Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes

 ,
,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
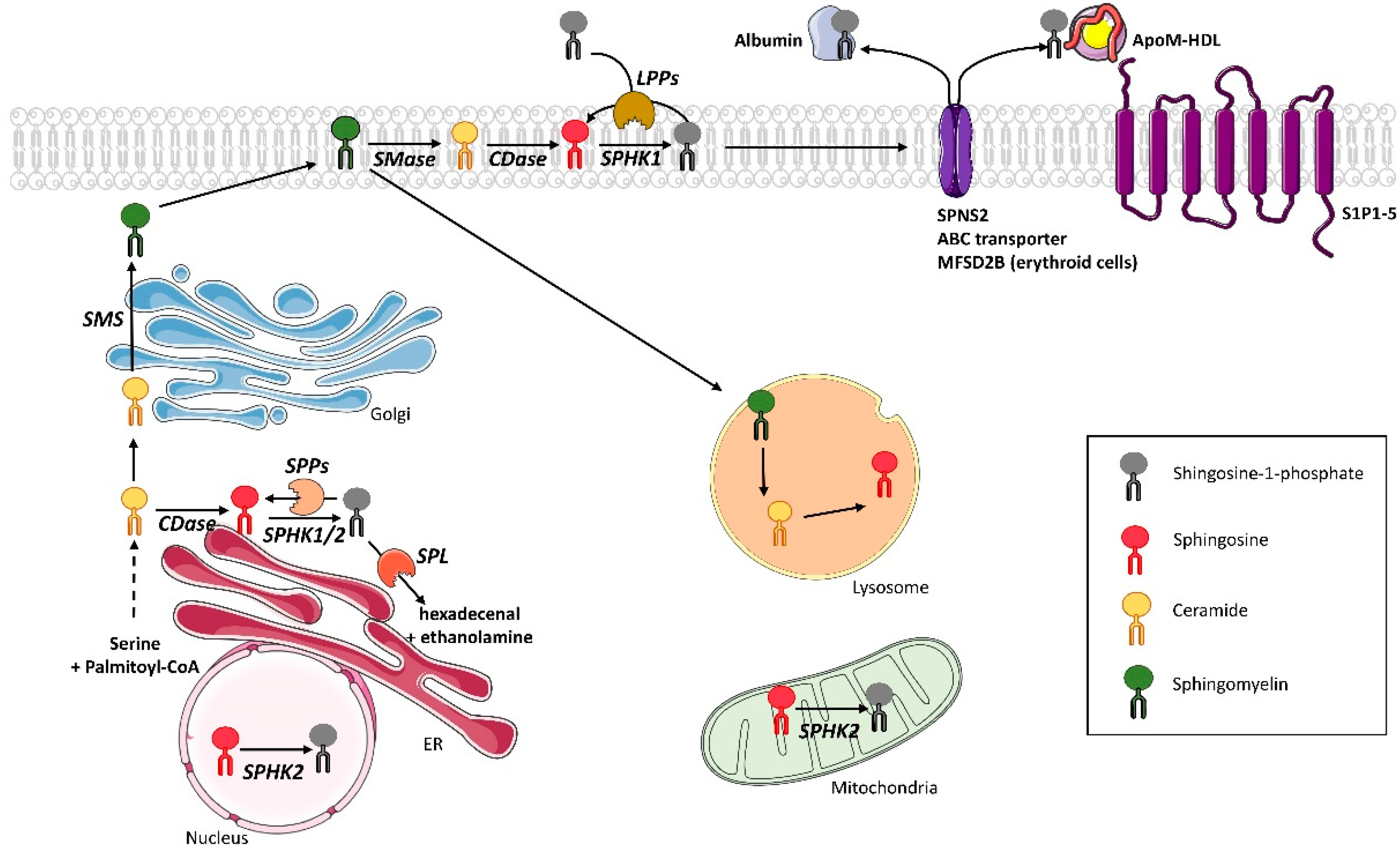
2. S1P Metabolism in Mammals
2.1. S1P Synthesis
2.2. S1P Recycling and Degradation
2.3. S1P Transport
2.4. S1P Receptors
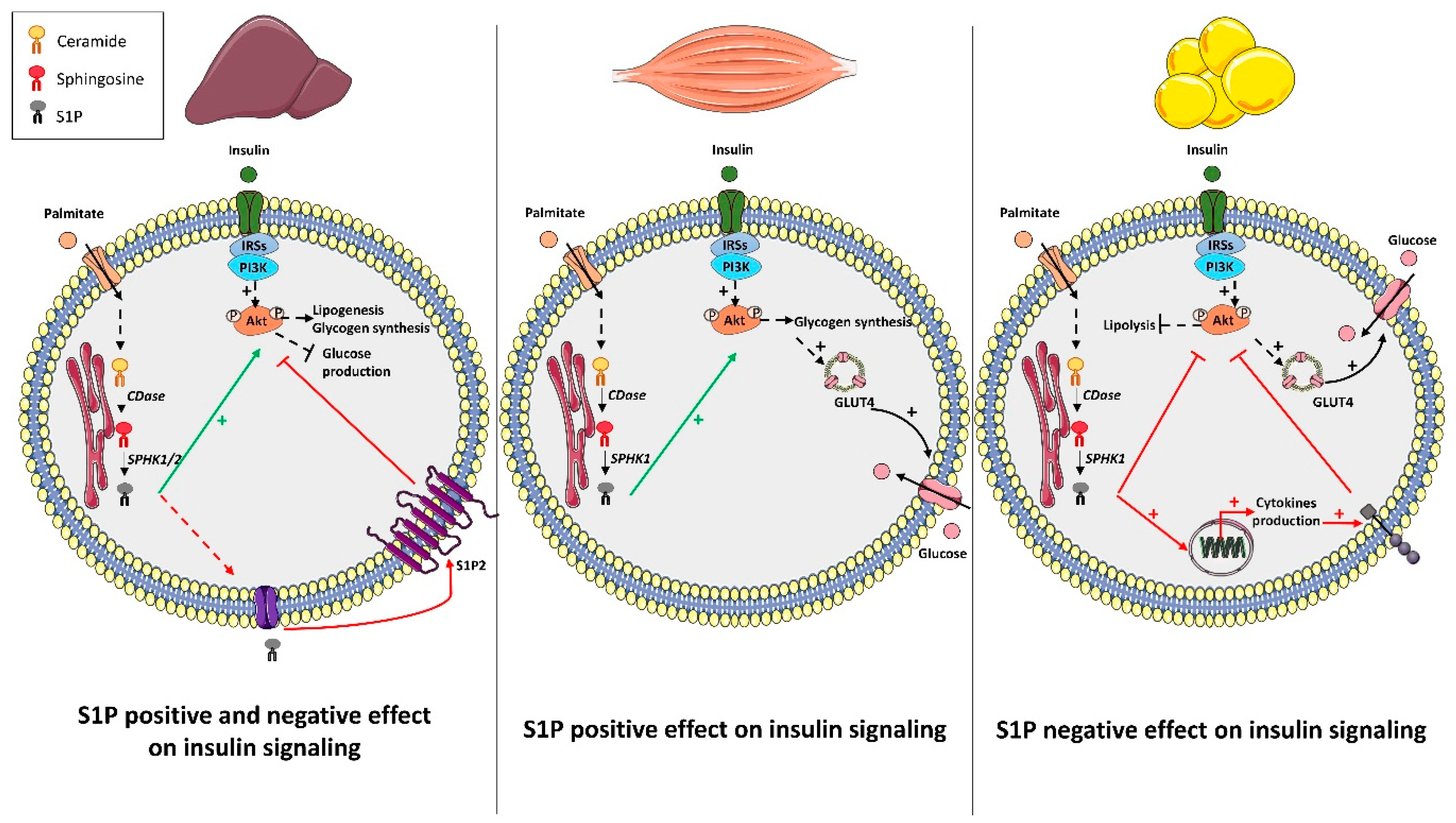
3. S1P Metabolism and Insulin Action: Muscle, Liver, and Adipose Tissue
3.1. Liver
3.2. Muscle
3.3. Adipose Tissue
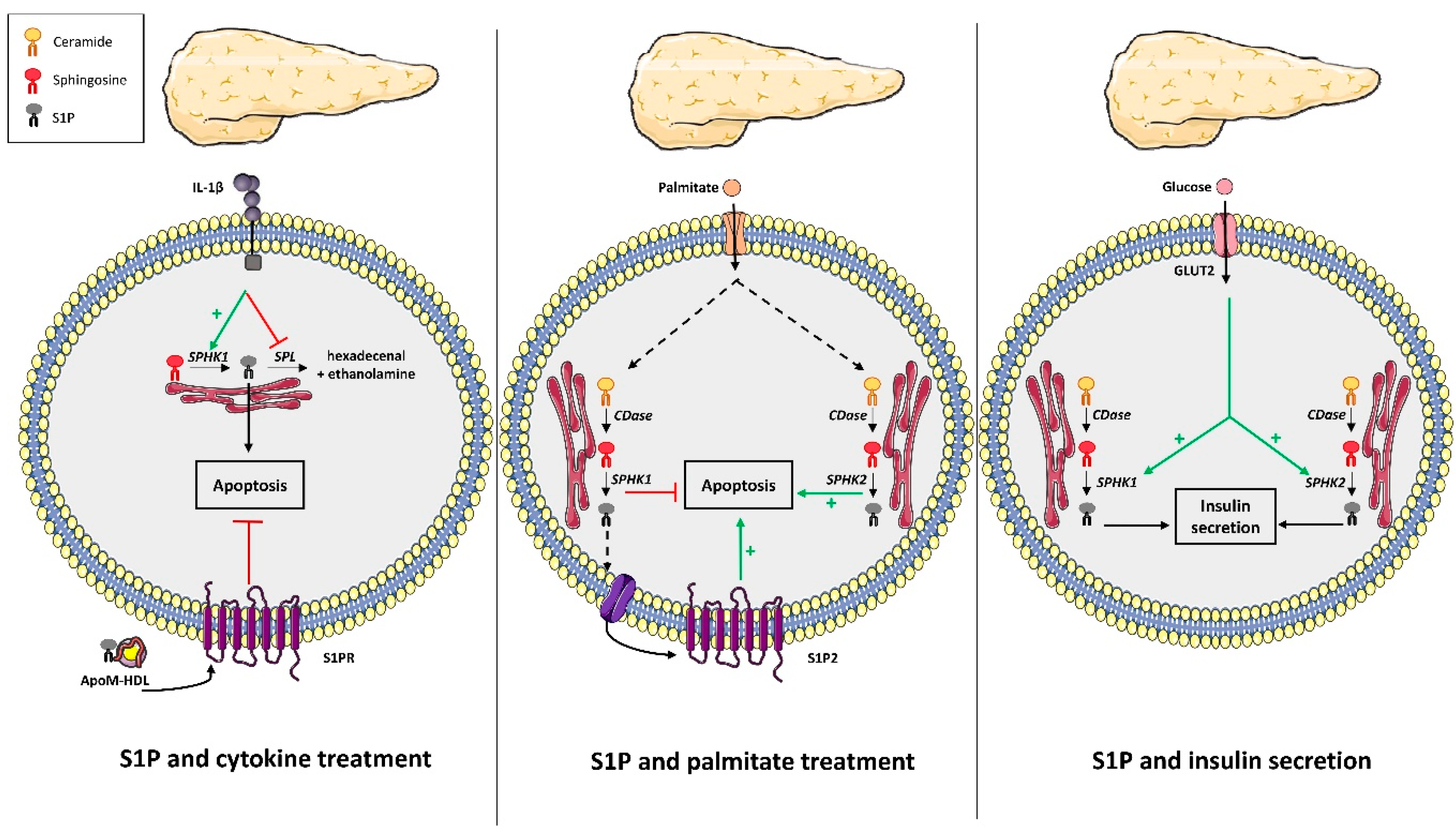
4. S1P Metabolism and Pancreatic β Cell Fate
5. S1P and the Hypothalamic Regulation of Body Weight and Energy Homeostasis
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galgani, J.E.; Moro, C.; Ravussin, E. Metabolic flexibility and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1009–E1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharewicz, E.; Hesselink, M.K.C.; Schrauwen, P. Exercise counteracts lipotoxicity by improving lipid turnover and lipid droplet quality. J. Intern. Med. 2018, 284, 505–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, M.K.; De Nardo, W.; Watt, M.J. Impact of Lipotoxicity on Tissue “Cross Talk” and Metabolic Regulation. Physiol. Bethesda Md. 2019, 34, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Torretta, E.; Barbacini, P.; Al-Daghri, N.M.; Gelfi, C. Sphingolipids in Obesity and Correlated Co-Morbidities: The Contribution of Gender, Age and Environment. Int. J. Mol. Sci. 2019, 20, 5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells during Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, O.P.; Dahl, D.B.; Brechtel, K.; Machann, J.; Haap, M.; Maier, T.; Loviscach, M.; Stumvoll, M.; Claussen, C.D.; Schick, F.; et al. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes 2001, 50, 2579–2584. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Ralston, J.C.; Nguyen-Tu, M.-S.; Lyons, C.L.; Cooke, A.A.; Murphy, A.M.; Falvey, A.; Finucane, O.M.; McGillicuddy, F.C.; Rutter, G.A.; Roche, H.M. Dietary substitution of SFA with MUFA within high-fat diets attenuates hyperinsulinaemia and pancreatic islet dysfunction. Br. J. Nutr. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Stunff, H.L.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ségui, B.; Andrieu-Abadie, N.; Jaffrézou, J.-P.; Benoist, H.; Levade, T. Sphingolipids as modulators of cancer cell death: Potential therapeutic targets. Biochim. Biophys. Acta 2006, 1758, 2104–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Hage Hassan, R.; Bourron, O.; Hajduch, E. Defect of insulin signal in peripheral tissues: Important role of ceramide. World J. Diabetes 2014, 5, 244–257. [Google Scholar] [CrossRef]
