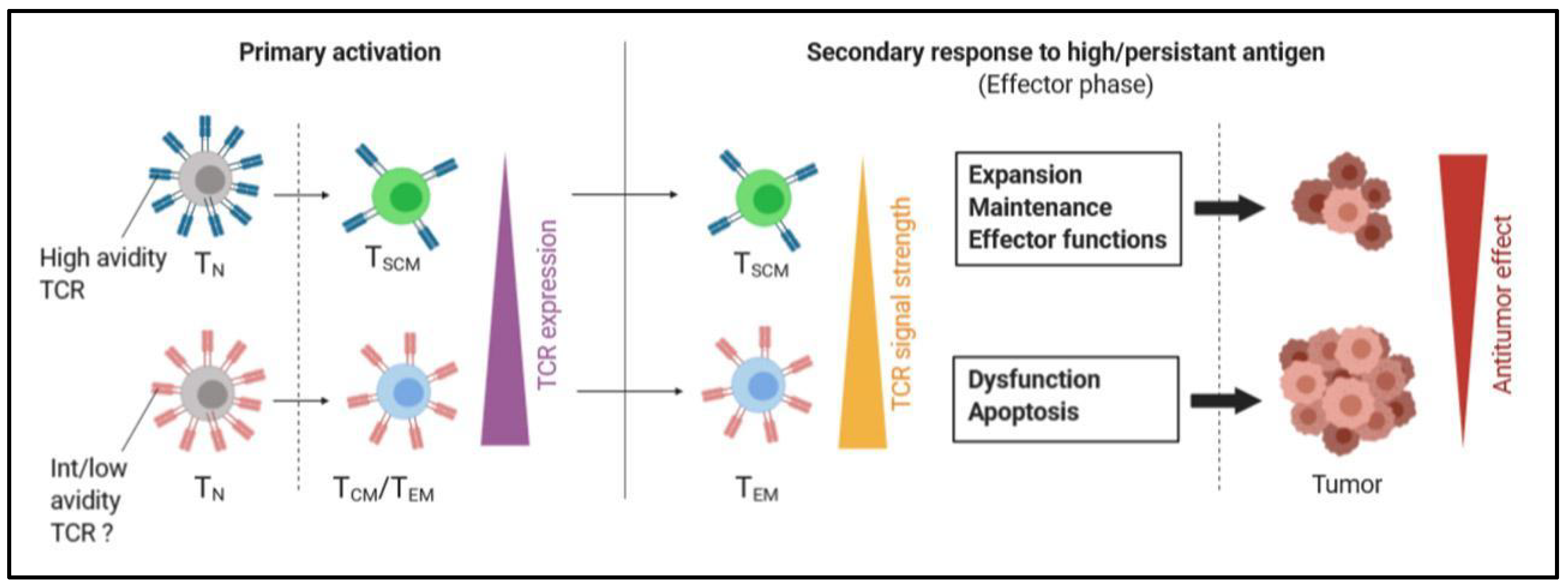
2.2.1. Stimulation of TSCM Cells
2.2.2. Harnessing TRM Cells in Cancer
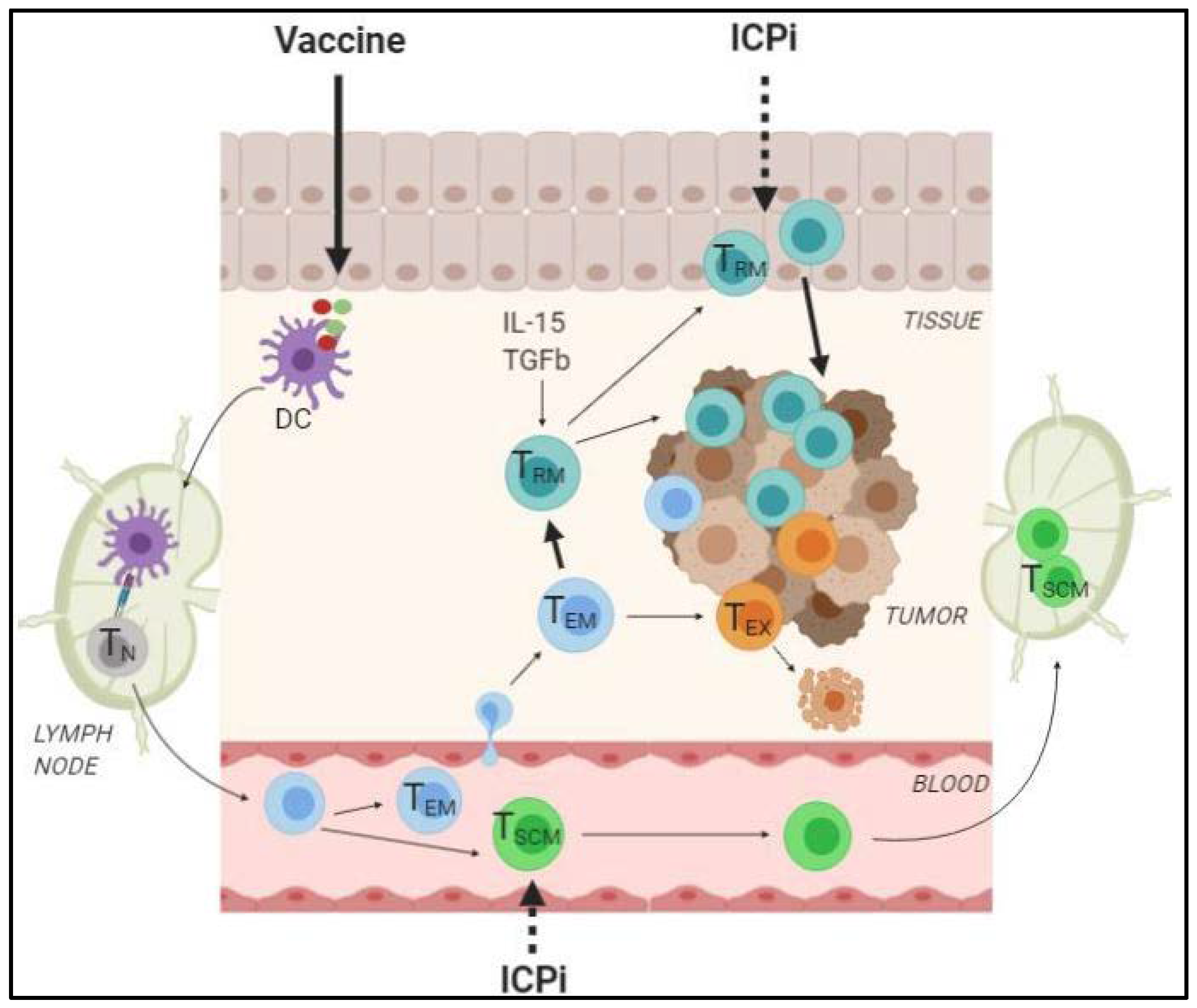
2.2.3. How can the Generation of TRM and TSCM Foster the Efficacy of ICPi?
3. Immunogenic Therapy and Immune Checkpoint Inhibitors: A Matter of Dose and Timing
3.1. ICPi Therapy: A Preventive rather than Curative Care for T Cell Dysfunction?
3.2. Concurrent Versus Sequential Combinations: A Balance between Antitumor Efficiency and Immune-Related Side Effects
3.3. Influence of the Dose on the Immunostimulatory Effect of Therapies
3.4. Defining Optimal Duration for ICPi Therapy: When to Stop and Replace?
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. CD4+ T lymphocytes: A critical component of antitumor immunity. Cancer Investig. 2005, 23, 413–419. [Google Scholar]
- Kennedy, R.; Celis, E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015, 6, 6072. [Google Scholar] [CrossRef]
- Schwartz, H.S.; Grindey, G.B. Adriamycin and daunorubicin: A comparison of antitumor activities and tissue uptake in mice following immunosuppression. Cancer Res. 1973, 33, 1837–1844. [Google Scholar] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29, 482–491. [Google Scholar] [CrossRef]
- Hato, S.V.; Khong, A.; de Vries, I.J.M.; Lesterhuis, W.J. Molecular pathways: The immunogenic effects of platinum-based chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Immunogenic cell death in radiation therapy. Oncoimmunology 2013, 2, e26536. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Merino, L.; Illescas-Vacas, A.; Grueso-López, A.; Barco-Sánchez, A.; Míguez-Sánchez, C. Cancer Immunotherapies Spanish Group (GETICA) Radiation for Awakening the Dormant Immune System, a Promising Challenge to be Explored. Front. Immunol. 2014, 5, 102. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362. [Google Scholar] [CrossRef]
- Rivera Vargas, T.; Apetoh, L. Can Immunogenic Chemotherapies Relieve Cancer Cell Resistance to Immune Checkpoint Inhibitors? Front. Immunol. 2019, 10, 1181. [Google Scholar] [CrossRef]
- Apetoh, L.; Smyth, M.J.; Drake, C.G.; Abastado, J.-P.; Apte, R.N.; Ayyoub, M.; Blay, J.-Y.; Bonneville, M.; Butterfield, L.H.; Caignard, A.; et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015, 4, e998538. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Lauret Marie-Joseph, E.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 2018, 7, e1433981. [Google Scholar] [CrossRef] [PubMed]
- Limagne, E.; Thibaudin, M.; Nuttin, L.; Spill, A.; Derangère, V.; Fumet, J.-D.; Amellal, N.; Peranzoni, E.; Cattan, V.; Ghiringhelli, F. Trifluridine/Tipiracil plus Oxaliplatin Improves PD-1 Blockade in Colorectal Cancer by Inducing Immunogenic Cell Death and Depleting Macrophages. Cancer Immunol. Res. 2019, 7, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Dudzinski, S.O.; Cameron, B.D.; Wang, J.; Rathmell, J.C.; Giorgio, T.D.; Kirschner, A.N. Combination immunotherapy and radiotherapy causes an abscopal treatment response in a mouse model of castration resistant prostate cancer. J. Immunother. Cancer 2019, 7, 218. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef]
