Calcium-Sensing Receptor and Regulation of WNK Kinases in the Kidney

 , , , and
, , , and 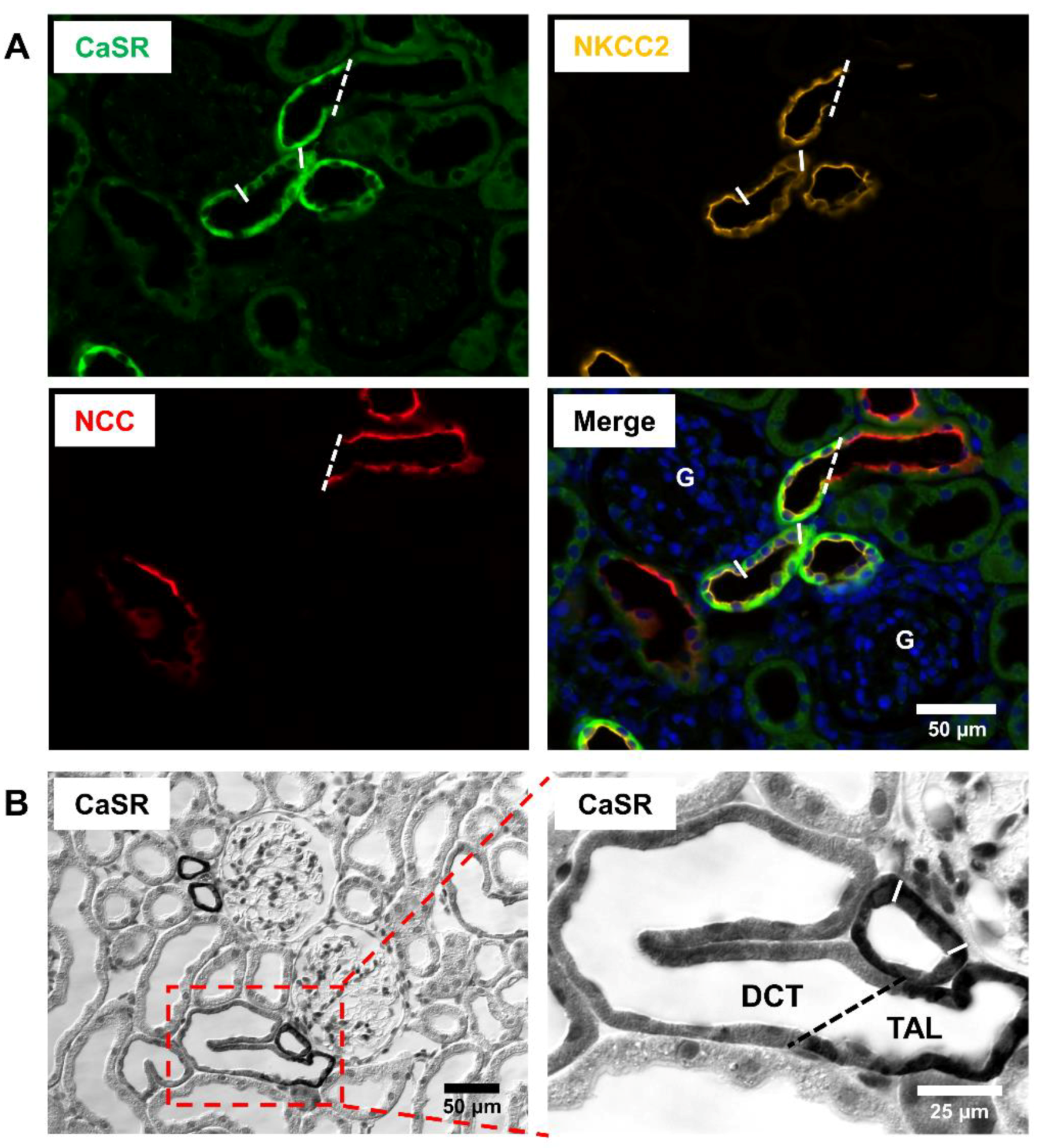{kind=link}
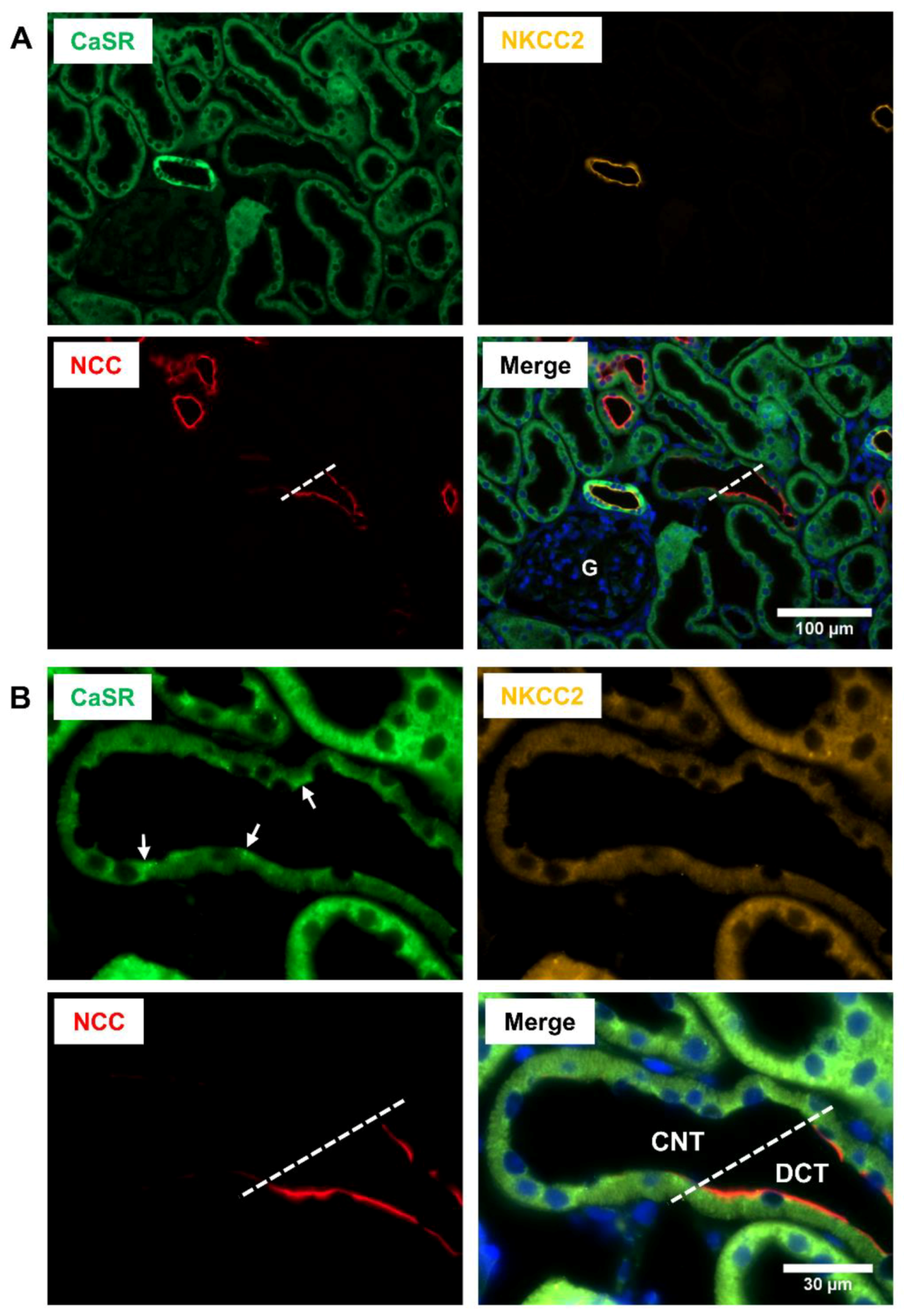{kind=link}
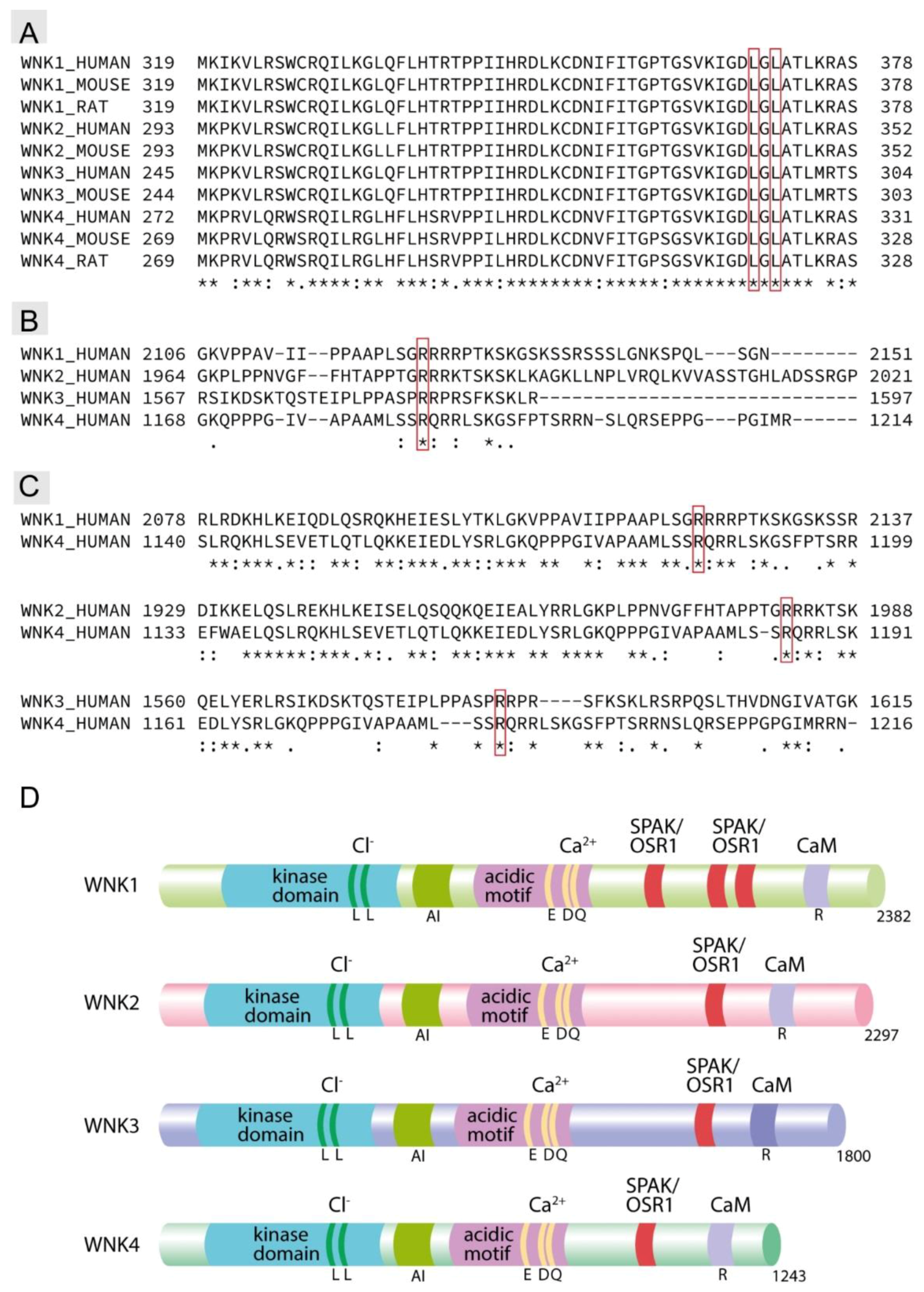{kind=link}
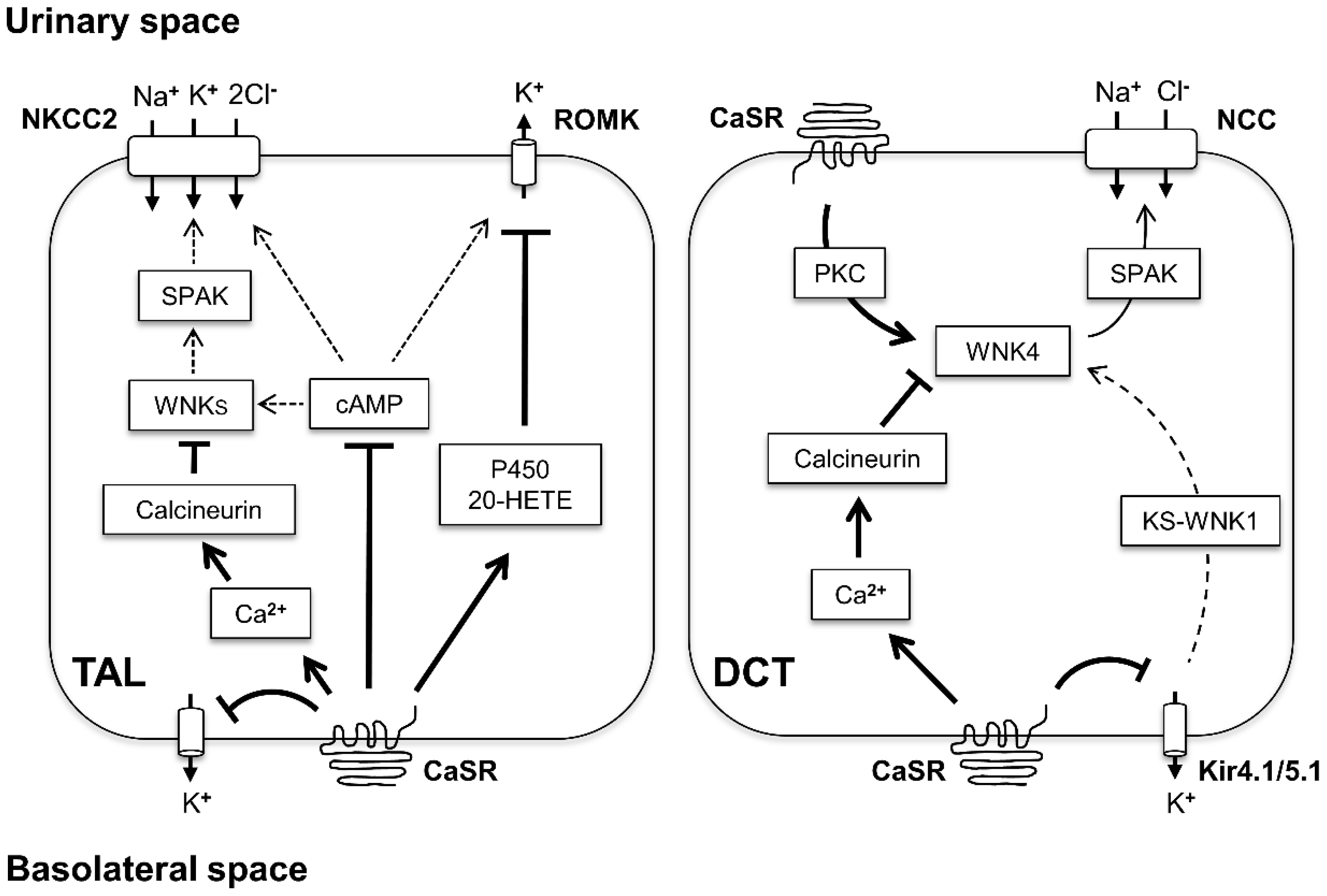{kind=link}
Abstract
1. Introduction
2. Renal Distribution of CaSR
3. CaSR Function in TAL
3.1. CaSR Inhibits the Transcellular NaCl Reabsorption
3.2. CaSR Inhibits the Paracellular Ca2+ and Mg2+ Reabsorption
4. CaSR Function in JGA
5. CaSR Function in DCT
6. CaSR Function in CNT and CD
7. Translational Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lee, K.; Brown, D.; Ureña, P.; Ardaillou, N.; Ardaillou, R.; Deeds, J.; Segre, G.V. Localization of parathyroid hormone/parathyroid hormone-related peptide receptor mRNA in kidney. Am. J. Physiol. 1996, 270, F186–F191. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Lee, W.S.; Lee, K.; Segre, G.V.; Brown, E.M.; Hebert, S.C. Localization of the extracellular Ca(2+)-sensing receptor and PTH/PTHrP receptor in rat kidney. Am. J. Physiol. 1996, 271, F951–F956. [Google Scholar] [CrossRef]
- Melamed, M.L.; Thadhani, R.I. Vitamin D therapy in chronic kidney disease and end stage renal disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Sexton, P.M.; Adam, W.R.; Moseley, J.M.; Martin, T.J.; Mendelsohn, F.A.O. Localization and characterization of renal calcitonin receptors by in vitro autoradiography. Kidney Int. 1987, 32, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Graca, J.A.Z.; Schepelmann, M.; Brennan, S.C.; Reens, J.; Chang, W.; Yan, P.; Toka, H.; Riccardi, D.; Price, S.A. Comparative expression of the extracellular calcium-sensing receptor in the mouse, rat, and human kidney. Am. J. Physiol. Ren. Physiol. 2016, 310, F518–F533. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Brown, E.M. Physiology and pathophysiology of the calcium-sensing receptor in the kidney. Am. J. Physiol. Ren. Physiol. 2010, 298, F485–F499. [Google Scholar] [CrossRef]
- Riccardi, D.; Valenti, G. Localization and function of the renal calcium-sensing receptor. Nat. Rev. Nephrol. 2016, 12, 414–425. [Google Scholar] [CrossRef]
- Wu, L.-G.; Hamid, E.; Shin, W.; Chiang, H.-C. Exocytosis and endocytosis: Modes, functions, and coupling mechanisms. Annu. Rev. Physiol. 2014, 76, 301–331. [Google Scholar] [CrossRef]
- Topala, C.N.; Schoeber, J.P.H.; Searchfield, L.E.; Riccardi, D.; Hoenderop, J.G.J.; Bindels, R.J.M. Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium 2009, 45, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Dimke, H.; Desai, P.; Borovac, J.; Lau, A.; Pan, W.; Alexander, R.T. Activation of the Ca(2+)-sensing receptor increases renal claudin-14 expression and urinary Ca(2+) excretion. Am. J. Physiol. Ren. Physiol. 2013, 304, F761–F769. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Hou, J. Claudin-14 Underlies Ca++-Sensing Receptor-Mediated Ca++ Metabolism via NFAT-microRNA-Based Mechanisms. J. Am. Soc. Nephrol. 2014, 25, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Gerbino, A.; Hendy, G.N.; Torretta, S.; Rizzo, F.; Debellis, L.; Procino, G.; Svelto, M. NKCC2 activity is inhibited by the Bartter’s syndrome type 5 gain-of-function CaR-A843E mutant in renal cells. Biol. Cell 2015, 107, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Bazúa-Valenti, S.; Rojas-Vega, L.; Castañeda-Bueno, M.; Barrera-Chimal, J.; Bautista, R.; Cervantes-Pérez, L.G.; Vázquez, N.; Plata, C.; Murillo-de-Ozores, A.R.; González-Mariscal, L.; et al. The calcium-sensing receptor increases activity of the renal NCC through the WNK4-SPAK pathway. J. Am. Soc. Nephrol. 2018, 29, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Smajilovic, S.; Tfelt-Hansen, J. Novel role of the calcium-sensing receptor in blood pressure modulation. Hypertension 2008, 52, 994–1000. [Google Scholar] [CrossRef]