- Campana, M.; Bellini, L.; Rouch, C.; Rachdi, L.; Coant, N.; Butin, N.; Bandet, C.L.; Philippe, E.; Meneyrol, K.; Kassis, N.; et al. Inhibition of central de novo ceramide synthesis restores insulin signaling in hypothalamus and enhances β-cell function of obese Zucker rats. Mol. Metab. 2018, 8, 23–36. [Google Scholar] [CrossRef]
- Bellini, L.; Campana, M.; Mahfouz, R.; Carlier, A.; Véret, J.; Magnan, C.; Hajduch, E.; Stunff, H.L. Targeting sphingolipid metabolism in the treatment of obesity/type 2 diabetes. Expert Opin. Ther. Targets 2015, 19, 1037–1050. [Google Scholar] [CrossRef]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-Phosphate Signaling and Its Role in Disease. Trends Cell Biol. 2012, 22, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma Sphingosine-1-Phosphate Is Elevated in Obesity. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.L.; Wadham, C.; Sukocheva, O.A. The role of sphingolipid signalling in diabetes-associated pathologies (Review). Int. J. Mol. Med. 2017, 39, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemany, R.; van Koppen, C.J.; Danneberg, K.; Ter Braak, M.; Meyer zu Heringdorf, D. Regulation and functional roles of sphingosine kinases. Naunyn. Schmiedebergs Arch. Pharmacol. 2007, 374, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, Sphingosine Kinase Isoenzymes with Opposing Functions in Sphingolipid Metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [Green Version]
- Lysophospholipid Receptors: Signaling and Biochemistry; Chun, J. (Ed.) Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-118-53130-3. [Google Scholar]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [Green Version]
- Kleuser, B. Divergent Role of Sphingosine 1-Phosphate in Liver Health and Disease. Int. J. Mol. Sci. 2018, 19, 722. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H.; Giussani, P.; Maceyka, M.; Lepine, S.; Milstien, S.; Spiegel, S. Recycling of Sphingosine Is Regulated by the Concerted Actions of Sphingosine-1-phosphate Phosphohydrolase 1 and Sphingosine Kinase 2. J. Biol. Chem. 2007, 282, 34372–34380. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, A.; Saba, J.D. Truth and consequences of sphingosine-1-phosphate lyase. Adv. Biol. Regul. 2012, 52, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Le Stunff, H. Sphingosine-1-phosphate and lipid phosphohydrolases. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2002, 1582, 8–17. [Google Scholar] [CrossRef]
