MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton


Abstract
1. Introduction
2. Regulation of Actin and Myosin Cytoskeleton
2.1. Actin Cytoskeleton
2.2. Myosin
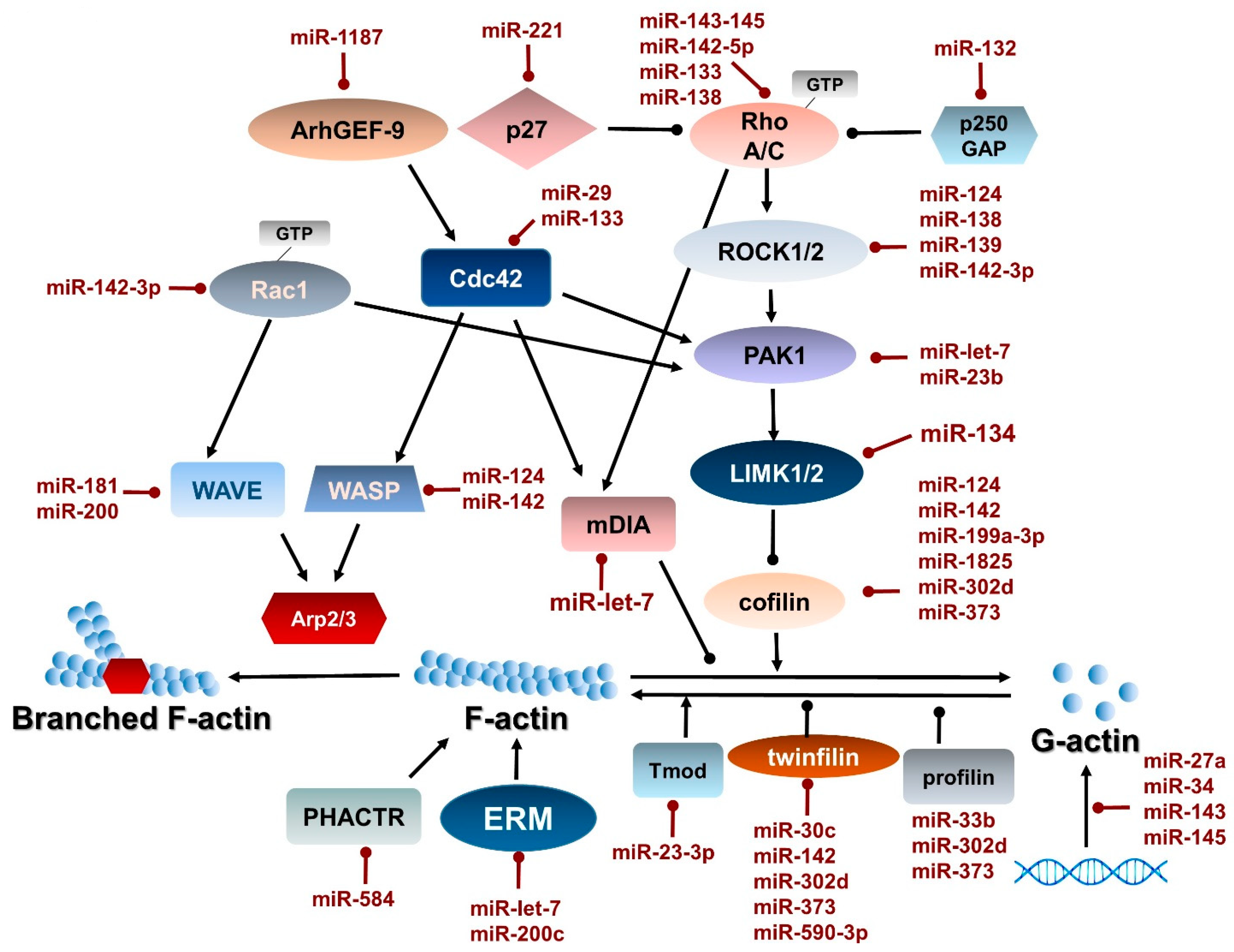
2.3. Regulation of Actin–Myosin Dynamics by Small GTPases
3. MicroRNA Overview
4. Direct Regulation of Actin and Myosin Gene Expression by miRNAs
4.1. Direct Regulation of Actin Gene Expression by miRNAs
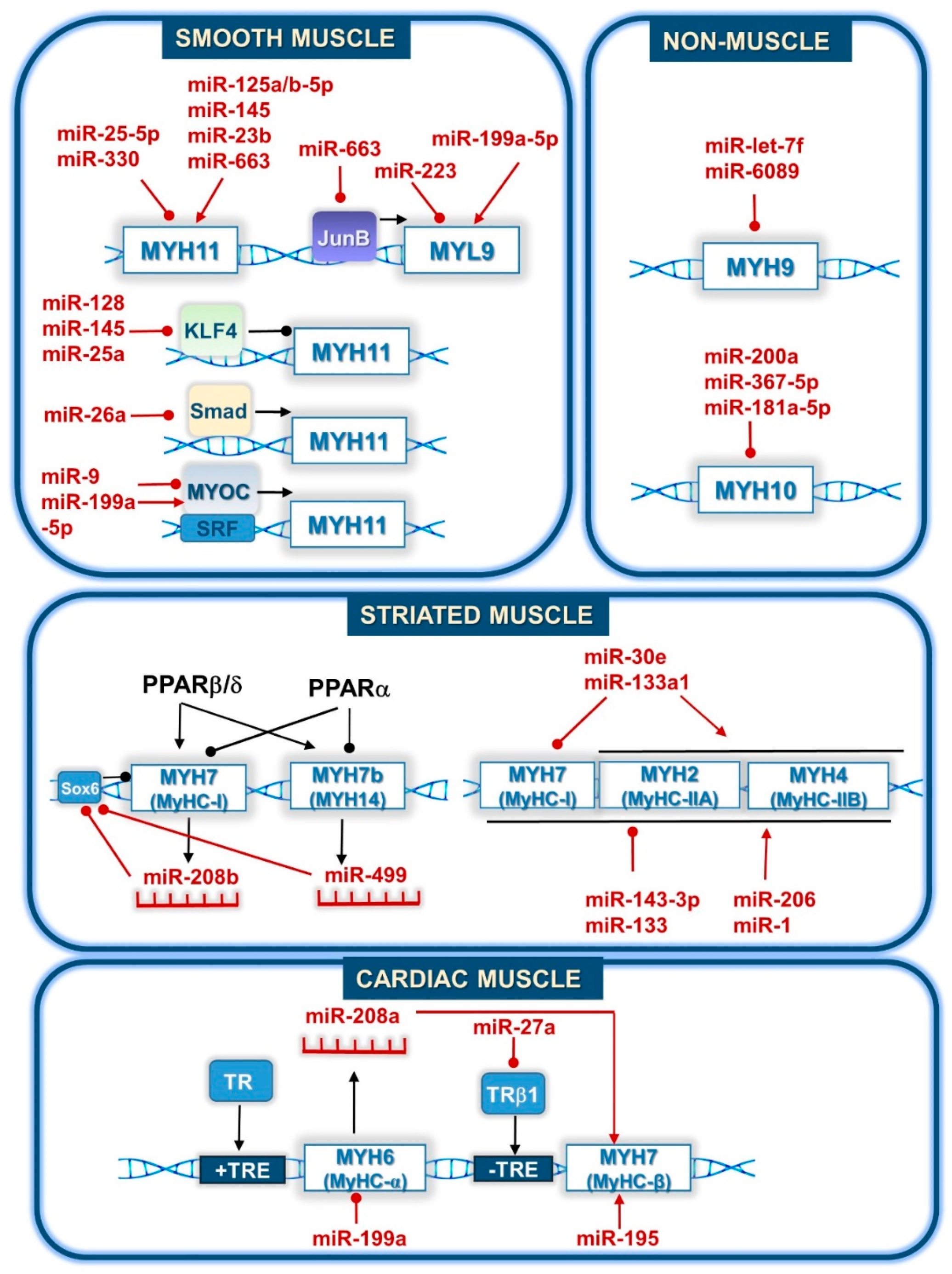
4.2. Direct Regulation of Myosin Gene Expression by miRNAs
5. Physiological and Pathological Processes Related to miRNA-Regulated Pathways
5.1. MiRNAs in Smooth Muscle Function and Diseases
5.2. MiRNAs in Cardiovascular Diseases
5.3. Regulation of Actin Cytoskeleton by miRNAs in the Hematopoietic System
5.4. Actin Regulation in Podocyte Biology
5.5. MiRNA Regulation of the Actin Cytoskeleton in Osteoblast Differentiation
5.6. MiRNAs as Key Regulators in Actin Reorganization in on Cilia Assembly
5.7. Signaling Pathways of miRNAs-Regulated Cancer Formation
5.8. MiRNAs in Actin Cytoskeleton Regulation: Leukocytes and Lymphocytes in Action
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ABLIM1 | actin binding LIM protein 1 |
| ABLIM3 | actin binding LIM protein 3 |
| ACTB | beta-actin |
| ADF/cofilin family | actin depolymerizing factor/cofilin family |
| apoE | apolipoprotein E |
| ARhGEF-9 | Cdc42 guanine nucleotide exchange factor 9 |
| ARP2/3 complex | actin-related protein 2/3 complex |
| ARPP-21 | cyclic AMP-regulated phosphoprotein of 21 kDa |
| BDNF | brain-derived neurotropic factor |
| BMP-II | bone morphogenic protein II |
| BMPR | bone morphogenic protein receptor |
| BRK1 | BRICK1 subunit of SCAR/WAVE actin-nucleating complex |
| CD25 | interleukin-2 receptor alpha chain |
| CFL2 | cofilin 2 |
| circACTA | circular α-actin |
| Clcn5 | voltage sensitive chloride channel 5 |
| CP110 | centriolar coiled-coil protein of 110 kDa |
| CPI17 | protein kinase C-potentiated inhibitor protein of 17 kDa |
| CXCL2 | chemokine ligand 2 |
| DIAPH2 | diaphanous-related formin 2 |
| Ena/VASP family | enabled/vasodilator-stimulated phosphoprotein family |
| E-Tmod41 | erythrocyte tropomodulin of 41 kDa |
| ETS-1 | E-twenty-six family member 1 |
| FGF9 | fibroblast growth factor 9 |
| FHOD1 | formin homology 2 domain containing 1 |
| GAP | GTPase activating protein |
| GEF | guanine nucleotide exchange factor |
| GMF | glial maturation factor |
| GRK6 | G-protein-coupled receptor kinase 6 |
| HCC | hepatocellular carcinoma |
| IKZF1 | Ikaros family zinc finger protein 1 |
| ILK | integrin-linked kinase |
| ITGB8 | integrin beta 8 |
| JG | juxtaglomerular |
| KLF4 | Krüppel-like factor 4 |
| KLF5 | Krüppel-like factor 5 |
| LIMK1 | LIM domain kinase 1 |
| MCC | multiciliated cell |
| MCK | muscle creatine kinase |
| MEKK1 | mitogen-activated protein kinase kinase 1 |
| miRNA | microRNA |
| miRNP | miRNA ribonucleoprotein complex |
| MLC | myosin light chain |
| MLCP | myosin light chain phosphatase |
| Mlph | melanophilin |
| MRTF-B | myocardin-related transcription factor B |
| MYH10 | myosin heavy chain 10 |
| MYH11 | myosin heavy chain 11 |
| MYH17 | myosin heavy chain 17 |
| MYH7b (MYH14) | myosin heavy chain 7b (myosin heavy chain 14) |
| MYH9 | myosin heavy chain 9 |
| MyHC | myosin heavy chain |
| MyHC-emb | embryonic myosin heavy chain |
| MyHC-IIA | myosin heavy chain-IIA |
| MyHC-IIB | myosin heavy chain-IIB |
| MyHC-IIX | myosin heavy chain-IIX |
| MyHC-slow(I) | myosin heavy chain slow type I |
| MyHC-α | myosin heavy chain alpha |
| MyHC-β | myosin heavy chain beta |
| MYL9 | myosin light chain 9 |
| MYLK | myosin light chain kinase |
| MYO1A | myosin 1A |
| MYO1B | myosin 1B |
| MYO1C | myosin 1C |
| MYO5A | myosin 5A |
| MYOCD | myocardin |
| MYPT1 | myosin phosphatase target subunit 1 |
| Mφ | macrophage |
| NPF | nucleation promoting factor |
| NRG-1 | neuroregulin-1 |
| NRVM | neonatal rat ventricular myocyte |
| p250GAP | brain-enriched GTPase-activating protein for Rho family GTPases |
| p27Kip1 | cyclin-dependent kinase inhibitor 1B |
| PAK | p21-activated kinase |
| PDGF-B | platelet-derived growth factor-B |
| PDGFRB | platelet-derived growth factor receptor B |
| Pgc1α | peroxisome proliferator-activated receptor gamma coactivator 1 alpha |
| PHACTR1 | protein phosphatase and actin regulator 1 |
| PIK3R3 | phosphoinositide-3-kinase regulatory subunit 3 |
| PKC | protein kinase C |
| PP1c | protein phosphatase 1 catalytic subunit |
| PPARα | peroxisome proliferator-activated receptor alpha |
| PPARβ/δ | peroxisome proliferator-activated receptor beta/delta |
| PPM1F | Mg2+/Mn2+-dependent protein phosphatase 1F |
| pre-miRNA | hairpin-like miRNA precursor |
| pri-miRNA | primary microRNA |
| R3HDM1 | R3H domain containing 1 |
| Rab27a | Ras-related protein Rab27a |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RCS | regulator of calmodulin signaling |
| RDX | radixin |
| RhoA | Ras homolog family member A |
| RhoB | Ras homolog family member B |
| RhoC | Ras homolog family member C |
| ROCK | Rho-associated protein kinase |
| ROCK2 | Rho-associated protein kinase 2 |
| R-Ras | Ras-related protein |
| SM22α | smooth muscle 22α |
| SMC | smooth muscle cell |
| SMTN | smoothelin |
| SOL | soleus muscle |
| Sox6 | SRY-related HMG box 6 |
| SRF | serum response factor |
| STAT3 | signal transducer and activator of transcription 3 |
| STTM | short tandem target mimic |
| TAB | thoracic aortic banding |
| TEAD1 | TEA domain family member 1 |
| TGF-β | transforming growth factor beta |
| TH | thyroid hormone |
| THR | thyroid hormone receptor |
| THRβ1 | thyroid hormone receptor beta 1 |
| TKS5 | tyrosine kinase substrate with 5 SH domains |
| TrkB | tropomyosin-related kinase B |
| TSCC | tongue squamous cell carcinoma |
| VSMC | vascular smooth muscle cell |
| WASF1 | WAS protein family member 1 |
| WASL | Wiscott–Aldrich syndrome protein |
| WAVE3 | WASP family protein member 3 |
| WT | wild type |
| YAP | Yes-associated protein |
| ZEB1 | zinc finger e-box binding homeobox 1 |
| ZIPK | zipper-interacting protein kinase |
| α-SMA | alpha smooth muscle actin |
References
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Kasza, K.E.; Supriyatno, S.; Zallen, J.A. Cellular defects resulting from disease-related myosin II mutations in Drosophila. Proc. Natl. Acad. Sci. USA 2019, 116, 22205–22211. [Google Scholar] [CrossRef] [PubMed]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef]