- Li, B.; VanRoey, M.; Wang, C.; Chen, T.T.; Korman, A.; Jooss, K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor--secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin. Cancer Res. 2009, 15, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Kleponis, J.; Skelton, R.; Zheng, L. Fueling the engine and releasing the break: Combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol. Med. 2015, 12, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Bartkowiak, T.; Singh, S.; Yang, G.; Galvan, G.; Haria, D.; Ai, M.; Allison, J.P.; Sastry, K.J.; Curran, M.A. Unique potential of 4-1BB agonist antibody to promote durable regression of HPV+ tumors when combined with an E6/E7 peptide vaccine. Proc. Natl. Acad. Sci. USA 2015, 112, E5290–E5299. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Ramamoorthi, G.; Snyder, C.; Basu, A.; Jia, Y.; Awshah, S.; Beyer, A.P.; Wiener, D.; Lam, L.; Zhang, H.; et al. Sequential Anti-PD1 Therapy Following Dendritic Cell Vaccination Improves Survival in a HER2 Mammary Carcinoma Model and Identifies a Critical Role for CD4 T Cells in Mediating the Response. Front. Immunol. 2019, 10, 1939. [Google Scholar] [CrossRef]
- Shahda, S.; Noonan, A.M.; Bekaii-Saab, T.S.; O’Neil, B.H.; Sehdev, A.; Shaib, W.L.; Helft, P.R.; Loehrer, P.J.; Tong, Y.; Liu, Z.; et al. A phase II study of pembrolizumab in combination with mFOLFOX6 for patients with advanced colorectal cancer. J. Clin. Oncol. 2017, 35, 3541. [Google Scholar] [CrossRef]
- Bendell, J.C.; Powderly, J.D.; Lieu, C.H.; Eckhardt, S.G.; Hurwitz, H.; Hochster, H.S.; Murphy, J.E.; Funke, R.P.; Rossi, C.; Wallin, J.; et al. Safety and efficacy of MPDL3280A (anti-PDL1) in combination with bevacizumab (bev) and/or FOLFOX in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 704. [Google Scholar] [CrossRef]
- Fumet, J.-D.; Isambert, N.; Hervieu, A.; Zanetta, S.; Guion, J.-F.; Hennequin, A.; Rederstorff, E.; Bertaut, A.; Ghiringhelli, F. Phase Ib/II trial evaluating the safety, tolerability and immunological activity of durvalumab (MEDI4736) (anti-PD-L1) plus tremelimumab (anti-CTLA-4) combined with FOLFOX in patients with metastatic colorectal cancer. Esmo Open 2018, 3, e000375. [Google Scholar] [CrossRef]
- Hadash-Bengad, R.; Hajaj, E.; Klein, S.; Merims, S.; Frank, S.; Eisenberg, G.; Yakobson, A.; Orevi, M.; Caplan, N.; Peretz, T.; et al. Immunotherapy Potentiates the Effect of Chemotherapy in Metastatic Melanoma-A Retrospective Study. Front. Oncol. 2020, 10, 70. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Addeo, A.; Banna, G.L.; Metro, G.; Di Maio, M. Chemotherapy in Combination With Immune Checkpoint Inhibitors for the First-Line Treatment of Patients With Advanced Non-small Cell Lung Cancer: A Systematic Review and Literature-Based Meta-Analysis. Front. Oncol. 2019, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Godet, Y.; Fabre, E.; Dosset, M.; Lamuraglia, M.; Levionnois, E.; Ravel, P.; Benhamouda, N.; Cazes, A.; Le Pimpec-Barthes, F.; Gaugler, B.; et al. Analysis of spontaneous tumor-specific CD4 T-cell immunity in lung cancer using promiscuous HLA-DR telomerase-derived epitopes: Potential synergistic effect with chemotherapy response. Clin. Cancer Res. 2012, 18, 2943–2953. [Google Scholar] [CrossRef]
- Godet, Y.; Dosset, M.; Borg, C.; Adotevi, O. Is preexisting antitumor CD4 T cell response indispensable for the chemotherapy induced immune regression of cancer? Oncoimmunology 2012, 1, 1617–1619. [Google Scholar] [CrossRef][Green Version]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Feng, L.; Lee, J.J.; Tran, H.; Kim, Y.U.; et al. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients With Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef]