- Sigal, Y.J.; McDermott, M.I.; Morris, A.J. Integral membrane lipid phosphatases/phosphotransferases: Common structure and diverse functions. Biochem. J. 2005, 387, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Sphingolipids as Signaling and Regulatory Molecules; Chalfant, C.; Del Poeta, M. (Eds.) Advances in Experimental Medicine and Biology; Springer Science+Business Media: New York, NY, USA; Landes Bioscience: Austin, TX, USA, 2010; ISBN 978-1-4419-6740-4. [Google Scholar]
- Lépine, S.; Allegood, J.C.; Park, M.; Dent, P.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011, 18, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.; Allende, M.L.; Mizukami, H.; Cook, E.K.; Gavrilova, O.; Tuymetova, G.; Clarke, B.A.; Chen, W.; Olivera, A.; Proia, R.L. Sphingosine-1-phosphate Phosphatase 2 Regulates Pancreatic Islet β-Cell Endoplasmic Reticulum Stress and Proliferation. J. Biol. Chem. 2016, 291, 12029–12038. [Google Scholar] [CrossRef] [Green Version]
- Reitsema, V.; Bouma, H.; Willem Kok, J. Sphingosine-1-phosphate transport and its role in immunology. AIMS Mol. Sci. 2014, 1, 183–201. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kawasaki-Nishi, S.; Otsuka, M.; Hisano, Y.; Yamaguchi, A.; Nishi, T. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Yatomi, Y. Plasma sphingosine 1-phosphate metabolism and analysis. Glycobiol. Sphingobiology 2008, 1780, 606–611. [Google Scholar] [CrossRef]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Lee, Y.-M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar] [CrossRef] [Green Version]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.; Lapergue, B.; Burillo, E.; Michel, J.; Levoye, A.; Martin-Ventura, J.; et al. HDL and endothelial protection: HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [Green Version]
- Poti, F.; Simoni, M.; Nofer, J.-R. Atheroprotective role of high-density lipoprotein (HDL)-associated sphingosine-1-phosphate (S1P). Cardiovasc. Res. 2014, 103, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O. Les récepteurs de la sphingosine 1-phosphate: De la biologie à la physiopathologie. Médecine/Sciences 2012, 28, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-J.; Im, D.-S. Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol. Ther. 2017, 25, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [Green Version]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef] [Green Version]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Bradham, C.A.; Plümpe, J.; Manns, M.P.; Brenner, D.A.; Trautwein, C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am. J. Physiol. 1998, 275, G387–G392. [Google Scholar] [CrossRef]