- Seidman, J.G.; Seidman, C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Jaeger, M.A.; Ommen, S.R.; Will, M.L.; Gersh, B.J.; Tajik, A.J.; Ackerman, M.J. Comprehensive Analysis of the Beta-Myosin Heavy Chain Gene in 389 Unrelated Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Fagnant, P.M.; Bookwalter, C.S.; Joel, P.; Trybus, K.M. Vascular disease-causing mutation R258C in ACTA2 disrupts actin dynamics and interaction with myosin. Proc. Natl. Acad. Sci. USA 2015, 112, E4168–E4177. [Google Scholar] [CrossRef]
- Regalado, E.S.; Guo, N.-C.; Prakash, S.; Bensend, T.A.; Flynn, K.; Estrera, A.; Safi, H.; Liang, D.; Hyland, J.; Child, A.; et al. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ. Cardiovasc. Genet. 2015, 8, 457–464. [Google Scholar] [CrossRef]
- Guo, D.-C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in Smooth Muscle Alpha-Actin (ACTA2) Cause Coronary Artery Disease, Stroke, and Moyamoya Disease, Along with Thoracic Aortic Disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef]
- Ostberg, N.P.; Zafar, M.A.; Ziganshin, B.A.; Elefteriades, J.A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 2020, 10, 182. [Google Scholar] [CrossRef]
- An, S.S.; Bai, T.R.; Bates, J.H.T.; Black, J.L.; Brown, R.H.; Brusasco, V.; Chitano, P.; Deng, L.; Dowell, M.; Eidelman, D.H.; et al. Airway smooth muscle dynamics: A common pathway of airway obstruction in asthma. Eur. Respir. J. 2007, 29, 834–860. [Google Scholar] [CrossRef] [PubMed]
- An, S.S.; Fredberg, J.J. Biophysical basis for airway hyperresponsiveness. Can. J. Physiol. Pharmacol. 2007, 85, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Parkos, C.A.; Nusrat, A. Cytoskeletal Regulation of Epithelial Barrier Function during Inflammation. Am. J. Pathol. 2010, 177, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Ouderkirk, J.L.; Krendel, M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton (Hoboken) 2014, 71, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D. The Dynamic Actin Cytoskeleton in Smooth Muscle. Adv. Pharmacol. 2018, 81, 1–38. [Google Scholar]
- Oda, T.; Iwasa, M.; Aihara, T.; Maéda, Y.; Narita, A. The nature of the globular- to fibrous-actin transition. Nature 2009, 457, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef]
- Skruber, K.; Read, T.-A.; Vitriol, E.A. Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell Sci. 2018, 131, jcs203760. [Google Scholar] [CrossRef]
- Rotty, J.D.; Wu, C.; Bear, J.E. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 7–12. [Google Scholar] [CrossRef]
- Boczkowska, M.; Rebowski, G.; Kast, D.J.; Dominguez, R. Structural analysis of the transitional state of Arp2/3 complex activation by two actin-bound WCAs. Nat. Commun. 2014, 5, 3308. [Google Scholar] [CrossRef]
- Breitsprecher, D.; Goode, B.L. Formins at a glance. J. Cell Sci. 2013, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Brühmann, S.; Ushakov, D.S.; Winterhoff, M.; Dickinson, R.B.; Curth, U.; Faix, J. Distinct VASP tetramers synergize in the processive elongation of individual actin filaments from clustered arrays. Proc. Natl. Acad. Sci. USA 2017, 114, E5815–E5824. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Beltzner, C.C.; Pollard, T.D. Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr. Biol. 2009, 19, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.S.; Chemeris, A.; Guo, S.; Alioto, S.L.; Gandhi, M.; Padrick, S.; Goode, B.L. Structural Basis of Arp2/3 Complex Inhibition by GMF, Coronin, and Arpin. J. Mol. Biol. 2017, 429, 237–248. [Google Scholar] [CrossRef]
- Peckham, M. How myosin organization of the actin cytoskeleton contributes to the cancer phenotype. Biochem. Soc. Trans. 2016, 44, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.F.; Langford, G. Myosin superfamily evolutionary history. Anat. Rec. 2002, 268, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Van Gele, M.; Dynoodt, P.; Lambert, J. Griscelli syndrome: A model system to study vesicular trafficking. Pigment. Cell Melanoma Res. 2009, 22, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.C.; Mammucari, C.; Argentini, C.; Reggiani, C.; Schiaffino, S. Two novel/ancient myosins in mammalian skeletal muscles: MYH14/7b and MYH15 are expressed in extraocular muscles and muscle spindles. J. Physiol. 2010, 588, 353–364. [Google Scholar] [CrossRef]
- McConnell, R.E.; Tyska, M.J. Leveraging the membrane—Cytoskeleton interface with myosin-1. Trends Cell Boil. 2010, 20, 418–426. [Google Scholar] [CrossRef]
- Almeida, C.G.; Yamada, A.; Tenza, D.; Louvard, D.; Raposo, G.; Coudrier, E. Myosin 1b promotes the formation of post-Golgi carriers by regulating actin assembly and membrane remodelling at the trans-Golgi network. Nat. Cell Biol. 2011, 13, 779–789. [Google Scholar] [CrossRef]
- Krendel, M.; Osterweil, E.K.; Mooseker, M.S. Myosin 1E interacts with synaptojanin-1 and dynamin and is involved in endocytosis. FEBS Lett. 2007, 581, 644–650. [Google Scholar] [CrossRef]
- Tokuo, H.; Coluccio, L.M. Myosin-1c regulates the dynamic stability of E-cadherin–based cell–cell contacts in polarized Madin–Darby canine kidney cells. Mol. Boil. Cell 2013, 24, 2820–2833. [Google Scholar] [CrossRef] [PubMed]
- Makowska, K.A.; Hughes, R.E.; White, K.J.; Wells, C.M.; Peckham, M. Specific Myosins Control Actin Organization, Cell Morphology, and Migration in Prostate Cancer Cells. Cell Rep. 2015, 13, 2118–2125. [Google Scholar] [CrossRef] [PubMed]
- Coluccio, L.M.; Geeves, M.A. Transient Kinetic Analysis of the 130-kDa Myosin I (MYR-1 Gene Product) from Rat Liver. A myosin I designed for maintenance of tension? J. Boil. Chem. 1999, 274, 21575–21580. [Google Scholar] [CrossRef]
- Tang, N.; Ostap, E.M. Motor domain-dependent localization of myo1b (myr-1). Curr. Boil. 2001, 11, 1131–1135. [Google Scholar] [CrossRef]
- Bose, A.; Robida, S.; Furcinitti, P.S.; Chawla, A.; Fogarty, K.; Corvera, S.; Czech, M.P. Unconventional Myosin Myo1c Promotes Membrane Fusion in a Regulated Exocytic Pathway. Mol. Cell. Boil. 2004, 24, 5447–5458. [Google Scholar] [CrossRef]
- Billington, N.; Wang, A.; Mao, J.; Adelstein, R.S.; Sellers, J.R. Characterization of Three Full-length Human Nonmuscle Myosin II Paralogs. J. Boil. Chem. 2013, 288, 33398–33410. [Google Scholar] [CrossRef] [PubMed]
- Shutova, M.S.; Spessott, W.A.; Giraudo, C.G.; Svitkina, T.M. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr. Boil. 2014, 24, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Heissler, S.M.; Manstein, D.J. Nonmuscle myosin-2: Mix and match. Cell. Mol. Life Sci. 2012, 70, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Wirth, A.; Schroeter, M.; Kock-Hauser, C.; Manser, E.; Chalovich, J.M.; De Lanerolle, P.; Pfitzer, G. Inhibition of Contraction and Myosin Light Chain Phosphorylation in Guinea-Pig Smooth Muscle by p21-Activated Kinase 1. J. Physiol. 2003, 549, 489–500. [Google Scholar] [CrossRef]