- Kusano, E.; Murayama, N.; Werness, J.L.; Christensen, S.; Homma, S.; Yusufi, A.N.; Dousa, T.P. Effects of calcium on the vasopressin-sensitive cAMP metabolism in medullary tubules. Am. J. Physiol. Ren. Physiol. 1985, 249, F956–F966. [Google Scholar] [CrossRef]
- Reilly, R.F.; Ellison, D.H. Mammalian distal tubule: Physiology, pathophysiology, and molecular anatomy. Physiol. Rev. 2000, 80, 277–313. [Google Scholar] [CrossRef]
- Madsen, K.M.; Tisher, C.C. Structural-functional relationships along the distal nephron. Am. J. Physiol. 1986, 250, F1–F15. [Google Scholar] [CrossRef]
- Chen, L.; Clark, J.Z.; Nelson, J.W.; Kaissling, B.; Ellison, D.H.; Knepper, M.A. Renal-Tubule epithelial cell nomenclature for single-cell RNA-sequencing studies. J. Am. Soc. Nephrol. 2019. [Google Scholar] [CrossRef]
- Gamba, G.; Friedman, P.A. Thick ascending limb: The Na+:K+:2Cl− co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflüg. Arch. Eur. J. Physiol. 2009, 458, 61–76. [Google Scholar] [CrossRef]
- Mount, D.B. Thick ascending limb of the loop of henle. Clin. J. Am. Soc. Nephrol. 2014, 9, 1974–1986. [Google Scholar] [CrossRef]
- Mutig, K.; Borowski, T.; Boldt, C.; Borschewski, A.; Paliege, A.; Popova, E.; Bader, M.; Bachmann, S. Demonstration of the functional impact of vasopressin signaling in the thick ascending limb by a targeted transgenic rat approach. Am. J. Physiol. Ren. Physiol. 2016, 311, F411–F423. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Regulation of the renal Na+-Cl− cotransporter by phosphorylation and ubiquitylation. Am. J. Physiol. Ren. Physiol. 2012, 303, F1573–F1583. [Google Scholar] [CrossRef] [PubMed]
- Terker, A.S.; Zhang, C.; McCormick, J.A.; Lazelle, R.A.; Zhang, C.; Meermeier, N.P.; Siler, D.A.; Park, H.J.; Fu, Y.; Cohen, D.M.; et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab. 2015, 21, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Ostrosky-Frid, M.; Castañeda-Bueno, M.; Gamba, G. Regulation of the renal NaCl cotransporter by the WNK/SPAK pathway: Lessons learned from genetically altered animals. Am. J. Physiol. Ren. Physiol. 2019, 316, F146–F158. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.T.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef]
- Simon, D.B.; Karet, F.E.; Hamdan, J.M.; DiPietro, A.; Sanjad, S.A.; Lifton, R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat. Genet. 1996, 13, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef]
- Watanabe, S.; Fukumoto, S.; Chang, H.; Takeuchi, Y.; Hasegawa, Y.; Okazaki, R.; Chikatsu, N.; Fujita, T. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet Lond. Engl. 2002, 360, 692–694. [Google Scholar] [CrossRef]
- Fujita, T.; Ando, K.; Sato, Y.; Yamashita, K.; Nomura, M.; Fukui, T. Independent roles of prostaglandins and the renin-angiotensin system in abnormal vascular reactivity in Bartter’s syndrome. Am. J. Med. 1982, 73, 71–76. [Google Scholar] [CrossRef]
- Toka, H.R.; Al-Romaih, K.; Koshy, J.M.; DiBartolo, S.; Kos, C.H.; Quinn, S.J.; Curhan, G.C.; Mount, D.B.; Brown, E.M.; Pollak, M.R. Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J. Am. Soc. Nephrol. 2012, 23, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Zacchia, M.; Capasso, G. The role of the kidney in salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Majid, D.S.A.; Prieto, M.C.; Navar, L.G. Salt-sensitive hypertension: Perspectives on intrarenal mechanisms. Curr. Hypertens. Rev. 2015, 11, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Hadchouel, J.; Ellison, D.H.; Gamba, G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 Kinases. Annu. Rev. Physiol. 2016, 78, 367–389. [Google Scholar] [CrossRef]