- Osawa, Y.; Banno, Y.; Nagaki, M.; Brenner, D.A.; Naiki, T.; Nozawa, Y.; Nakashima, S.; Moriwaki, H. TNF-α-Induced Sphingosine 1-Phosphate Inhibits Apoptosis Through a Phosphatidylinositol 3-Kinase/Akt Pathway in Human Hepatocytes. J. Immunol. 2001, 167, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Uchinami, H.; Bielawski, J.; Schwabe, R.F.; Hannun, Y.A.; Brenner, D.A. Roles for C16-ceramide and Sphingosine 1-Phosphate in Regulating Hepatocyte Apoptosis in Response to Tumor Necrosis Factor-α. J. Biol. Chem. 2005, 280, 27879–27887. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Feng, Q.; Shi, Y.; Zhou, J.; Wang, Q.; Zhong, L. Hepatic insulin resistance induced by mitochondrial oxidative stress can be ameliorated by sphingosine 1-phosphate. Mol. Cell. Endocrinol. 2020, 501, 110660. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.M.; Chen, J.L.; Wang, G.G.; Wang, H.; Lu, Y.; Li, J.F.; Yi, J.; Yuan, Y.J.; Zhang, Q.W.; Mi, J.; et al. Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia 2007, 50, 891–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, Y.; Seki, E.; Kodama, Y.; Suetsugu, A.; Miura, K.; Adachi, M.; Ito, H.; Shiratori, Y.; Banno, Y.; Olefsky, J.M.; et al. Acid sphingomyelinase regulates glucose and lipid metabolism in hepatocytes through AKT activation and AMP-activated protein kinase suppression. FASEB J. 2011, 25, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.; Qi, Y.; Kaczorowski, D.; McCaughan, G.W.; Gamble, J.R.; Don, A.S.; Gao, X.; Vadas, M.A.; Xia, P. Deletion of sphingosine kinase 1 ameliorates hepatic steatosis in diet-induced obese mice: Role of PPARγ. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2016, 1861, 138–147. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Hong, I.-K.; Kim, B.-R.; Shim, S.-M.; Sung Lee, J.; Lee, H.-Y.; Soo Choi, C.; Kim, B.-K.; Park, T.-S. Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice: LEE, HONG, ET AL. Hepatology 2015, 62, 135–146. [Google Scholar] [CrossRef]
- Fayyaz, S.; Henkel, J.; Japtok, L.; Krämer, S.; Damm, G.; Seehofer, D.; Püschel, G.P.; Kleuser, B. Involvement of sphingosine 1-phosphate in palmitate-induced insulin resistance of hepatocytes via the S1P2 receptor subtype. Diabetologia 2014, 57, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Chen, J.; Lay, A.; Don, A.; Vadas, M.; Xia, P. Loss of sphingosine kinase 1 predisposes to the onset of diabetes via promoting pancreatic β-cell death in diet-induced obese mice. FASEB J. 2013, 27, 4294–4304. [Google Scholar] [CrossRef]