- Perrino, B.A. Calcium Sensitization Mechanisms in Gastrointestinal Smooth Muscles. J. Neurogastroenterol. Motil. 2016, 22, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.T.; Sutherland, C.; Brautigan, D.L.; Eto, M.; Walsh, M.P. Phosphorylation of the myosin phosphatase inhibitors, CPI-17 and PHI-1, by integrin-linked kinase. Biochem. J. 2002, 367 Pt 2, 517–524. [Google Scholar] [CrossRef]
- Kitazawa, T.; Eto, M.; Woodsome, T.P.; Khalequzzaman, M.D. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J. Physiol. 2003, 546 Pt 3, 879–889. [Google Scholar] [CrossRef]
- Koyama, M.; Ito, M.; Feng, J.; Seko, T.; Shiraki, K.; Takase, K.; Hartshorne, D.J.; Nakano, T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000, 475, 197–200. [Google Scholar] [CrossRef]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adhes. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; A Grässer, F.; Lenhof, H.-P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Deschamps-Francoeur, G.; Scott, M.S. Protein coding genes as hosts for noncoding RNA expression. Semin. Cell Dev. Boil. 2018, 75, 3–12. [Google Scholar] [CrossRef]
- Nilsen, T.W. Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet. 2007, 23, 243–249. [Google Scholar] [CrossRef]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Issabekova, A.; Berillo, O.; Régnier, M.; Anatoly, I. Interactions of intergenic microRNAs with mRNAs of genes involved in carcinogenesis. Bioinformation 2012, 8, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Godnic, I.; Zorc, M.; Skok, D.J.; Calin, G.A.; Horvat, S.; Dovč, P.; Kovač, M.; Kunej, T. Genome-Wide and Species-Wide In Silico Screening for Intragenic MicroRNAs in Human, Mouse and Chicken. PLoS ONE 2013, 8, e65165. [Google Scholar] [CrossRef] [PubMed]
- Schanen, B.C.; Li, X. Transcriptional regulation of mammalian miRNA genes. Genomics 2011, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Boil. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Mourelatos, Z. Site-specific crosslinking of human microRNPs to RNA targets. RNA 2008, 14, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear Export of MicroRNA Precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef]
- Koscianska, E.; Starega-Roslan, J.; Krzyzosiak, W.J. The Role of Dicer Protein Partners in the Processing of MicroRNA Precursors. PLoS ONE 2011, 6, e28548. [Google Scholar] [CrossRef] [PubMed]
- Maniataki, E.; Mourelatos, Z. A human, ATP-independent, RISC assembly machine fueled by pre-miRNA. Genome Res. 2005, 19, 2979–2990. [Google Scholar] [CrossRef]
- Lee, Y.; Hur, I.; Park, S.-Y.; Kim, Y.-K.; Suh, M.R.; Kim, V.N. The role of PACT in the RNA silencing pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Ladomery, M.R.; Maddocks, D.G.; Wilson, I.D. MicroRNAs: Their discovery, biogenesis, function and potential use as biomarkers in non-invasive prenatal diagnostics. Int. J. Mol. Epidemiol. Genet. 2011, 2, 253–260. [Google Scholar] [PubMed]
- Valastyan, S.; A Weinberg, R. Roles for microRNAs in the regulation of cell adhesion molecules. J. Cell Sci. 2011, 124, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Jacks, T. MicroRNAs and cancer: Short RNAs go a long way. Cell 2009, 136, 586–591. [Google Scholar] [CrossRef]
- An, L.; Liu, Y.; Wu, A.; Guan, Y. MicroRNA-124 Inhibits Migration and Invasion by Down-Regulating ROCK1 in Glioma. PLoS ONE 2013, 8, e69478. [Google Scholar] [CrossRef]
- Ghosh, T.; Soni, K.; Scaria, V.; Halimani, M.; Bhattacharjee, C.; Pillai, B. MicroRNA-mediated up-regulation of an alternatively polyadenylated variant of the mouse cytoplasmic β-actin gene. Nucleic Acids Res. 2008, 36, 6318–6332. [Google Scholar] [CrossRef]
- Wang, T.; Chen, K.; Hsu, P.; Lin, H.-F.; Wang, Y.-S.; Chen, C.-Y.; Liao, Y.; Juo, S.-H.H. MicroRNA let-7g suppresses PDGF-induced conversion of vascular smooth muscle cell into the synthetic phenotype. J. Cell. Mol. Med. 2017, 21, 3592–3601. [Google Scholar] [CrossRef]
- Han, M.; Dong, L.-H.; Zheng, B.; Shi, J.-H.; Wen, J.-K.; Cheng, Y. Smooth muscle 22 alpha maintains the differentiated phenotype of vascular smooth muscle cells by inducing filamentous actin bundling. Life Sci. 2009, 84, 394–401. [Google Scholar] [CrossRef]
- Zhang, J.C.L.; Kim, S.; Helmke, B.P.; Yu, W.W.; Du, K.L.; Lu, M.M.; Strobeck, M.; Yu, Q.-C.; Parmacek, M.S. Analysis of SM22α-Deficient Mice Reveals Unanticipated Insights into Smooth Muscle Cell Differentiation and Function. Mol. Cell. Boil. 2001, 21, 1336–1344. [Google Scholar] [CrossRef]
- Sun, G.; Song, H.; Wu, S. miR-19a promotes vascular smooth muscle cell proliferation, migration and invasion through regulation of Ras homolog family member B. Int. J. Mol. Med. 2019, 44, 1991–2002. [Google Scholar] [CrossRef]
- Han, H.; Yang, S.; Liang, Y.; Zeng, P.; Liu, L.; Yang, X.; Duan, Y.; Han, J.; Chen, Y. Teniposide regulates the phenotype switching of vascular smooth muscle cells in a miR-21-dependent manner. Biochem. Biophys. Res. Commun. 2018, 506, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Iaconetti, C.; De Rosa, S.; Polimeni, A.; Sorrentino, S.; Gareri, C.; Carino, A.; Sabatino, J.; Colangelo, M.; Curcio, A.; Indolfi, C. Down-regulation of miR-23b induces phenotypic switching of vascular smooth muscle cells in vitro and in vivo. Cardiovasc. Res. 2015, 107, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Gareri, C.; Iaconetti, C.; Sorrentino, S.; Covello, C.; De Rosa, S.; Indolfi, C. MiR-125a-5p Modulates Phenotypic Switch of Vascular Smooth Muscle Cells by Targeting ETS-1. J. Mol. Boil. 2017, 429, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Dong, M.; Wen, H.; Liu, X.; Zhang, M.; Ma, L.; Zhang, C.; Luan, X.; Lu, H.; Zhang, Y. MiR-26a contributes to the PDGF-BB-induced phenotypic switch of vascular smooth muscle cells by suppressing Smad1. Oncotarget 2017, 8, 75844–75853. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-M.; Deng, H.-Y.; Li, H.-H. MicroRNA-27a regulates angiotensin II-induced vascular smooth muscle cell proliferation and migration by targeting α-smooth muscle-actin in vitro. Biochem. Biophys. Res. Commun. 2019, 509, 973–977. [Google Scholar] [CrossRef]