- Hong, H.; Gu, Y.; Sheng, S.Y.; Lu, C.G.; Zou, J.Y.; Wu, C.Y. The Distribution of Human Stem Cell-like Memory T Cell in Lung Cancer. J. Immunother. 2016, 39, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Chang, J.; Song, X.; Yan, F.; Yu, W.; An, Y.; Wei, F.; Yang, L.; Ren, X. Memory stem T cells generated by Wnt signaling from blood of human renal clear cell carcinoma patients. Cancer Biol. Med. 2019, 16, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.Y.; Gu, Y.; Lu, C.G.; Tang, Y.Y.; Zou, J.Y.; Zhang, Y.Q.; Wang, R.F.; Hong, H. The Characteristics of Naive-like T Cells in Tumor-infiltrating Lymphocytes from Human Lung Cancer. J. Immunother. 2017, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vahidi, Y.; Faghih, Z.; Talei, A.-R.; Doroudchi, M.; Ghaderi, A. Memory CD4+ T cell subsets in tumor draining lymph nodes of breast cancer patients: A focus on T stem cell memory cells. Cell Oncol. 2018, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211.e10. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Ji, Y.; Restifo, N.P. Wnt/beta-catenin signaling in T-cell immunity and cancer immunotherapy. Clin. Cancer Res. 2010, 16, 4695–4701. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Flynn, J.K.; Gorry, P.R. Stem memory T cells (TSCM)-their role in cancer and HIV immunotherapies. Clin. Transl. Immunol. 2014, 3, e20. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Magun, A.M.; Reiner, S.L. TCF1 expression marks self-renewing human CD8+ T cells. Blood Adv. 2018, 2, 1685–1690. [Google Scholar] [CrossRef]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T cell, exhaustion, and tumor immunity. Immunol. Med. 2020, 43, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jaafoura, S.; de Goër de Herve, M.G.; Hernandez-Vargas, E.A.; Hendel-Chavez, H.; Abdoh, M.; Mateo, M.C.; Krzysiek, R.; Merad, M.; Seng, R.; Tardieu, M.; et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4+ memory T Cells. Nat. Commun. 2014, 5, 5407. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Lugli, E.; Dominguez, M.H.; Gattinoni, L.; Chattopadhyay, P.K.; Bolton, D.L.; Song, K.; Klatt, N.R.; Brenchley, J.M.; Vaccari, M.; Gostick, E.; et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Investig. 2013, 123, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhu, W.; Peng, Y.; Wang, L.; Hong, Y.; Huang, L.; Dong, D.; Xie, J.; Merchen, T.; Kruse, E.; et al. The Antitumor Effects of Vaccine-Activated CD8+ T Cells Associate with Weak TCR Signaling and Induction of Stem-Like Memory T Cells. Cancer Immunol. Res. 2017, 5, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Biasco, L.; Scala, S.; Basso Ricci, L.; Dionisio, F.; Baricordi, C.; Calabria, A.; Giannelli, S.; Cieri, N.; Barzaghi, F.; Pajno, R.; et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci. Transl. Med. 2015, 7, 273ra13. [Google Scholar] [CrossRef] [PubMed]
- Kurtulus, S.; Madi, A.; Escobar, G.; Klapholz, M.; Nyman, J.; Christian, E.; Pawlak, M.; Dionne, D.; Xia, J.; Rozenblatt-Rosen, O.; et al. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1-CD8+ Tumor-Infiltrating T Cells. Immunity 2019, 50, 181–194.e6. [Google Scholar] [CrossRef]
- Gannon, P.O.; Baumgaertner, P.; Huber, A.; Iancu, E.M.; Cagnon, L.; Abed Maillard, S.; Maby-El Hajjami, H.; Speiser, D.E.; Rufer, N. Rapid and Continued T-Cell Differentiation into Long-term Effector and Memory Stem Cells in Vaccinated Melanoma Patients. Clin. Cancer Res. 2017, 23, 3285–3296. [Google Scholar] [CrossRef]
- Blaeschke, F.; Stenger, D.; Kaeuferle, T.; Willier, S.; Lotfi, R.; Kaiser, A.D.; Assenmacher, M.; Döring, M.; Feucht, J.; Feuchtinger, T. Induction of a central memory and stem cell memory phenotype in functionally active CD4+ and CD8+ CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19+ acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2018, 67, 1053–1066. [Google Scholar] [CrossRef]