- Kowalski, G.M.; Kloehn, J.; Burch, M.L.; Selathurai, A.; Hamley, S.; Bayol, S.A.M.; Lamon, S.; Watt, M.J.; Lee-Young, R.S.; McConville, M.J.; et al. Overexpression of sphingosine kinase 1 in liver reduces triglyceride content in mice fed a low but not high-fat diet. Biochim. Biophys. Acta 2015, 1851, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Pacana, T.; Sanyal, A.J. Recent advances in understanding/management of non-alcoholic steatohepatitis. F1000prime Rep. 2015, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatol. Baltim. Md. 2015, 61, 1216–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, C.-Y.; Song, M.J.; Gao, Y.; Mauer, A.S.; Revzin, A.; Malhi, H. Hepatocyte-Derived Lipotoxic Extracellular Vesicle Sphingosine 1-Phosphate Induces Macrophage Chemotaxis. Front. Immunol. 2018, 9, 2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, T.; Sutter, A.; Harland, M.D.; Law, B.A.; Ross, J.S.; Lewin, D.; Palanisamy, A.; Russo, S.B.; Chavin, K.D.; Cowart, L.A. SphK1 mediates hepatic inflammation in a mouse model of NASH induced by high saturated fat feeding and initiates proinflammatory signaling in hepatocytes. J. Lipid Res. 2015, 56, 2359–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Ferrannini, E.; Sato, Y.; Felig, P.; Wahren, J. Synergistic interaction between exercise and insulin on peripheral glucose uptake. J. Clin. Investig. 1981, 68, 1468–1474. [Google Scholar] [CrossRef]
- Hu, W.; Bielawski, J.; Samad, F.; Merrill, A.H.; Cowart, L.A. Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J. Lipid Res. 2009, 50, 1852–1862. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Hu, W.; Rosen, B.; Snider, A.J.; Obeid, L.M.; Cowart, L.A. Sphingosine Kinase 1 Is Regulated by Peroxisome Proliferator-activated Receptor α in Response to Free Fatty Acids and Is Essential for Skeletal Muscle Interleukin-6 Production and Signaling in Diet-induced Obesity. J. Biol. Chem. 2013, 288, 22193–22206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapizzi, E.; Taddei, M.L.; Fiaschi, T.; Donati, C.; Bruni, P.; Chiarugi, P. Sphingosine 1-phosphate increases glucose uptake through trans-activation of insulin receptor. Cell. Mol. Life Sci. 2009, 66, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Kowalski, G.M.; Selathurai, A.; Lee-Young, R.S.; Weir, J.M.; Yoshioka, K.; Takuwa, Y.; et al. Overexpression of Sphingosine Kinase 1 Prevents Ceramide Accumulation and Ameliorates Muscle Insulin Resistance in High-Fat Diet-Fed Mice. Diabetes 2012, 61, 3148–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, C.R.; Risis, S.; Babb, J.R.; Yang, C.; Lee-Young, R.S.; Henstridge, D.C.; Febbraio, M.A. The sphingosine-1-phosphate analog FTY720 reduces muscle ceramide content and improves glucose tolerance in high fat-fed male mice. Endocrinology 2013, 154, 65–76. [Google Scholar] [CrossRef]