- Wang, L.; Bao, H.; Wang, K.-X.; Zhang, P.; Yao, Q.-P.; Chen, X.-H.; Huang, K.; Qi, Y.-X.; Jiang, Z.-L. Secreted miR-27a Induced by Cyclic Stretch Modulates the Proliferation of Endothelial Cells in Hypertension via GRK6. Sci. Rep. 2017, 7, 41058. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genome Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Xie, B.D.; Sun, L.; Chen, W.; Jiang, S.L.; Liu, W.; Li, R.K. Phenotypic switching of vascular smooth muscle cells in the ‘normal region’of aorta from atherosclerosis patients is regulated by miR-145. J. Cell Mol. Med. 2016, 20, 1049–1061. [Google Scholar] [CrossRef]
- Dong, N.; Wang, W.; Tian, J.; Xie, Z.; Lv, B.; Dai, J.; Li, H. MicroRNA-182 prevents vascular smooth muscle cell dedifferentiation via FGF9/PDGFRbeta signaling. Int. J. Mol. Med. 2017, 39, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Medrano, S.; Monteagudo, M.C.; Sequeira-Lopez, M.L.S.; Pentz, E.S.; Gomez, R.A. Two microRNAs, miR-330 and miR-125b-5p, mark the juxtaglomerular cell and balance its smooth muscle phenotype. Am. J. Physiol. Physiol. 2012, 302, F29–F37. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Z.; Zheng, B.; Zhang, X.H.; Zhang, M.L.; Zhao, X.S.; Wen, J.K. A Novel Regulatory Mechanism of Smooth Muscle alpha-Actin Expression by NRG-1/circACTA2/miR-548f-5p Axis. Circ. Res. 2017, 121, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhu, N.; Yi, B.; Wang, N.; Chen, M.; You, X.; Zhao, X.; Solomides, C.C.; Qin, Y.; Sun, J. MicroRNA-663 regulates human vascular smooth muscle cell phenotypic switch and vascular neointimal formation. Circ. Res. 2013, 113, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Boil. 2006, 174, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; E Callis, T.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Du, J.; Zhang, Y.; Shen, L.; Luo, J.; Lei, H.; Zhang, P.; Pu, Q.; Liu, Y.; Shuai, S.; Li, Q.; et al. Effect of miR-143-3p on C2C12 myoblast differentiation. Biosci. Biotechnol. Biochem. 2016, 80, 706–711. [Google Scholar] [CrossRef]
- Jia, H.; Zhao, Y.; Li, T.; Zhang, Y.; Zhu, D. Mir-30e is negatively regulated by myostatin in skeletal muscle and is functionally related to fiber-type composition. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 392–399. [Google Scholar] [CrossRef]
- Nie, Y.; Sato, Y.; Wang, C.; Yue, F.; Kuang, S.; Gavin, T.P. Impaired exercise tolerance, mitochondrial biogenesis, and muscle fiber maintenance in miR-133a–deficient mice. FASEB J. 2016, 30, 3745–3758. [Google Scholar] [CrossRef]
- Mizbani, A.; Luca, E.; Rushing, E.J.; Krützfeldt, J. MicroRNA deep sequencing in two adult stem cell populations identifies miR-501 as a novel regulator of myosin heavy chain during muscle regeneration. Development 2016, 143, 4137–4148. [Google Scholar] [CrossRef]
- Nishi, H.; Ono, K.; Horie, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Watanabe, S.; Takaya, T.; Tamaki, Y.; Takanabe-Mori, R.; et al. MicroRNA-27a Regulates Beta Cardiac Myosin Heavy Chain Gene Expression by Targeting Thyroid Hormone Receptor β1 in Neonatal Rat Ventricular Myocytes. Mol. Cell. Boil. 2011, 31, 744–755. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, X.; Li, Y.; Zhao, L.; Lu, M.; Yao, X.; Xia, H.; Wang, Y.-C.; Liu, M.-F.; Jiang, J.; et al. Thyroid hormone regulates muscle fiber type conversion via miR-133a1. J. Cell Boil. 2014, 207, 753–766. [Google Scholar] [CrossRef]
- Qi, S.; Liu, B.; Zhang, J.; Liu, X.; Dong, C.; Fan, R. Knockdown of microRNA-143-5p by STTM technology affects eumelanin and pheomelanin production in melanocytes. Mol. Med. Rep. 2019, 20, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Dynoodt, P.; Mestdagh, P.; Van Peer, G.; Vandesompele, J.; Goossens, K.; Peelman, L.J.; Geusens, B.; Speeckaert, R.M.; Lambert, J.L.; Van Gele, M. Identification of miR-145 as a Key Regulator of the Pigmentary Process. J. Investig. Dermatol. 2013, 133, 201–209. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef]
- Song, X.-W.; Li, Q.; Lin, L.; Wang, X.-C.; Li, D.-F.; Wang, G.-K.; Ren, A.-J.; Wang, Y.-R.; Qin, Y.-W.; Yuan, W.-J.; et al. MicroRNAs are dynamically regulated in hypertrophic hearts, and miR-199a is essential for the maintenance of cell size in cardiomyocytes. J. Cell. Physiol. 2010, 225, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Rumsey, J.; Hazen, B.C.; Lai, L.; Leone, T.C.; Vega, R.B.; Xie, H.; Conley, K.E.; Auwerx, J.; Smith, S.R.; et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J. Clin. Investig. 2013, 123, 2564–2575. [Google Scholar] [CrossRef]
- Bell, M.L.; Buvoli, M.; Leinwand, L.A. Uncoupling of Expression of an Intronic MicroRNA and Its Myosin Host Gene by Exon Skipping. Mol. Cell. Boil. 2010, 30, 1937–1945. [Google Scholar] [CrossRef]
- Rawal, S.; Nagesh, P.T.; Coffey, S.; Van Hout, I.; Galvin, I.F.; Bunton, R.W.; Davis, P.; Williams, M.J.; Katare, R. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc. Diabetol. 2019, 18, 13. [Google Scholar] [CrossRef]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M.L. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef]
- Liang, S.; He, L.; Zhao, X.; Miao, Y.; Gu, Y.; Guo, C.; Xue, Z.; Dou, W.; Hu, F.; Wu, K.; et al. MicroRNA Let-7f Inhibits Tumor Invasion and Metastasis by Targeting MYH9 in Human Gastric Cancer. PLoS ONE 2011, 6, e18409. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ning, Y.; Yi, J.; Yuan, J.; Fang, W.; Lin, Z.; Zeng, Z. MiR-6089/MYH9/beta-catenin/c-Jun negative feedback loop inhibits ovarian cancer carcinogenesis and progression. Biomed. Pharmacother. 2020, 125, 109865. [Google Scholar] [CrossRef] [PubMed]
- Senol, O.; Schaaij-Visser, T.B.M.; Erkan, E.P.; Dorfer, C.; Lewandrowski, G.; Pham, T.V.; Piersma, S.R.; Peerdeman, S.M.; Strobel, T.; Tannous, B.; et al. MiR-200a-mediated suppression of non-muscle heavy chain IIb inhibits meningioma cell migration and tumor growth in vivo. Oncogene 2015, 34, 1790–1798. [Google Scholar] [CrossRef]
- Liu, W.; Cai, T.; Li, L.; Chen, H.; Chen, R.; Zhang, M.; Zhang, W.; Zhao, L.; Xiong, H.; Qin, P.; et al. MiR-200a Regulates Nasopharyngeal Carcinoma Cell Migration and Invasion by Targeting MYH10. J. Cancer 2020, 11, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Saha, S.; Das, S.; Sen, R.; Goswami, S.; Jana, S.S.; Chakrabarti, J. MiRepress: Modelling gene expression regulation by microRNA with non-conventional binding sites. Sci. Rep. 2016, 6, 22334. [Google Scholar] [CrossRef] [PubMed]