- Gargett, T.; Brown, M.P. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 2015, 17, 487–495. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.-F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; van Gisbergen, K.P.J.M.; Hombrink, P.; van Lier, R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018, 19, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, C.S.; Bar, N.; Kadaveru, K.; Krauthammer, M.; Pornputtapong, N.; Mai, Z.; Ariyan, S.; Narayan, D.; Kluger, H.; Deng, Y.; et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 2016, 1, e88955. [Google Scholar] [CrossRef] [PubMed]
- Shwetank, N.; Abdelsamed, H.A.; Frost, E.L.; Schmitz, H.M.; Mockus, T.E.; Youngblood, B.A.; Lukacher, A.E. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell Biol. 2017, 95, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef]
- Szabo, P.A.; Miron, M.; Farber, D.L. Location, location, location: Tissue resident memory T cells in mice and humans. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Carpenter, D.J.; Chait, M.; Dogra, P.; Gartrell-Corrado, R.D.; Chen, A.X.; Campbell, S.; Liu, W.; Saraf, P.; Snyder, M.E.; et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 2019, 29, 3916–3932.e5. [Google Scholar] [CrossRef]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma-immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef]
- Purwar, R.; Campbell, J.; Murphy, G.; Richards, W.G.; Clark, R.A.; Kupper, T.S. Resident memory T cells (T(RM)) are abundant in human lung: Diversity, function, and antigen specificity. PLoS ONE 2011, 6, e16245. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.C.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Gehad, A.; Yang, C.; Scott, L.L.; Teague, J.E.; Schlapbach, C.; Elco, C.P.; Huang, V.; Matos, T.R.; Kupper, T.S.; et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015, 7, 279ra39. [Google Scholar] [CrossRef] [PubMed]
- Thome, J.J.C.; Farber, D.L. Emerging concepts in tissue-resident T cells: Lessons from humans. Trends Immunol. 2015, 36, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Díaz, X.; Saavedra-Almarza, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef]
- Dumauthioz, N.; Labiano, S.; Romero, P. Tumor Resident Memory T Cells: New Players in Immune Surveillance and Therapy. Front. Immunol. 2018, 9, 2076. [Google Scholar] [CrossRef]
- Wong, M.T.; Ong, D.E.H.; Lim, F.S.H.; Teng, K.W.W.; McGovern, N.; Narayanan, S.; Ho, W.Q.; Cerny, D.; Tan, H.K.K.; Anicete, R.; et al. A High-Dimensional Atlas of Human T Cell Diversity Reveals Tissue-Specific Trafficking and Cytokine Signatures. Immunity 2016, 45, 442–456. [Google Scholar] [CrossRef]
- Beura, L.K.; Fares-Frederickson, N.J.; Steinert, E.M.; Scott, M.C.; Thompson, E.A.; Fraser, K.A.; Schenkel, J.M.; Vezys, V.; Masopust, D. CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med. 2019, 216, 1214–1229. [Google Scholar] [CrossRef]
- Brizić, I.; Hiršl, L.; Šustić, M.; Golemac, M.; Britt, W.J.; Krmpotić, A.; Jonjić, S. CD4 T cells are required for maintenance of CD8 TRM cells and virus control in the brain of MCMV-infected newborn mice. Med. Microbiol. Immunol. 2019, 208, 487–494. [Google Scholar] [CrossRef]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.-L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Smazynski, J.; Webb, J.R. Resident Memory-Like Tumor-Infiltrating Lymphocytes (TILRM): Latest Players in the Immuno-Oncology Repertoire. Front. Immunol 2018, 9, 1741. [Google Scholar] [CrossRef] [PubMed]