- Wilson, F.H.; Disse-Nicodème, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef]
- Shibata, S.; Zhang, J.; Puthumana, J.; Stone, K.L.; Lifton, R.P. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc. Natl. Acad. Sci. USA 2013, 110, 7838–7843. [Google Scholar] [CrossRef] [PubMed]
- Mu, S.; Shimosawa, T.; Ogura, S.; Wang, H.; Uetake, Y.; Kawakami-Mori, F.; Marumo, T.; Yatomi, Y.; Geller, D.S.; Tanaka, H.; et al. Epigenetic modulation of the renal β-adrenergic–WNK4 pathway in salt-sensitive hypertension. Nat. Med. 2011, 17, 573–580. [Google Scholar] [CrossRef] [PubMed]
- San-Cristobal, P.; Pacheco-Alvarez, D.; Richardson, C.; Ring, A.M.; Vazquez, N.; Rafiqi, F.H.; Chari, D.; Kahle, K.T.; Leng, Q.; Bobadilla, N.A.; et al. Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4384–4389. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Walsh, S.B.; McCormick, J.A.; Fürstenberg, A.; Yang, C.-L.; Roeschel, T.; Paliege, A.; Howie, A.J.; Conley, J.; Bachmann, S.; et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat. Med. 2011, 17, 1304–1309. [Google Scholar] [CrossRef]
- Borschewski, A.; Himmerkus, N.; Boldt, C.; Blankenstein, K.I.; McCormick, J.A.; Lazelle, R.; Willnow, T.E.; Jankowski, V.; Plain, A.; Bleich, M.; et al. Calcineurin and sorting-related receptor with a-type repeats interact to regulate the renal Na+-K+-2Cl− Cotransporter. J. Am. Soc. Nephrol. 2016, 27, 107–119. [Google Scholar] [CrossRef]
- Blankenstein, K.I.; Borschewski, A.; Labes, R.; Paliege, A.; Boldt, C.; McCormick, J.A.; Ellison, D.H.; Bader, M.; Bachmann, S.; Mutig, K. Calcineurin inhibitor cyclosporine a activates renal Na-(K)-Cl Cotransporters via Local and Systemic Mechanisms. Am. J. Physiol. Ren. Physiol. 2016, 312, F489–F501. [Google Scholar] [CrossRef]
- Ishizawa, K.; Wang, Q.; Li, J.; Yamazaki, O.; Tamura, Y.; Fujigaki, Y.; Uchida, S.; Lifton, R.P.; Shibata, S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc. Natl. Acad. Sci. USA 2019, 116, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Hatton, D.C.; McCarron, D.A. Dietary calcium and blood pressure in experimental models of hypertension. A review. Hypertension 1994, 23, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sano, H.; Furuta, Y.; Yamanishi, J.; Omatsu, T.; Ito, Y.; Fukuzaki, H. Calcium supplementation in salt-dependent hypertension. Contrib. Nephrol. 1991, 90, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Sano, H.; Furuta, Y.; Fukuzaki, H. Effect of oral calcium on blood pressure response in salt-loaded borderline hypertensive patients. Hypertens. Dallas Tex 1979 1989, 13, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.H.; Wagner, U.L.; Fineberg, N.S. The blood pressure effects of calcium supplementation in humans of known sodium responsiveness. Am. J. Hypertens. 1993, 6, 799–805. [Google Scholar] [CrossRef]
- Riccardi, D.; Park, J.; Lee, W.S.; Gamba, G.; Brown, E.M.; Hebert, S.C. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 131–135. [Google Scholar] [CrossRef]
- Yang, T.; Hassan, S.; Huang, Y.G.; Smart, A.M.; Briggs, J.P.; Schnermann, J.B. Expression of PTHrP, PTH/PTHrP receptor, and Ca(2+)-sensing receptor mRNAs along the rat nephron. Am. J. Physiol. 1997, 272, F751–F758. [Google Scholar] [CrossRef]
- Riccardi, D.; Hall, A.E.; Chattopadhyay, N.; Xu, J.Z.; Brown, E.M.; Hebert, S.C. Localization of the extracellular Ca2+/polyvalent cation-sensing protein in rat kidney. Am. J. Physiol. 1998, 274, F611–F622. [Google Scholar] [CrossRef]