- Alajbegovic, A.; Turczyńska, K.M.; Hien, T.T.; Cidad, P.; Swärd, K.; Hellstrand, P.; Della Corte, A.; Forte, A.; Albinsson, S. Regulation of microRNA expression in vascular smooth muscle by MRTF-A and actin polymerization. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Skoura, A.; Yu, J.; Di Lorenzo, A.; Fernandez-Hernando, C.; Offermanns, S.; Miano, J.M.; Sessa, W.C. Smooth Muscle miRNAs Are Critical for Post-Natal Regulation of Blood Pressure and Vascular Function. PLoS ONE 2011, 6, e18869. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Suarez, Y.; Skoura, A.; Offermanns, S.; Miano, J.M.; Sessa, W.C. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arter. Thromb. Vasc. Boil. 2010, 30, 1118–1126. [Google Scholar] [CrossRef]
- Boettger, T.; Beetz, N.; Kostin, S.; Schneider, J.; Krüger, M.; Hein, L.; Braun, T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J. Clin. Investig. 2009, 119, 2634–2647. [Google Scholar] [CrossRef]
- Bruno, I.G.; Karam, R.; Huang, L.; Bhardwaj, A.; Lou, C.H.; Shum, E.Y.; Song, H.-W.; Corbett, M.A.; Gifford, W.D.; Gecz, J.; et al. Identification of a MicroRNA that Activates Gene Expression by Repressing Nonsense-Mediated RNA Decay. Mol. Cell 2011, 42, 500–510. [Google Scholar] [CrossRef]
- Chiba, Y.; Tanabe, M.; Goto, K.; Sakai, H.; Misawa, M. Down-Regulation of miR-133a Contributes to Up-Regulation of RhoA in Bronchial Smooth Muscle Cells. Am. J. Respir. Crit. Care Med. 2009, 180, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.M.; Hall, I.F.; Serio, S.; Zani, S.M.; Climent, M.; Salvarani, N.; Carullo, P.; Civilini, E.; Condorelli, G.; Elia, L.; et al. MiR-128-3p Is a Novel Regulator of Vascular Smooth Muscle Cell Phenotypic Switch and Vascular Diseases. Circ. Res. 2020, 126. [Google Scholar] [CrossRef] [PubMed]
- Gheinani, A.H.; Burkhard, F.C.; Rehrauer, H.; Fournier, C.A.; Monastyrskaya, K. MicroRNA MiR-199a-5p Regulates Smooth Muscle Cell Proliferation and Morphology by Targeting WNT2 Signaling Pathway. J. Boil. Chem. 2015, 290, 7067–7086. [Google Scholar] [CrossRef] [PubMed]
- Hien, T.T.; Turczyńska, K.M.; Dahan, D.; Ekman, M.; Grossi, M.; Sjögren, J.; Nilsson, J.; Braun, T.; Boettger, T.; Vaz, E.G.; et al. Elevated Glucose Levels Promote Contractile and Cytoskeletal Gene Expression in Vascular Smooth Muscle via Rho/Protein Kinase C and Actin Polymerization. J. Boil. Chem. 2016, 291, 3552–3568. [Google Scholar] [CrossRef] [PubMed]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Nicoletti, A.; Li, Z.; Michel, J.-B. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc. Res. 2012, 95, 194–204. [Google Scholar] [CrossRef]
- Luo, J.; Liu, L.; Wu, Z.; Chen, G.; Li, E.; Luo, L.; Li, F.; Zhao, S.; Wei, A.; Zhao, Z. The effects of miRNA-145 on the phenotypic modulation of rat corpus cavernosum smooth muscle cells. Int. J. Impot. Res. 2017, 29, 229–234. [Google Scholar] [CrossRef]
- McDonald, R.A.; Hata, A.; MacLean, M.R.; Morrell, N.W.; Baker, A.H. MicroRNA and vascular remodelling in acute vascular injury and pulmonary vascular remodelling. Cardiovasc. Res. 2011, 93, 594–604. [Google Scholar] [CrossRef]
- Medjkane, S.; Pérez-Sánchez, C.; Gaggioli, C.; Sahai, E.; Treisman, R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat. Cell Biol. 2009, 11, 257–268. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Moore-Olufemi, S.D.; Olsen, A.B.; Hook-Dufresne, D.M.; Bandla, V.; Cox, C.S. Transforming Growth Factor-Beta 3 Alters Intestinal Smooth Muscle Function: Implications for Gastroschisis-Related Intestinal Dysfunction. Dig. Dis. Sci. 2015, 60, 1206–1214. [Google Scholar] [CrossRef]
- Park, C.; Hennig, G.W.; Sanders, K.M.; Cho, J.H.; Hatton, W.J.; Redelman, D.; Park, J.K.; Ward, S.M.; Miano, J.M.; Yan, W.; et al. Serum Response Factor–Dependent MicroRNAs Regulate Gastrointestinal Smooth Muscle Cell Phenotypes. Gastroenterology 2011, 141, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Yan, W.; Ward, S.M.; Hwang, S.J.; Wu, Q.; Hatton, W.J.; Park, J.; Sanders, K.M.; Ro, S. MicroRNAs Dynamically Remodel Gastrointestinal Smooth Muscle Cells. PLoS ONE 2011, 6, e18628. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.E.; Hernández, J.Á.; Benito, R.; Gutiérrez, N.C.; García, J.L.; Hernández-Sánchez, M.; de Coca, A.G. Molecular characterization of chronic lymphocytic leukemia patients with a high number of losses in 13q14. PLoS ONE 2012, 7, e48485. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Suto, W.; Kai, Y.; Chiba, Y. Mechanisms underlying the pathogenesis of hyper-contractility of bronchial smooth muscle in allergic asthma. J. Smooth Muscle Res. 2017, 53, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, M.; Lecuona, E.; Angulo, M.; Homma, T.; Rodríguez, D.A.; Gonzalez-Gonzalez, F.J.; Welch, L.C.; Amarelle, L.; Kim, S.-J.; Kaminski, N.; et al. Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling. Sci. Transl. Med. 2018, 10, eaat1662. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, X.-J.; Ding, Y.-M.; Jiang, J.-X. MiR-1181 inhibits invasion and proliferation via STAT3 in pancreatic cancer. World J. Gastroenterol. 2017, 23, 1594–1601. [Google Scholar] [CrossRef]
- Yu, B.; Qian, T.; Wang, Y.; Zhou, S.; Ding, G.; Ding, F.; Gu, X. MiR-182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res. 2012, 40, 10356–10365. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Kang, K.; Chen, J.; Wu, Z.; Huang, J.; Lu, W.; Chen, Y.; Zhang, J.; Wang, Z.; et al. MicroRNA-223 Attenuates Hypoxia-induced Vascular Remodeling by Targeting RhoB/MLC2 in Pulmonary Arterial Smooth Muscle Cells. Sci. Rep. 2016, 6, 24900. [Google Scholar] [CrossRef]
- Bender, M.; Eckly, A.; Hartwig, J.H.; Elvers, M.; Pleines, I.; Gupta, S.; Krohne, G.; Jeanclos, E.; Gohla, A.; Gurniak, C.; et al. ADF/n-cofilin–dependent actin turnover determines platelet formation and sizing. Blood 2010, 116, 1767–1775. [Google Scholar] [CrossRef]
- Chapnik, E.; Rivkin, N.; Mildner, A.; Beck, G.; Pasvolsky, R.; Metzl-Raz, E.; Birger, Y.; Amir, G.; Tirosh, I.; Porat, Z.; et al. MiR-142 orchestrates a network of actin cytoskeleton regulators during megakaryopoiesis. Elife 2014, 3, e01964. [Google Scholar] [CrossRef] [PubMed]
- Gauwerky, C.E.; Huebner, K.; Isobe, M.; Nowell, P.C.; Croce, C.M. Activation of MYC in a masked t(8;17) translocation results in an aggressive B-cell leukemia. Proc. Natl. Acad. Sci. USA 1989, 86, 8867–8871. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-J.; Chau, J.; Ebert, P.J.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. MiR-181a Is an Intrinsic Modulator of T Cell Sensitivity and Selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef]
- Mayoral, R.J.; Dehò, L.; Rusca, N.; Bartonicek, N.; Saini, H.; Enright, A.J.; Monticelli, S. MiR-221 Influences Effector Functions and Actin Cytoskeleton in Mast Cells. PLoS ONE 2011, 6, e26133. [Google Scholar] [CrossRef] [PubMed]