- Van Braeckel-Budimir, N.; Varga, S.M.; Badovinac, V.P.; Harty, J.T. Repeated Antigen Exposure Extends the Durability of Influenza-Specific Lung-Resident Memory CD8+ T Cells and Heterosubtypic Immunity. Cell Rep. 2018, 24, 3374–3382.e3. [Google Scholar] [CrossRef]
- Wilk, M.M.; Mills, K.H.G. CD4 TRM Cells Following Infection and Immunization: Implications for More Effective Vaccine Design. Front. Immunol. 2018, 9, 1860. [Google Scholar] [CrossRef]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.-F.; Freyburger, L.; Clement, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Sci. Transl. Med. 2013, 5, 172ra20. [Google Scholar] [CrossRef]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Gálvez-Cancino, F.; López, E.; Menares, E.; Díaz, X.; Flores, C.; Cáceres, P.; Hidalgo, S.; Chovar, O.; Alcántara-Hernández, M.; Borgna, V.; et al. Vaccination-induced skin-resident memory CD8+ T cells mediate strong protection against cutaneous melanoma. Oncoimmunology 2018, 7, e1442163. [Google Scholar] [CrossRef]
- Sun, Y.-Y.; Peng, S.; Han, L.; Qiu, J.; Song, L.; Tsai, Y.; Yang, B.; Roden, R.B.S.; Trimble, C.L.; Hung, C.-F.; et al. Local HPV Recombinant Vaccinia Boost Following Priming with an HPV DNA Vaccine Enhances Local HPV-Specific CD8+ T-cell-Mediated Tumor Control in the Genital Tract. Clin. Cancer Res. 2016, 22, 657–669. [Google Scholar] [CrossRef]
- Iborra, S.; Martínez-López, M.; Khouili, S.C.; Enamorado, M.; Cueto, F.J.; Conde-Garrosa, R.; Del Fresno, C.; Sancho, D. Optimal Generation of Tissue-Resident but Not Circulating Memory T Cells during Viral Infection Requires Crosspriming by DNGR-1+ Dendritic Cells. Immunity 2016, 45, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Kumamoto, Y.; Gopinath, S.; Iwasaki, A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat. Commun. 2016, 7, 13346. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.I.; Becker, C.; Wang, Y.; Marches, F.; Helft, J.; Leboeuf, M.; Anguiano, E.; Pourpe, S.; Goller, K.; Pascual, V.; et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-β. Immunity 2013, 38, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15-Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef]
- Arina, A.; Beckett, M.; Fernandez, C.; Zheng, W.; Pitroda, S.; Chmura, S.J.; Luke, J.J.; Forde, M.; Hou, Y.; Burnette, B.; et al. Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 2019, 10, 3959. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef]
- Laheurte, C.; Dosset, M.; Vernerey, D.; Boullerot, L.; Gaugler, B.; Gravelin, E.; Kaulek, V.; Jacquin, M.; Cuche, L.; Eberst, G.; et al. Distinct prognostic value of circulating anti-telomerase CD4+ Th1 immunity and exhausted PD-1+/TIM-3+ T cells in lung cancer. Br. J. Cancer 2019, 121, 405–416. [Google Scholar] [CrossRef]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255. [Google Scholar] [CrossRef]
- Singer, M.; Wang, C.; Cong, L.; Marjanovic, N.D.; Kowalczyk, M.S.; Zhang, H.; Nyman, J.; Sakuishi, K.; Kurtulus, S.; Gennert, D.; et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 2016, 166, 1500–1511. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.-C.; Ferté, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, K.H.; Kang, J.; Borcoman, E.; Saada-Bouzid, E.; Kronbichler, A.; Hong, S.H.; de Rezende, L.F.M.; Ogino, S.; Keum, N.; et al. Hyperprogressive Disease during Anti-PD-1 (PDCD1)/PD-L1 (CD274) Therapy: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1699. [Google Scholar] [CrossRef] [PubMed]