- Kendall, M. FTY720, a sphingosine-1-phosphate receptor modulator, reverses high-fat diet-induced weight gain, insulin resistance and adipose tissue inflammation in C57BL/6 mice. Diabetes Obes. Metab. 2008, 10, 802–805. [Google Scholar] [CrossRef]
- Brinkmann, V. Sphingosine 1-phosphate receptors in health and disease: Mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol. Ther. 2007, 115, 84–105. [Google Scholar] [CrossRef]
- Berdyshev, E.V.; Gorshkova, I.; Skobeleva, A.; Bittman, R.; Lu, X.; Dudek, S.M.; Mirzapoiazova, T.; Garcia, J.G.N.; Natarajan, V. FTY720 Inhibits Ceramide Synthases and Up-regulates Dihydrosphingosine 1-Phosphate Formation in Human Lung Endothelial Cells. J. Biol. Chem. 2009, 284, 5467–5477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorena, K.; Jachimowicz-Duda, O.; Ślęzak, D.; Robakowska, M.; Mrugacz, M. Adipokines and Obesity. Potential Link to Metabolic Disorders and Chronic Complications. Int. J. Mol. Sci. 2020, 21, 3570. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jönsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106 (Suppl. 3), S5–S78. [Google Scholar] [CrossRef] [PubMed]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of adipose tissue inflammation to the development of type 2 diabetes mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef]
- Hashimoto, T.; Igarashi, J.; Kosaka, H. Sphingosine kinase is induced in mouse 3T3-L1 cells and promotes adipogenesis. J. Lipid Res. 2009, 50, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol.—Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef] [Green Version]
- Blachnio-Zabielska, A.U.; Koutsari, C.; Tchkonia, T.; Jensen, M.D. Sphingolipid content of human adipose tissue: Relationship to adiponectin and insulin resistance. Obes. Silver Spring Md. 2012, 20, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhao, J.; Meng, H.; Zhang, X. Adipose Tissue-Resident Immune Cells in Obesity and Type 2 Diabetes. Front. Immunol. 2019, 10, 1173. [Google Scholar] [CrossRef]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.-J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, T.L.; Mirzaian, M.; Hooibrink, B.; Ottenhoff, R.; van Roomen, C.; Aerts, J.M.F.G.; van Eijk, M. Induction of Sphk1 activity in obese adipose tissue macrophages promotes survival. PLoS ONE 2017, 12, e0182075. [Google Scholar] [CrossRef] [Green Version]
- Mastrandrea, L.D.; Sessanna, S.M.; Laychock, S.G. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: Response to cytokines. Diabetes 2005, 54, 1429–1436. [Google Scholar] [CrossRef] [Green Version]