- Mildner, A.; Chapnik, E.; Manor, O.; Yona, S.; Kim, K.-W.; Aychek, T.; Varol, D.; Beck, G.; Itzhaki, Z.B.; Feldmesser, E.; et al. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood 2013, 121, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Wang, X.; Zhang, X.; Zhu, S.; Sun, D.; Ka, W.; Sung, L.A.; Yao, W. Fluid Shear Stress Upregulates E-Tmod41 via miR-23b-3p and Contributes to F-Actin Cytoskeleton Remodeling during Erythropoiesis. PLoS ONE 2015, 10, e0136607. [Google Scholar] [CrossRef] [PubMed]
- Undi, R.B.; Kandi, R.; Gutti, R.K. MicroRNAs as Haematopoiesis Regulators. Adv. Hematol. 2013, 2013, 695754. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Boil. 2007, 17, 428–437. [Google Scholar] [CrossRef]
- Li, M.; Armelloni, S.; Zennaro, C.; Wei, C.; Corbelli, A.; Ikehata, M.; Berra, S.; Giardino, L.; Mattinzoli, D.; Watanabe, S.; et al. BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J. Pathol. 2015, 235, 731–744. [Google Scholar] [CrossRef]
- John, A.A.; Prakash, R.; Kureel, J.; Singh, D. Identification of novel microRNA inhibiting actin cytoskeletal rearrangement thereby suppressing osteoblast differentiation. J. Mol. Med. (Berl.) 2018, 96, 427–444. [Google Scholar] [CrossRef]
- Okamoto, H.; Matsumi, Y.; Hoshikawa, Y.; Takubo, K.; Ryoke, K.; Shiota, G. Involvement of MicroRNAs in Regulation of Osteoblastic Differentiation in Mouse Induced Pluripotent Stem Cells. PLoS ONE 2012, 7, e43800. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, Y.; Fukuhara, S.; Ando, K.; Matsuda, M.; Mochizuki, N. Cdc42 Mediates Bmp-Induced Sprouting Angiogenesis through Fmnl3-Driven Assembly of Endothelial Filopodia in Zebrafish. Dev. Cell 2015, 32, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, T.; Frank, D.; Kuwahara, K.; Bezprozvannaya, S.; Pipes, G.C.T.; Bassel-Duby, R.; Richardson, J.A.; Katus, H.A.; Olson, E.N.; Frey, N. Two Novel Members of the ABLIM Protein Family, ABLIM-2 and -3, Associate with STARS and Directly Bind F-actin. J. Boil. Chem. 2007, 282, 8393–8403. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Shen, Y.; Zhu, L.; Xu, Y.; Zhou, Y.; Wu, Z.; Li, Y.; Yan, X.; Zhu, X. MiR-129-3p controls cilia assembly by regulating CP110 and actin dynamics. Nat. Cell Biol. 2012, 14, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, B.; Adamiok, A.; Mercey, O.; Revinski, D.R.; Zaragosi, L.E.; Pasini, A.; Marcet, B. MiR-34/449 control apical actin network formation during multiciliogenesis through small GTPase pathways. Nat. Commun. 2015, 6, 8386. [Google Scholar] [CrossRef]
- Dawe, H.R.; Farr, H.; Gull, K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J. Cell Sci. 2007, 120, 7–15. [Google Scholar] [CrossRef]
- Mercey, O.; Kodjabachian, L.; Barbry, P.; Marcet, B. MicroRNAs as key regulators of GTPase-mediated apical actin reorganization in multiciliated epithelia. Small GTPases 2016, 7, 54–58. [Google Scholar] [CrossRef]
- Bockhorn, J.; Yee, K.; Chang, Y.-F.; Prat, A.; Huo, D.; Nwachukwu, C.; Dalton, R.; Huang, S.; Swanson, K.E.; Perou, C.M.; et al. MicroRNA-30c targets cytoskeleton genes involved in breast cancer cell invasion. Breast Cancer Res. Treat. 2013, 137, 373–382. [Google Scholar] [CrossRef]
- Fils-Aimé, N.; Dai, M.; Guo, J.; El-Mousawi, M.; Kahramangil, B.; Neel, J.-C.; Lebrun, J.-J. MicroRNA-584 and the Protein Phosphatase and Actin Regulator 1 (PHACTR1), a New Signaling Route through Which Transforming Growth Factor-β Mediates the Migration and Actin Dynamics of Breast Cancer Cells. J. Boil. Chem. 2013, 288, 11807–11823. [Google Scholar] [CrossRef]
- Fu, Y.-T.; Zhang, D.-Q.; Zhou, L.; Li, S.-J.; Sun, H.; Liu, X.-L.; Zheng, H.-B. Has-MiR-196a-2 is up-regulated and acts as an independent unfavorable prognostic factor in thyroid carcinoma. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 2707–2714. [Google Scholar] [PubMed]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes. Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Howe, E.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13, R45. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Guo, J.; Zheng, L.; Li, C.; Zheng, T.M.; Tanyi, J.L.; Liang, S.; Benedetto, C.; Mitidieri, M.; Katsaros, D.; et al. The Heterochronic microRNA let-7 Inhibits Cell Motility by Regulating the Genes in the Actin Cytoskeleton Pathway in Breast Cancer. Mol. Cancer Res. 2013, 11, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, X.; Kolokythas, A.; Yu, J.; Wang, A.; Heidbreder, C.E.; Shi, F.; Zhou, X. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138-mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int. J. Cancer 2010, 127, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Jurmeister, S.; Baumann, M.; Balwierz, A.; Keklikoglou, I.; Ward, A.; Uhlmann, S.; Zhang, J.D.; Wiemann, S.; Şahin, Ö. MicroRNA-200c represses migration and invasion of breast cancer cells by targeting actin-regulatory proteins FHOD1 and PPM1F. Mol. Cell. Boil. 2012, 32, 633–651. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.-G.; Tan, E.-J.; Manser, E.; Lim, L. The p21-activated kinase PAK is negatively regulated by POPX1 and POPX2, a pair of serine/threonine phosphatases of the PP2C family. Curr. Boil. 2002, 12, 317–321. [Google Scholar] [CrossRef]
- Kong, D.; Li, Y.; Wang, Z.; Banerjee, S.; Ahmad, A.; Kim, H.-R.C.; Sarkar, F.H. MiR-200 Regulates PDGF-D-Mediated Epithelial-Mesenchymal Transition, Adhesion, and Invasion of Prostate Cancer Cells. Stem Cells 2009, 27, 1712–1721. [Google Scholar] [CrossRef]
- Yang, S.; Mo, P. The store-operated calcium channels in cancer metastasis from cell migration, invasion to metastatic colonization. Front. Biosci. (Landmark Ed.) 2018, 23, 1241–1256. [Google Scholar] [CrossRef]
- Moore, L.D.; Isayeva, T.; Siegal, G.P.; Ponnazhagan, S. Silencing of transforming growth factor-beta1 in situ by RNA interference for breast cancer: Implications for proliferation and migration in vitro and metastasis in vivo. Clin. Cancer Res. 2008, 14, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.S.; Maeda, L.S.; Ioannidis, J.P. Clinical Outcome Prediction by MicroRNAs in Human Cancer: A Systematic Review. J. Natl. Cancer Inst. 2012, 104, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, L.; Stebbing, J.; Braga, V.M.; Frampton, A.E.; Jacob, J.; Buluwela, L.; Jiao, L.R.; Periyasamy, M.; Madsen, C.D.; Caley, M.P.; et al. MiR-23b regulates cytoskeletal remodeling, motility and metastasis by directly targeting multiple transcripts. Nucleic Acids Res. 2013, 41, 5400–5412. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.-B.; Xue, L.; Ma, A.-H.; Tepper, C.G.; Gandour-Edwards, R.; Kung, H.-J.; White, R.W.D. Tumor suppressive miR-124 targets androgen receptor and inhibits proliferation of prostate cancer cells. Oncogene 2013, 32, 4130–4138. [Google Scholar] [CrossRef] [PubMed]