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008. [Google Scholar] [CrossRef]
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Salmons, J.; Nowak, A.K.; Rozali, E.N.; Khong, A.; Dick, I.M.; Harken, J.A.; Robinson, B.W.; Lake, R.A. Synergistic effect of CTLA-4 blockade and cancer chemotherapy in the induction of anti-tumor immunity. PLoS ONE 2013, 8, e61895. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- McKee, S.J.; Doff, B.L.; Soon, M.S.F.; Mattarollo, S.R. Therapeutic Efficacy of 4-1BB Costimulation Is Abrogated by PD-1 Blockade in a Model of Spontaneous B-cell Lymphoma. Cancer Immunol. Res. 2017, 5, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Messenheimer, D.J.; Jensen, S.M.; Afentoulis, M.E.; Wegmann, K.W.; Feng, Z.; Friedman, D.J.; Gough, M.J.; Urba, W.J.; Fox, B.A. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clin. Cancer Res. 2017, 23, 6165–6177. [Google Scholar] [CrossRef]
- Sun, D.; Ma, J.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Anti-PD-1 therapy combined with chemotherapy in patients with advanced biliary tract cancer. Cancer Immunol. Immunother. 2019, 68, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Langer, C.J.; Gadgeel, S.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. 24-Month Overall Survival from KEYNOTE-021 Cohort G: Pemetrexed and Carboplatin with or without Pembrolizumab as First-Line Therapy for Advanced Nonsquamous Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 124–129. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Lu, C.-S.; Liu, J.-H. Pneumonitis in cancer patients receiving anti-PD-1 and radiotherapies: Three case reports. Medicine 2017, 96, e5747. [Google Scholar] [CrossRef]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Vacchelli, E.; Bloy, N.; Aranda, F.; Buqué, A.; Cremer, I.; Demaria, S.; Eggermont, A.; Formenti, S.C.; Fridman, W.H.; Fucikova, J.; et al. Trial Watch: Immunotherapy plus radiation therapy for oncological indications. Oncoimmunology 2016, 5, e1214790. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bondarenko, I.; Luft, A.; Serwatowski, P.; Barlesi, F.; Chacko, R.; Sebastian, M.; Neal, J.; Lu, H.; Cuillerot, J.-M.; et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: Results from a randomized, double-blind, multicenter phase II study. J. Clin. Oncol. 2012, 30, 2046–2054. [Google Scholar] [CrossRef]
- Vera Aguilera, J.; Paludo, J.; Bangalore, A.; Failing, J.; McWilliams, R.R.; Kottschade, L.A.; Block, M.S.; Markovic, S.; Dronca, R.S.; Yan, Y. Chemoimmunotherapy combination after PD-1 inhibitor failure to improve clinical outcomes in metastatic melanoma patients. J. Clin. Oncol. 2018, 36, 9558. [Google Scholar] [CrossRef]
- Gabri, M.R.; Mazorra, Z.; Ripoll, G.V.; Mesa, C.; Fernandez, L.E.; Gomez, D.E.; Alonso, D.F. Complete antitumor protection by perioperative immunization with GM3/VSSP vaccine in a preclinical mouse melanoma model. Clin. Cancer Res. 2006, 12, 7092–7098. [Google Scholar] [CrossRef] [PubMed]
- Czajka, H.; Unal, S.; Ulusoy, S.; Usluer, G.; Strus, A.; Sennaroglu, E.; Guzik, J.; Topeli Iskit, A.; Dargiewicz, A.; Musial, D.; et al. A phase II, randomised clinical trial to demonstrate the non-inferiority of low-dose MF59-adjuvanted pre-pandemic A/H5N1 influenza vaccine in adult and elderly subjects. J. Prev. Med. Hyg. 2012, 53, 136–142. [Google Scholar] [PubMed]