- Hasan, N.M.; Longacre, M.J.; Stoker, S.; Kendrick, M.A.; Druckenbrod, N.R.; Laychock, S.G.; Mastrandrea, L.D.; MacDonald, M.J. Sphingosine Kinase 1 Knockdown Reduces Insulin Synthesis and Secretion in a Rat Insulinoma Cell Line. Arch. Biochem. Biophys. 2012, 518, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, J.C.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Özcan, S. Sphingosine 1-Phosphate (S1P) Regulates Glucose-stimulated Insulin Secretion in Pancreatic Beta Cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, C.; Tyka, K.; Saba, J.D.; Lenzen, S.; Gurgul-Convey, E. Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 2017, 292, 20292–20304. [Google Scholar] [CrossRef] [Green Version]
- Laychock, S.G.; Sessanna, S.M.; Lin, M.-H.; Mastrandrea, L.D. Sphingosine 1-phosphate affects cytokine-induced apoptosis in rat pancreatic islet beta-cells. Endocrinology 2006, 147, 4705–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rütti, S.; Ehses, J.A.; Sibler, R.A.; Prazak, R.; Rohrer, L.; Georgopoulos, S.; Meier, D.T.; Niclauss, N.; Berney, T.; Donath, M.Y.; et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 2009, 150, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Véret, J.; Coant, N.; Gorshkova, I.A.; Giussani, P.; Fradet, M.; Riccitelli, E.; Skobeleva, A.; Goya, J.; Kassis, N.; Natarajan, V.; et al. Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 β-cell survival. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2013, 1831, 251–262. [Google Scholar] [CrossRef]
- Japtok, L.; Schmitz, E.I.; Fayyaz, S.; Krämer, S.; Hsu, L.J.; Kleuser, B. Sphingosine 1-phosphate counteracts insulin signaling in pancreatic β-cells via the sphingosine 1-phosphate receptor subtype 2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 3357–3369. [Google Scholar] [CrossRef] [Green Version]
- Lacaná, E.; Maceyka, M.; Milstien, S.; Spiegel, S. Cloning and Characterization of a Protein Kinase A Anchoring Protein (AKAP)-related Protein That Interacts with and Regulates Sphingosine Kinase 1 Activity. J. Biol. Chem. 2002, 277, 32947–32953. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Harashima, S.; Wang, Y.; Suzuki, K.; Tokumoto, S.; Usui, R.; Tatsuoka, H.; Tanaka, D.; Yabe, D.; Harada, N.; et al. Sphingosine kinase 1–interacting protein is a dual regulator of insulin and incretin secretion. FASEB J. 2019, 33, 6239–6253. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A.H.; Tang, J.; Yadev, A.K.; Saghafi, S.T.; Kibbe, C.R.; Linnemann, A.K.; Merrins, M.J.; Davis, D.B. Intra-islet GLP-1, but Not CCK, Is Necessary for β-Cell Function in Mouse and Human Islets. Available online: https://pubmed.ncbi.nlm.nih.gov/32071395/ (accessed on 20 May 2020).