- Saritas, T.; Borschewski, A.; McCormick, J.A.; Paliege, A.; Dathe, C.; Uchida, S.; Terker, A.; Himmerkus, N.; Bleich, M.; Demaretz, S.; et al. SPAK differentially mediates vasopressin effects on sodium cotransporters. J. Am. Soc. Nephrol. 2013, 24, 407–418. [Google Scholar] [CrossRef]
- Günzel, D.; Yu, A.S.L. Function and regulation of claudins in the thick ascending limb of Henle. Pflug. Arch. 2009, 458, 77–88. [Google Scholar] [CrossRef]
- Ferreri, N.R.; Hao, S.; Pedraza, P.L.; Escalante, B.; Vio, C.P. Eicosanoids and tumor necrosis factor-alpha in the kidney. Prostaglandins Other Lipid Mediat. 2012, 98, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Lu, M.; Hebert, S.C. Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am. J. Physiol. Cell Physiol. 1996, 271, C103–C111. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, M.; Balazy, M.; Hebert, S.C. Phospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TAL. Am. J. Physiol. Ren. Physiol. 1997, 273, F421–F429. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Zhang, C.; Li, W.; Wang, L.; Luan, H.; Wang, W.-H.; Gu, R. Stimulation of Ca2+-sensing receptor inhibits the basolateral 50-pS K channels in the thick ascending limb of rat kidney. Biochim. Biophys. Acta BBA Mol. Cell Res. 2012, 1823, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.-K.; Huang, C.; Ding, Y.; Qi, X.; Huang, C.-L.; Miller, R.T. Calcium-sensing receptor decreases cell surface expression of the inwardly rectifying K+ channel Kir4.1. J. Biol. Chem. 2011, 286, 1828–1835. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Sindic, A.; Hill, C.E.; Hujer, K.M.; Chan, K.W.; Sassen, M.; Wu, Z.; Kurachi, Y.; Nielsen, S.; Romero, M.F.; et al. Interaction of the Ca2+-sensing receptor with the inwardly rectifying potassium channels Kir4.1 and Kir4.2 results in inhibition of channel function. Am. J. Physiol. Ren. Physiol. 2007, 292, F1073–F1081. [Google Scholar] [CrossRef]
- Su, X.-T.; Wang, W.-H. The expression, regulation, and function of Kir4.1 (Kcnj10) in the mammalian kidney. Am. J. Physiol. Ren. Physiol. 2016, 311, F12–F15. [Google Scholar] [CrossRef]
- Abdullah, H.I.; Pedraza, P.L.; McGiff, J.C.; Ferreri, N.R. Calcium-sensing receptor signaling pathways in medullary thick ascending limb cells mediate COX-2-derived PGE2 production: Functional significance. Am. J. Physiol. Ren. Physiol. 2008, 295, F1082–F1089. [Google Scholar] [CrossRef]
- Reinalter, S.C.; Jeck, N.; Brochhausen, C.; Watzer, B.; Nüsing, R.M.; Seyberth, H.W.; Kömhoff, M. Role of cyclooxygenase-2 in hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2002, 62, 253–260. [Google Scholar] [CrossRef]
- Kammerl, M.C.; Nüsing, R.M.; Richthammer, W.; Krämer, B.K.; Kurtz, A. Inhibition of COX-2 counteracts the effects of diuretics in rats. Kidney Int. 2001, 60, 1684–1691. [Google Scholar] [CrossRef]
- Lazelle, R.A.; McCully, B.H.; Terker, A.S.; Himmerkus, N.; Blankenstein, K.I.; Mutig, K.; Bleich, M.; Bachmann, S.; Yang, C.-L.; Ellison, D.H. Renal deletion of 12 kDa FK506-binding protein attenuates tacrolimus-induced hypertension. J. Am. Soc. Nephrol. 2016, 27, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Terker, A.S.; Zhang, C.; Erspamer, K.J.; Gamba, G.; Yang, C.-L.; Ellison, D.H. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016, 89, 127–134. [Google Scholar] [CrossRef] [PubMed]
- de Ferreira, M.C.J.; Héliès-Toussaint, C.; Imbert-Teboul, M.; Bailly, C.; Verbavatz, J.-M.; Bellanger, A.-C.; Chabardès, D. Co-expression of a Ca2+ -inhibitable adenylyl cyclase and of a Ca2+ -sensing receptor in the cortical thick ascending limb cell of the rat kidney: Inhibition of hormone-dependent cAMP accumulation by extracellular Ca2+. J. Biol. Chem. 1998, 273, 15192–15202. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, R.; Braucht, D.W.W.; Rinschen, M.M.; Chou, C.-L.; Hoffert, J.D.; Pisitkun, T.; Knepper, M.A. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15653–15658. [Google Scholar] [CrossRef]