- John, T.; Voysey, M.; Yu, L.M.; McCarthy, N.; Baudin, M.; Richard, P.; Fiquet, A.; Kitchin, N.; Pollard, A.J. Immunogenicity of a low-dose diphtheria, tetanus and acellular pertussis combination vaccine with either inactivated or oral polio vaccine compared to standard-dose diphtheria, tetanus, acellular pertussis when used as a pre-school booster in UK children: A 5-year follow-up of a randomised controlled study. Vaccine 2015, 33, 4579–4585. [Google Scholar] [CrossRef]
- Marlow, R.; Kuriyakose, S.; Mesaros, N.; Han, H.H.; Tomlinson, R.; Faust, S.N.; Snape, M.D.; Pollard, A.J.; Finn, A. A phase III, open-label, randomised multicentre study to evaluate the immunogenicity and safety of a booster dose of two different reduced antigen diphtheria-tetanus-acellular pertussis-polio vaccines, when co-administered with measles-mumps-rubella vaccine in 3 and 4-year-old healthy children in the UK. Vaccine 2018, 36, 2300–2306. [Google Scholar] [CrossRef]
- Melief, C.J.M.; Welters, M.J.P.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Tjalma, W.A.A.; Denys, H.; van Poelgeest, M.I.E.; Nijman, H.W.; et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Peng, S.; Lyford-Pike, S.; Akpeng, B.; Wu, A.; Hung, C.-F.; Hannaman, D.; Saunders, J.R.; Wu, T.-C.; Pai, S.I. Low-dose cyclophosphamide administered as daily or single dose enhances the antitumor effects of a therapeutic HPV vaccine. Cancer Immunol. Immunother. 2013, 62, 171–182. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8+ T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef]
- Wada, S.; Yoshimura, K.; Hipkiss, E.L.; Harris, T.J.; Yen, H.-R.; Goldberg, M.V.; Grosso, J.F.; Getnet, D.; Demarzo, A.M.; Netto, G.J.; et al. Cyclophosphamide augments antitumor immunity: Studies in an autochthonous prostate cancer model. Cancer Res. 2009, 69, 4309–4318. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Lasalvia-Prisco, E.; Goldschmidt, P.; Galmarini, F.; Cucchi, S.; Vázquez, J.; Aghazarian, M.; Lasalvia-Galante, E.; Golomar, W.; Gordon, W. Addition of an induction regimen of antiangiogenesis and antitumor immunity to standard chemotherapy improves survival in advanced malignancies. Med. Oncol. 2012, 29, 3626–3633. [Google Scholar] [CrossRef] [PubMed]
- Ellebaek, E.; Engell-Noerregaard, L.; Iversen, T.Z.; Froesig, T.M.; Munir, S.; Hadrup, S.R.; Andersen, M.H.; Svane, I.M. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: Results from a phase II trial. Cancer Immunol. Immunother. 2012, 61, 1791–1804. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Boustani, J.; Grapin, M.; Laurent, P.-A.; Apetoh, L.; Mirjolet, C. The 6th R of Radiobiology: Reactivation of Anti-Tumor Immune Response. Cancers 2019, 11, 860. [Google Scholar] [CrossRef] [PubMed]
- Grapin, M.; Richard, C.; Limagne, E.; Boidot, R.; Morgand, V.; Bertaut, A.; Derangere, V.; Laurent, P.-A.; Thibaudin, M.; Fumet, J.D.; et al. Optimized fractionated radiotherapy with anti-PD-L1 and anti-TIGIT: A promising new combination. J. Immunother. Cancer 2019, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Kusunoki, Y.; Akiyama, M. Radiosensitivity of CD4 or CD8 positive human T-lymphocytes by an in vitro colony formation assay. Radiat. Res. 1990, 123, 224–227. [Google Scholar] [CrossRef]
- Yovino, S.; Kleinberg, L.; Grossman, S.A.; Narayanan, M.; Ford, E. The etiology of treatment-related lymphopenia in patients with malignant gliomas: Modeling radiation dose to circulating lymphocytes explains clinical observations and suggests methods of modifying the impact of radiation on immune cells. Cancer Investig. 2013, 31, 140–144. [Google Scholar] [CrossRef]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef]