- Sossey-Alaoui, K.; Bialkowska, K.; Plow, E.F. The miR200 Family of MicroRNAs Regulates WAVE3-dependent Cancer Cell Invasion. J. Boil. Chem. 2009, 284, 33019–33029. [Google Scholar] [CrossRef] [PubMed]
- Sossey-Alaoui, K.; Downs-Kelly, E.; Das, M.; Izem, L.; Tubbs, R.; Plow, E.F. WAVE3, an actin remodeling protein, is regulated by the metastasis suppressor microRNA, miR-31, during the invasion-metastasis cascade. Int. J. Cancer 2011, 129, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, V.; Gengenbacher, N.; Stemmler, M.P.; Kleemann, J.A.; Brabletz, T.; Brabletz, S. The ZEB1/miR-200c feedback loop regulates invasion via actin interacting proteins MYLK and TKS5. Oncotarget 2015, 6, 27083–27096. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, H.; Wei, Z.; Jia, H.; Liu, Y.; Liu, J. Systematic analysis of the molecular mechanism of microRNA-124 in hepatoblastoma cells. Oncol. Lett. 2017, 14, 7161–7170. [Google Scholar] [CrossRef]
- Wong, C.C.-L.; Wong, C.-M.; Tung, E.K.; Au, S.L.; Lee, J.M.; Poon, R.T.; Man, K.; Ng, I.O.-L. The MicroRNA miR-139 Suppresses Metastasis and Progression of Hepatocellular Carcinoma by Down-regulating Rho-Kinase 2. Gastroenterology 2011, 140, 322–331. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Li, Z.; Chen, R.; Guo, N.; Zhou, J.; Zhou, Q.; Lin, Q.; Cheng, D.; Liao, Q.; Zheng, L.; et al. Epigenetic regulation of miR-124 by Hepatitis C Virus core protein promotes migration and invasion of intrahepatic cholangiocarcinoma cells by targeting SMYD3. FEBS Lett. 2012, 586, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Liao, Y.; Cai, M.-Y.; Liu, Y.-H.; Liu, T.-H.; Chen, S.-P.; Bian, X.-W.; Guan, X.-Y.; Lin, M.C.-M.; Zeng, Y.-X.; et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut 2012, 61, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Song, F.; Zhang, L.; Yang, D.; Ji, P.; Wang, Y.; Zhang, W. Genetic variants at the miR-124 binding site on the cytoskeleton-organizing IQGAP1 gene confer differential predisposition to breast cancer. Int. J. Oncol. 2011, 38, 1153–1161. [Google Scholar] [CrossRef]
- Bettencourt, P.; Marion, S.; Pires, D.; Santos, L.F.; Lastrucci, C.; Carmo, N.; Blake, J.; Beneš, V.; Griffiths, G.; Neyrolles, O.; et al. Actin-binding protein regulation by microRNAs as a novel microbial strategy to modulate phagocytosis by host cells: The case of N-Wasp and miR-142-3p. Front. Cell Infect. Microbiol. 2013, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Vergne, I.; Chua, J.; Master, S.; Singh, S.B.; Fazio, J.; Kyei, G. Endosomal membrane traffic: Convergence point targeted by Mycobacterium tuberculosis and HIV. Cell. Microbiol. 2004, 6, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Lahoute, C.; Herbin, O.; Mallat, Z.; Tedgui, A. Adaptive immunity in atherosclerosis: Mechanisms and future therapeutic targets. Nat. Rev. Cardiol. 2011, 8, 348–358. [Google Scholar] [CrossRef]
- Liu, J.; Li, W.; Wang, S.; Wu, Y.; Li, Z.; Wang, W.; Liu, R.; Ou, J.; Zhang, C.; Wang, S. MiR-142-3p Attenuates the Migration of CD4+ T Cells through Regulating Actin Cytoskeleton via RAC1 and ROCK2 in Arteriosclerosis Obliterans. PLoS ONE 2014, 9, e95514. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kim, S.E.; Yano, H.; Matsumoto, G.; Ohuchida, R.; Ishikura, Y.; Araki, M.; Araki, K.; Park, S.; Komatsu, T.; et al. MiR-142 Is Required for Staphylococcus aureus Clearance at Skin Wound Sites via Small GTPase-Mediated Regulation of the Neutrophil Actin Cytoskeleton. J. Investig. Dermatol. 2017, 137, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Caré, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Torrini, C.; Cubero, R.J.; Dirkx, E.; Braga, L.; Ali, H.; Prosdocimo, G.; Gutierrez, M.I.; Collesi, C.; Licastro, D.; Zentilin, L.; et al. Common Regulatory Pathways Mediate Activity of MicroRNAs Inducing Cardiomyocyte Proliferation. Cell Rep. 2019, 27, 2759–2771.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Si, M.-L.; Wu, H.; Mo, Y.-Y. MicroRNA-21 Targets the Tumor Suppressor Gene Tropomyosin 1 (TPM1). J. Boil. Chem. 2007, 282, 14328–14336. [Google Scholar] [CrossRef] [PubMed]
- Riches, K.; Alshanwani, A.R.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Wood, I.C.; Porter, K.E. Elevated expression levels of miR-143/5 in saphenous vein smooth muscle cells from patients with Type 2 diabetes drive persistent changes in phenotype and function. J. Mol. Cell. Cardiol. 2014, 74, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, F.; Xin, M.; Li, G.; Luna, C.; Li, G.; Gonzalez, P. Regulation of intraocular pressure by microRNA cluster miR-143/145. Sci. Rep. 2017, 7, 915. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.P.; Ioannou, N.; Ramsay, A.; Darling, D.; Gäken, J.; Mufti, G.J. MiR-181c -BRK1 axis plays a key role in actin cytoskeleton-dependent T cell function. J. Leukoc. Boil. 2018, 103, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.; Li, C.; Zhao, D.; Li, S.; Ma, L.; Cui, Y.; Wei, X.; Zhao, Y.; Gao, Y. MicroRNA-92a promotes vascular smooth muscle cell proliferation and migration through the ROCK/MLCK signalling pathway. J. Cell. Mol. Med. 2019, 23, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.; Raff, J.W. Centrioles, Centrosomes, and Cilia in Health and Disease. Cell 2009, 139, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Kalluri, R. Molecular pathways: microRNAs as cancer therapeutics. Clin. Cancer Res. 2012, 18, 4234–4239. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Han, H.D.; Lopez-Berestein, G.; Sood, A.K. MicroRNA therapeutics: Principles, expectations, and challenges. Chin. J. Cancer. 2011, 30, 368–370. [Google Scholar] [CrossRef]
- Van Rooij, E.; Olson, E.N. MicroRNAs: Powerful new regulators of heart disease and provocative therapeutic targets. J. Clin. Investig. 2007, 117, 2369–2376. [Google Scholar] [CrossRef]
- Balacescu, O.; Visan, S.; Baldasici, O.; Balacescu, L.; Vlad, C.; Achimas-Cadariu, P. MiRNA-based therapeutics in oncology, realities, and challenges. In Antisense Therapy; Sharad, S., Kapur, S., Eds.; Intech Open: London, UK, 2019. [Google Scholar]
- Bansal, P.; Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| miRNA | Physiology | Regulation | Regulated Transcript | Citation |
|---|---|---|---|---|
| miR-let-7g | smooth muscle contraction | downregulation | α-SMA | [67] |