- Gerbino, A.; Ruder, W.C.; Curci, S.; Pozzan, T.; Zaccolo, M.; Hofer, A.M. Termination of cAMP signals by Ca2+ and Gαi via extracellular Ca2+ sensors. J. Cell Biol. 2005, 171, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Na, T.; Wu, G.; Peng, J.-B. Disease-causing mutations in the acidic motif of WNK4 impair the sensitivity of WNK4 kinase to calcium ions. Biochem. Biophys. Res. Commun. 2012, 419, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, M.; Mori, T.; Isobe, K.; Sohara, E.; Susa, K.; Araki, Y.; Chiga, M.; Kikuchi, E.; Nomura, N.; Mori, Y.; et al. Impaired KLHL3-Mediated Ubiquitination of WNK4 Causes Human Hypertension. Cell Rep. 2013, 3, 858–868. [Google Scholar] [CrossRef]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Na, T.; Wu, G.; Zhang, W.; Dong, W.-J.; Peng, J.-B. Disease-causing R1185C mutation of WNK4 disrupts a regulatory mechanism involving calmodulin binding and SGK1 phosphorylation sites. Am. J. Physiol. Ren. Physiol. 2013, 304, F8–F18. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Renigunta, V.; Himmerkus, N.; Zhang, J.; Renigunta, A.; Bleich, M.; Hou, J. Claudin-14 regulates renal Ca++ transport in response to CaSR signalling via a novel microRNA pathway: Claudin-14 function and regulation. EMBO J. 2012, 31, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; MacGregor, G.G.; Wilson, F.H.; Van Hoek, A.N.; Brown, D.; Ardito, T.; Kashgarian, M.; Giebisch, G.; Hebert, S.C.; Boulpaep, E.L.; et al. Paracellular Cl- permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 14877–14882. [Google Scholar] [CrossRef]
- Ohta, A.; Yang, S.-S.; Rai, T.; Chiga, M.; Sasaki, S.; Uchida, S. Overexpression of human WNK1 increases paracellular chloride permeability and phosphorylation of claudin-4 in MDCKII cells. Biochem. Biophys. Res. Commun. 2006, 349, 804–808. [Google Scholar] [CrossRef]
- Yamauchi, K.; Rai, T.; Kobayashi, K.; Sohara, E.; Suzuki, T.; Itoh, T.; Suda, S.; Hayama, A.; Sasaki, S.; Uchida, S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 2004, 101, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Hou, J. Claudins in barrier and transport function-the kidney. Pflug. Arch. 2017, 469, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Karger, C.; Machura, K.; Schneider, A.; Hugo, C.; Todorov, V.T.; Kurtz, A. COX-2-derived PGE2 triggers hyperplastic renin expression and hyperreninemia in aldosterone synthase-deficient mice. Pflug. Arch. 2018, 470, 1127–1137. [Google Scholar] [CrossRef]
- Vargas-Poussou, R. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a bartter-like syndrome. J. Am. Soc. Nephrol. 2002, 13, 2259–2266. [Google Scholar] [CrossRef] [PubMed]
- Atchison, D.K.; Ortiz-Capisano, M.C.; Beierwaltes, W.H. Acute activation of the calcium-sensing receptor inhibits plasma renin activity in vivo. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1020–R1026. [Google Scholar] [CrossRef][Green Version]
- Maillard, M.P.; Tedjani, A.; Perregaux, C.; Burnier, M. Calcium-sensing receptors modulate renin release in vivo and in vitro in the rat. J. Hypertens. 2009, 27, 1980–1987. [Google Scholar] [CrossRef]
- Kurtz, A.; Della Bruna, R.; Kühn, K. Cyclosporine A enhances renin secretion and production in isolated juxtaglomerular cells. Kidney Int. 1988, 33, 947–953. [Google Scholar] [CrossRef]