- Song, Z.; Wang, W.; Li, N.; Yan, S.; Rong, K.; Lan, T.; Xia, P. Sphingosine kinase 2 promotes lipotoxicity in pancreatic β-cells and the progression of diabetes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 3636–3646. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Jeong, J.H.; Lee, D.K.; Jo, Y.-H. Cholinergic neurons in the dorsomedial hypothalamus regulate food intake. Mol. Metab. 2017, 6, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Horvath, T.L. Molecular and cellular regulation of hypothalamic melanocortin neurons controlling food intake and energy metabolism. Mol. Psychiatry 2014, 19, 752–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.H.; Lee, D.K.; Liu, S.-M.; Chua, S.C.; Schwartz, G.J.; Jo, Y.-H. Activation of temperature-sensitive TRPV1-like receptors in ARC POMC neurons reduces food intake. PLoS Biol. 2018, 16, e2004399. [Google Scholar] [CrossRef] [Green Version]
- Picard, A.; Moullé, V.S.; Le Foll, C.; Cansell, C.; Véret, J.; Coant, N.; Le Stunff, H.; Migrenne, S.; Luquet, S.; Cruciani-Guglielmacci, C.; et al. Physiological and pathophysiological implications of lipid sensing in the brain. Diabetes Obes. Metab. 2014, 16 (Suppl. 1), 49–55. [Google Scholar] [CrossRef]
- Le Stunff, H.; Coant, N.; Migrenne, S.; Magnan, C. Targeting lipid sensing in the central nervous system: New therapy against the development of obesity and type 2 diabetes. Expert Opin. Ther. Targets 2013, 17, 545–555. [Google Scholar] [CrossRef]
- Silva, V.R.R.; Micheletti, T.O.; Pimentel, G.D.; Katashima, C.K.; Lenhare, L.; Morari, J.; Mendes, M.C.S.; Razolli, D.S.; Rocha, G.Z.; de Souza, C.T.; et al. Hypothalamic S1P/S1PR1 axis controls energy homeostasis. Nat. Commun. 2014, 5, 4859. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Iwaki, S.; Koike, K.; Yuda, Y.; Nagasaki, A.; Ohkawa, R.; Yatomi, Y.; Furumoto, T.; Tsutsui, H.; Sobel, B.E.; et al. Increased plasma sphingosine-1-phosphate in obese individuals and its capacity to increase the expression of plasminogen activator inhibitor-1 in adipocytes. Coron. Artery Dis. 2013, 24, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef] [Green Version]
- Dorfman, M.D.; Thaler, J.P. Hypothalamic inflammation and gliosis in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Mendes, N.F.; Kim, Y.-B.; Velloso, L.A.; Araújo, E.P. Hypothalamic Microglial Activation in Obesity: A Mini-Review. Front. Neurosci. 2018, 12, 846. [Google Scholar] [CrossRef] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Schwartz, M.W. Minireview: Inflammation and obesity pathogenesis: The hypothalamus heats up. Endocrinology 2010, 151, 4109–4115. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [Green Version]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Assi, E.; Cazzato, D.; De Palma, C.; Perrotta, C.; Clementi, E.; Cervia, D. Sphingolipids and brain resident macrophages in neuroinflammation: An emerging aspect of nervous system pathology. Clin. Dev. Immunol. 2013, 2013, 309302. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2016, 65, 551–562. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.-C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate—A double edged sword in the brain. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Federspiel, C.K.; Borup, A.; Christensen, P.M.; Madsen, A.N.; Heine, M.; Nielsen, C.H.; Kjaer, A.; Holst, B.; Heeren, J.; et al. The Apolipoprotein M/S1P Axis Controls Triglyceride Metabolism and Brown Fat Activity. Cell Rep. 2018, 22, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. https://doi.org/10.3390/cells9071682
Guitton J, Bandet CL, Mariko ML, Tan-Chen S, Bourron O, Benomar Y, Hajduch E, Le Stunff H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells. 2020; 9(7):1682. https://doi.org/10.3390/cells9071682
Chicago/Turabian StyleGuitton, Jeanne, Cécile L. Bandet, Mohamed L. Mariko, Sophie Tan-Chen, Olivier Bourron, Yacir Benomar, Eric Hajduch, and Hervé Le Stunff. 2020. "Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes" Cells 9, no. 7: 1682. https://doi.org/10.3390/cells9071682
APA StyleGuitton, J., Bandet, C. L., Mariko, M. L., Tan-Chen, S., Bourron, O., Benomar, Y., Hajduch, E., & Le Stunff, H. (2020). Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells, 9(7), 1682. https://doi.org/10.3390/cells9071682