- Filatenkov, A.; Baker, J.; Mueller, A.M.S.; Kenkel, J.; Ahn, G.-O.; Dutt, S.; Zhang, N.; Kohrt, H.; Jensen, K.; Dejbakhsh-Jones, S.; et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin. Cancer Res. 2015, 21, 3727–3739. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Schadendorf, D.; Wolchok, J.D.; Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Chesney, J.; et al. Efficacy and Safety Outcomes in Patients With Advanced Melanoma Who Discontinued Treatment With Nivolumab and Ipilimumab Because of Adverse Events: A Pooled Analysis of Randomized Phase II and III Trials. J. Clin. Oncol. 2017, 35, 3807–3814. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Rozeman, E.A.; Mason, R.; Goldinger, S.M.; Geukes Foppen, M.H.; Hoejberg, L.; Schmidt, H.; van Thienen, J.V.; Haanen, J.B.A.G.; Tiainen, L.; et al. Discontinuation of anti-PD-1 antibody therapy in the absence of disease progression or treatment limiting toxicity: Clinical outcomes in advanced melanoma. Ann. Oncol. 2019, 30, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Betof Warner, A.; Palmer, J.S.; Shoushtari, A.N.; Goldman, D.A.; Panageas, K.S.; Hayes, S.A.; Bajwa, R.; Momtaz, P.; Callahan, M.K.; Wolchok, J.D.; et al. Long-Term Outcomes and Responses to Retreatment in Patients With Melanoma Treated With PD-1 Blockade. J. Clin. Oncol. 2020, 38, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, H.; Li, G.; Zhou, X.; Shi, Y.; Zou, F.; Chen, Y.; Gao, J.; Yang, S.; Wu, S.; et al. Increased Tim-3 expression on TILs during treatment with the Anchored GM-CSF vaccine and anti-PD-1 antibodies is inversely correlated with response in prostate cancer. J. Cancer 2020, 11, 648–656. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations with Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol 2019, 10, 467. [Google Scholar] [CrossRef]
- Curran, M.A.; Glisson, B.S. New Hope for Therapeutic Cancer Vaccines in the Era of Immune Checkpoint Modulation. Annu. Rev. Med. 2019, 70, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Yoo, G.S.; Cho, W.K.; Park, H.C. Optimizing radiotherapy with immune checkpoint blockade in hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Manukian, G.; Bar-Ad, V.; Lu, B.; Argiris, A.; Johnson, J.M. Combining Radiation and Immune Checkpoint Blockade in the Treatment of Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, N.; Baldin, P.; Van den Eynde, M. Immunotherapy with immune checkpoint inhibitors in colorectal cancer: What is the future beyond deficient mismatch-repair tumours? Gastroenterol. Rep. 2020, 8, 11–24. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dosset, M.; Joseph, E.L.-M.; Rivera Vargas, T.; Apetoh, L. Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors. Cells 2020, 9, 1727. https://doi.org/10.3390/cells9071727
Dosset M, Joseph EL-M, Rivera Vargas T, Apetoh L. Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors. Cells. 2020; 9(7):1727. https://doi.org/10.3390/cells9071727
Chicago/Turabian StyleDosset, Magalie, Elodie Lauret-Marie Joseph, Thaiz Rivera Vargas, and Lionel Apetoh. 2020. "Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors" Cells 9, no. 7: 1727. https://doi.org/10.3390/cells9071727
APA StyleDosset, M., Joseph, E. L.-M., Rivera Vargas, T., & Apetoh, L. (2020). Modulation of Determinant Factors to Improve Therapeutic Combinations with Immune Checkpoint Inhibitors. Cells, 9(7), 1727. https://doi.org/10.3390/cells9071727