- Madsen, K.; Friis, U.G.; Gooch, J.L.; Hansen, P.B.; Holmgaard, L.; Skøtt, O.; Jensen, B.L. Inhibition of calcineurin phosphatase promotes exocytosis of renin from juxtaglomerular cells. Kidney Int. 2010, 77, 110–117. [Google Scholar] [CrossRef]
- McCormick, J.A.; Ellison, D.H. Distal convoluted tubule. Compr. Physiol. 2015, 5, 45–98. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bostanjoglo, M.; Schmitt, R.; Ellison, D.H. Sodium transport-related proteins in the mammalian distal nephron-distribution, ontogeny and functional aspects. Anat. Embryol. 1999, 200, 447–468. [Google Scholar] [CrossRef] [PubMed]
- Mistry, A.C.; Wynne, B.M.; Yu, L.; Tomilin, V.; Yue, Q.; Zhou, Y.; Al-Khalili, O.; Mallick, R.; Cai, H.; Alli, A.A.; et al. The sodium chloride cotransporter (NCC) and epithelial sodium channel (ENaC) associate. Biochem. J. 2016, 473, 3237–3252. [Google Scholar] [CrossRef] [PubMed]
- Wynne, B.M.; Mistry, A.C.; Al-Khalili, O.; Mallick, R.; Theilig, F.; Eaton, D.C.; Hoover, R.S. Aldosterone modulates the association between NCC and ENaC. Sci. Rep. 2017, 7, 4149. [Google Scholar] [CrossRef] [PubMed]
- Dimke, H.; Hoenderop, J.G.J.; Bindels, R.J.M. Molecular basis of epithelial Ca2+ and Mg2+ transport: Insights from the TRP channel family. J. Physiol. 2011, 589, 1535–1542. [Google Scholar] [CrossRef]
- Loffing, J.; Loffing-Cueni, D.; Valderrabano, V.; Kläusli, L.; Hebert, S.C.; Rossier, B.C.; Hoenderop, J.G.J.; Bindels, R.J.M.; Kaissling, B. Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am. J. Physiol. Ren. Physiol. 2001, 281, F1021–F1027. [Google Scholar] [CrossRef]
- Bostanjoglo, M.; Reeves, W.B.; Reilly, R.F.; Velázquez, H.; Robertson, N.; Litwack, G.; Morsing, P.; Dørup, J.; Bachmann, S.; Ellison, D.H.; et al. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J. Am. Soc. Nephrol. 1998, 9, 1347–1358. [Google Scholar]
- Hoover, R.S.; Tomilin, V.; Hanson, L.; Pochynyuk, O.; Ko, B. PTH modulation of NCC activity regulates TRPV5 Ca2+ reabsorption. Am. J. Physiol. Ren. Physiol. 2016, 310, F144–F151. [Google Scholar] [CrossRef]
- Reilly, R.F.; Huang, C.-L. The mechanism of hypocalciuria with NaCl cotransporter inhibition. Nat. Rev. Nephrol. 2011, 7, 669–674. [Google Scholar] [CrossRef]
- Nijenhuis, T.; Vallon, V.; van der Kemp, A.W.C.M.; Loffing, J.; Hoenderop, J.G.J.; Bindels, R.J.M. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J. Clin. Invest. 2005, 115, 1651–1658. [Google Scholar] [CrossRef]
- Castañeda-Bueno, M.; Arroyo, J.P.; Zhang, J.; Puthumana, J.; Yarborough, O.; Shibata, S.; Rojas-Vega, L.; Gamba, G.; Rinehart, J.; Lifton, R.P. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. USA 2017, 114, E879–E886. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Uchida, K.; Ohashi, T.; Nitta, K.; Ohta, A.; Chiga, M.; Sasaki, S.; Uchida, S. Immunolocalization of WNK4 in mouse kidney. Histochem. Cell Biol. 2011, 136, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Terker, A.S.; Castañeda-Bueno, M.; Ferdaus, M.Z.; Cornelius, R.J.; Erspamer, K.J.; Su, X.-T.; Miller, L.N.; McCormick, J.A.; Wang, W.-H.; Gamba, G.; et al. With no lysine kinase 4 modulates sodium potassium 2 chloride cotransporter activity in vivo. Am. J. Physiol. Ren. Physiol. 2018, 315, F781–F790. [Google Scholar] [CrossRef] [PubMed]
- Murillo-de-Ozores, A.R.; Rodríguez-Gama, A.; Bazúa-Valenti, S.; Leyva-Ríos, K.; Vázquez, N.; Pacheco-Álvarez, D.; De La Rosa-Velázquez, I.A.; Wengi, A.; Stone, K.L.; Zhang, J.; et al. C-terminally truncated, kidney-specific variants of the WNK4 kinase lack several sites that regulate its activity. J. Biol. Chem. 2018, 293, 12209–12221. [Google Scholar] [CrossRef]