| miR-19a | normal smooth muscle contractility | upregulation | α-SMA | [68,69,70] |
| miR-21 | smooth muscle contraction | upregulation | α-SMA | [71] |
| miR-23b | vascular smooth muscle contraction | upregulation | α-SMA | [72] |
| miR-25a-5p | vascular smooth muscle contraction | upregulation | α-SMA | [73] |
| miR-26a | vascular smooth muscle contraction | downregulation | α-SMA | [74] |
| miR-27a | primary vascular smooth muscle cells | downregulation | α-SMA | [75,76] |
| miR-34/34b-5p | normal cytoskeletal function | upregulation | α-actin | [66] |
| miR-143/145 | vascular smooth muscle | upregulation | α-actin | [77,78] |
| miR-182 | vascular smooth muscle | upregulation | α-SMA | [79] |
| miR-330 | vascular smooth muscle contraction | downregulation | α-SMA | [80] |
| miR-548-f | normal smooth muscle contractility | upregulation | α-SMA | [81] |
| miR-663 | vascular smooth muscle contraction | upregulation | α-SMA | [82] |
| miRNA | Physiological Effect | Regulation | Regulated Transcript | Citation |
|---|---|---|---|---|
| miR-1 | myoblast differentiation | upregulation | MYH (MyHC) | [84] |
| miR-23b | vascular smooth muscle contraction | upregulation | MYH11 | [72] |
| miR-26a | vascular smooth muscle contraction | downregulation | MYH11 | [74] |
| miR-27a | heart | upregulation | MYH7 (MyHC-) | [89] |
| miR-30e | skeletal muscle fiber type formation | upregulation | MYH2, MYH1, MYH4 (MyHC-IIA,IIX,IIB) | [86] |
| miR-125a-5p | vascular smooth muscle contraction | upregulation | MYH11 | [73] |
| miR-133 | myoblast differentiation | downregulation | MYH (MyHC) | [84] |
| miR-133a | skeletal muscle fiber type switch | upregulation | MYH4 (MyHC-IIB) | [87] |
| miR-133a1 | skeletal muscle fiber type formation | downregulation | MYH7 (MyHC-I) | [90] |
| miR-143-3p | myoblast differentiation | downregulation | MYH (MyHC) | [85] |
| miR-143-5p | melanin secretion | downregulation | MYO5A (myosin 5A) | [91] |
| miR-145 | melanosome transport | downregulation | MYO5A (myosin 5A) | [92] |
| miR-195 | cardiac remodeling | upregulation | MYH7 (MyHC-β) | [93] |
| miR-199a | cardiomyocyte contraction | downregulation | MYH6 (MyHC-α) | [94] |
| miR-206 | myoblast differentiation | upregulation | MYH (MyHC) | [83] |
| miR-208a | cardiac remodeling | upregulation | MYH7 (MyHC-β) | [95] |
| miR-208b | skeletal muscle fiber type and energy metabolism | upregulation | MYH7 (MyHC-I) | [96] |
| miR-330 | vascular smooth muscle contraction | downregulation | MYH11 | [80] |
| miR-499 | striated muscle | upregulation | MYH7b/MYH14 (MyHC-14) | [97] |
| miR-501 | skeletal muscle regeneration | upregulation | MYH3 (MyHC-emb) | [88] |
| miR-663 | vascular smooth muscle contraction | upregulation | MYH11 MYL9 | [82] |
| miRNA | Function/Pathology | Regulation | Regulated Transcript | Citation |
|---|---|---|---|---|
| miR-1 | cardiomyocyte hypertrophy | downregulation | Rho A, CDC42 | [179] |
| miR-let-7 | breast cancer | downregulation | PAK1, DIAPH2, RDX,ITGB8 | [155] |
| miR-let-7g | smooth muscle contractility | upregulation | calponin | [67] |
| miR-19a | smooth muscle contractility | upregulation | SM22α | [68,69] |
| miR-21 | smooth muscle contractility | downregulation | SM22α tropomyosin | [71,182] |
| miR-23b | breast cancer vascular smooth muscle | downregulation upregulation | PAK2, LIMK2 SM22α | [163] |
| miR-23b-3p | erythropoiesis | downregulation | E-Tmod41 | [136] |
| miR-26a | vascular smooth muscle contractility | downregulation | SM22α | [74] |
| miR-30c | breast cancer | downregulation | twinfilin 1, vimentin | [149] |
| miR-31 | tumorigenesis, metastasis formation | downregulation | RhoA, WAVE3 | [165,166] |
| miR-34/449 | ciliation | downregulation | R-Ras CP110 | [146,148] |
| miR-124 | liver cancer glioblastoma | downregulation | ROCK2, CRL, WARP, cofilin ROCK1 | [168] [65] |
| miR-128 | vascular smooth muscle contractility | upregulation | KLF4 | [112] |
| miR-129-3p | multiciliogenesis | [144,145] | ||
| miR-132 | podocyte formation | downregulation | LIMK1 | [139] |
| miR-133 | cardiomyocyte hypertrophy | downregulation | RhoA, CDC42 | [179] |
| miR-133a | airway smooth muscle | upregulation | RhoA, Cdc42 | [179] |
| miR-134 | podocyte | upregulation | P250GAP | [139] |
| miR-138 | tongue squamous cell carcinoma | upregulation | Rho C, ROCK2 | [156] |
| miR-139 | hepatocellular carcinoma | downregulation | ROCK2 | [169] |
| miR-142 | megakaryopoiesis | Needs to be active for normal | Cfl-2, Wasl, twinfilin, integrin α | [131] |
| miR-142-3p | phagocytosis T cell activation | upregulation downregulation | WASL, Cfl-2 Rac, ROCK2 | [174] [178] [177] |
| miR-142-5p | T cell activation | upregulation | RhoA | [178] |
| miR-143/145 | vascular smooth muscle atherosclerosis glaucoma | upregulation | MRTF-B, actin, Cfl-2, KLF5, MYOCD, ROCK1 Myosin VI, MYLK | [77,78] [183] [184] |
| miR-181c | T cell activation | downregulation | BRK1 | [185] |
| miR-182 | vascular smooth muscle contractility | upregulation | SM22α, calponin | [79] |
| Has-miR-196a-2 | thyroid cancer | upregulation | WNT | [151] |
| miR-200c | breast cancer tumorigenesis | down | ERM, fibronectin FHOD1, PPM1F TKS5, MYLK WAVE3 ZEB | [154,157,167] [153,165] |
| miR-221 | mast cell homeostasis and stimulation | upregulation | p27Kip1, CD23 | [134] |
| miR-223 | vascular smooth muscle contractility | downregulation | RhoB | [78] |
| miR-330 | vascular smooth muscle contractility | downregulation | calbindin, smoothelin, renin | [80] |
| miR-584 | breast cancer | downregulation | PHACTR | [150] |
| miR-663 | vascular smooth muscle contractility | upregulation | SM22α | [82] |
| miR-1181 | pancreatic cancer | downregulation | β-tubulin STAT3 | [127] |
| miR-1187 | osteoblast | downregulation | BMPR-II, ArhGEF-9 | [140] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uray, K.; Major, E.; Lontay, B. MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton. Cells 2020, 9, 1649. https://doi.org/10.3390/cells9071649
Uray K, Major E, Lontay B. MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton. Cells. 2020; 9(7):1649. https://doi.org/10.3390/cells9071649
Chicago/Turabian StyleUray, Karen, Evelin Major, and Beata Lontay. 2020. "MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton" Cells 9, no. 7: 1649. https://doi.org/10.3390/cells9071649
APA StyleUray, K., Major, E., & Lontay, B. (2020). MicroRNA Regulatory Pathways in the Control of the Actin–Myosin Cytoskeleton. Cells, 9(7), 1649. https://doi.org/10.3390/cells9071649