- Boyd-Shiwarski, C.R.; Shiwarski, D.J.; Roy, A.; Namboodiri, H.N.; Nkashama, L.J.; Xie, J.; McClain, K.L.; Marciszyn, A.; Kleyman, T.R.; Tan, R.J.; et al. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol. Biol. Cell 2018, 29, 499–509. [Google Scholar] [CrossRef]
- Thomson, M.N.; Cuevas, C.A.; Bewarder, T.M.; Dittmayer, C.; Miller, L.N.; Si, J.; Cornelius, R.J.; Su, X.-T.; Yang, C.-L.; McCormick, J.A.; et al. WNK bodies cluster WNK4 and SPAK/OSR1 to promote NCC activation in hypokalemia. Am. J. Physiol. Ren. Physiol. 2019. [Google Scholar] [CrossRef]
- Thomson, M.N.; Schneider, W.; Mutig, K.; Ellison, D.H.; Kettritz, R.; Bachmann, S. Patients with hypokalemia develop WNK bodies in the distal convoluted tubule of the kidney. Am. J. Physiol. Ren. Physiol. 2019, 316, F292–F300. [Google Scholar] [CrossRef]
- Subramanya, A.R.; Yang, C.-L.; Zhu, X.; Ellison, D.H. Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am. J. Physiol. Ren. Physiol. 2006, 290, F619–F624. [Google Scholar] [CrossRef]
- Argaiz, E.R.; Chavez-Canales, M.; Ostrosky-Frid, M.; Rodríguez-Gama, A.; Vázquez, N.; Gonzalez-Rodriguez, X.; Garcia-Valdes, J.; Hadchouel, J.; Ellison, D.; Gamba, G. Kidney-specific WNK1 isoform (KS-WNK1) is a potent activator of WNK4 and NCC. Am. J. Physiol. Ren. Physiol. 2018, 315, F734–F745. [Google Scholar] [CrossRef]
- Cuevas, C.A.; Su, X.-T.; Wang, M.-X.; Terker, A.S.; Lin, D.-H.; McCormick, J.A.; Yang, C.-L.; Ellison, D.H.; Wang, W.-H. Potassium Sensing by Renal Distal Tubules Requires Kir4.1. J. Am. Soc. Nephrol. 2017, 28, 1814–1825. [Google Scholar] [CrossRef]
- Wang, M.-X.; Cuevas, C.A.; Su, X.-T.; Wu, P.; Gao, Z.-X.; Lin, D.-H.; McCormick, J.A.; Yang, C.-L.; Wang, W.-H.; Ellison, D.H. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018, 93, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Ferdaus, M.Z.; Gratreak, B.D.K.; Miller, L.; Si, J.; McCormick, J.A.; Yang, C.; Ellison, D.H.; Terker, A.S. WNK4 limits distal calcium losses following acute furosemide treatment. Physiol. Rep. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cong, P.; Williams, S.R.; Zhang, W.; Na, T.; Ma, H.-P.; Peng, J.-B. WNK4 regulates the secretory pathway via which TRPV5 is targeted to the plasma membrane. Biochem. Biophys. Res. Commun. 2008, 375, 225–229. [Google Scholar] [CrossRef]
- Cha, S.-K.; Huang, C.-L. WNK4 kinase stimulates caveola-mediated endocytosis of TRPV5 amplifying the dynamic range of regulation of the channel by Protein Kinase C. J. Biol. Chem. 2010, 285, 6604–6611. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ferguson, W.B.; Peng, J.-B. WNK4 enhances TRPV5-mediated calcium transport: Potential role in hypercalciuria of familial hyperkalemic hypertension caused by gene mutation of WNK4. Am. J. Physiol. Ren. Physiol. 2007, 292, F545–F554. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Na, T.; Zhang, W.; Wu, G.; Liu, C.; Peng, J.-B. Concerted actions of NHERF2 and WNK4 in regulating TRPV5. Biochem. Biophys. Res. Commun. 2011, 404, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.T.F.; Wu, X.-R.; Huang, C.-L. Uromodulin upregulates TRPV5 by impairing caveolin-mediated endocytosis. Kidney Int. 2013, 84, 130–137. [Google Scholar] [CrossRef]
- Breton, S.; Lisanti, M.P.; Tyszkowski, R.; McLaughlin, M.; Brown, D. Basolateral distribution of caveolin-1 in the kidney: Absence from H+ -ATPase-coated endocytic vesicles in intercalated cells. J. Histochem. Cytochem. 1998, 46, 205–214. [Google Scholar] [CrossRef]
- Willière, Y.; Borschewski, A.; Patzak, A.; Nikitina, T.; Dittmayer, C.; Daigeler, A.L.; Schuelke, M.; Bachmann, S.; Mutig, K. Caveolin 1 promotes renal water and salt reabsorption. Sci. Rep. 2018, 8, 545. [Google Scholar] [CrossRef]
- Păunescu, T.G.; Lu, H.A.J.; Russo, L.M.; Pastor-Soler, N.M.; McKee, M.; McLaughlin, M.M.; Bartlett, B.E.; Breton, S.; Brown, D. Vasopressin induces apical expression of caveolin in rat kidney collecting duct principal cells. Am. J. Physiol. Ren. Physiol. 2013, 305, F1783–F1795. [Google Scholar] [CrossRef]
- Roy, A.; Al-bataineh, M.M.; Pastor-Soler, N.M. Collecting duct intercalated cell function and regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.Y.; Velic, A.; Dijkman, H.B.; Verkaart, S.; van der Kemp, A.W.; Nowik, M.; Timmermans, K.; Doucet, A.; Wagner, C.A.; Bindels, R.J.; et al. The calcium-sensing receptor promotes urinary acidification to prevent nephrolithiasis. J. Am. Soc. Nephrol. 2009, 20, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Giesecke, T.; Himmerkus, N.; Leipziger, J.; Bleich, M.; Koshimizu, T.-A.; Fähling, M.; Smorodchenko, A.; Shpak, J.; Knappe, C.; Isermann, J.; et al. Vasopressin increases urinary acidification via v1a receptors in collecting duct intercalated cells. J. Am. Soc. Nephrol. 2019, 30, 946–961. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Liu, Z.; Huang, C.-L. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc. Natl. Acad. Sci. USA 2006, 103, 1615–1620. [Google Scholar] [CrossRef]
- Hadchouel, J.; Soukaseum, C.; Büsst, C.; Zhou, X.; Baudrie, V.; Zürrer, T.; Cambillau, M.; Elghozi, J.-L.; Lifton, R.P.; Loffing, J.; et al. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc. Natl. Acad. Sci. USA 2010, 107, 18109–18114. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, G.; Macrina, L.; Magni, G.; Arcidiacono, T. Calcium-sensing receptor: Evidence and hypothesis for its role in nephrolithiasis. Urolithiasis 2019, 47, 23–33. [Google Scholar] [CrossRef]
- Pollak, M.R.; Brown, E.M.; Chou, Y.-H.W.; Hebert, S.C.; Marx, S.J.; Stelnmann, B.; Levi, T.; Seidman, C.E.; Seidman, J.G. Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993, 75, 1297–1303. [Google Scholar] [CrossRef]
- Pollak, M.R.; Chou, Y.H.; Marx, S.J.; Steinmann, B.; Cole, D.E.; Brandi, M.L.; Papapoulos, S.E.; Menko, F.H.; Hendy, G.N.; Brown, E.M. Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J. Clin. Invest. 1994, 93, 1108–1112. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Conner, D.A.; Pollak, M.R.; Ladd, D.J.; Kifor, O.; Warren, H.B.; Brown, E.M.; Seidman, J.G.; Seidman, C.E. A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat. Genet. 1995, 11, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.C. Therapeutic use of calcimimetics. Annu. Rev. Med. 2006, 57, 349–364. [Google Scholar] [CrossRef]
- Torres, P.A.U.; De Broe, M. Calcium-sensing receptor, calcimimetics, and cardiovascular calcifications in chronic kidney disease. Kidney Int. 2012, 82, 19–25. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ostroverkhova, D.S.; Hu, J.; Tarasov, V.V.; Melnikova, T.I.; Porozov, Y.B.; Mutig, K. Calcium-Sensing Receptor and Regulation of WNK Kinases in the Kidney. Cells 2020, 9, 1644. https://doi.org/10.3390/cells9071644
Ostroverkhova DS, Hu J, Tarasov VV, Melnikova TI, Porozov YB, Mutig K. Calcium-Sensing Receptor and Regulation of WNK Kinases in the Kidney. Cells. 2020; 9(7):1644. https://doi.org/10.3390/cells9071644
Chicago/Turabian StyleOstroverkhova, Daria S., Junda Hu, Vadim V. Tarasov, Tatiana I. Melnikova, Yuri B. Porozov, and Kerim Mutig. 2020. "Calcium-Sensing Receptor and Regulation of WNK Kinases in the Kidney" Cells 9, no. 7: 1644. https://doi.org/10.3390/cells9071644
APA StyleOstroverkhova, D. S., Hu, J., Tarasov, V. V., Melnikova, T. I., Porozov, Y. B., & Mutig, K. (2020). Calcium-Sensing Receptor and Regulation of WNK Kinases in the Kidney. Cells, 9(7), 1644. https://doi.org/10.3390/cells9071644