The Vasopressin Receptor 2 Mutant R137L Linked to the Nephrogenic Syndrome of Inappropriate Antidiuresis (NSIAD) Signals through an Alternative Pathway that Increases AQP2 Membrane Targeting Independently of S256 Phosphorylation

 ,
, 
 and
and 
Abstract
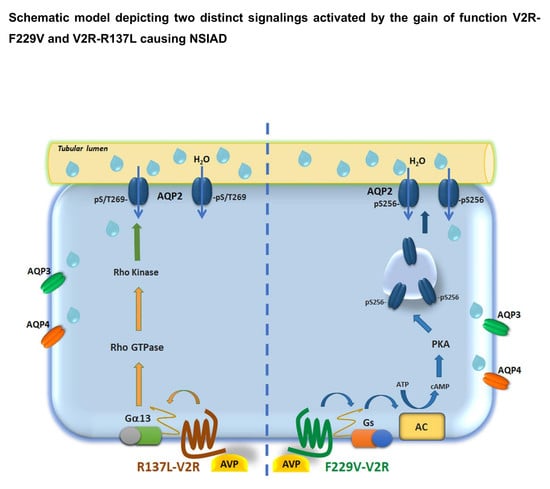
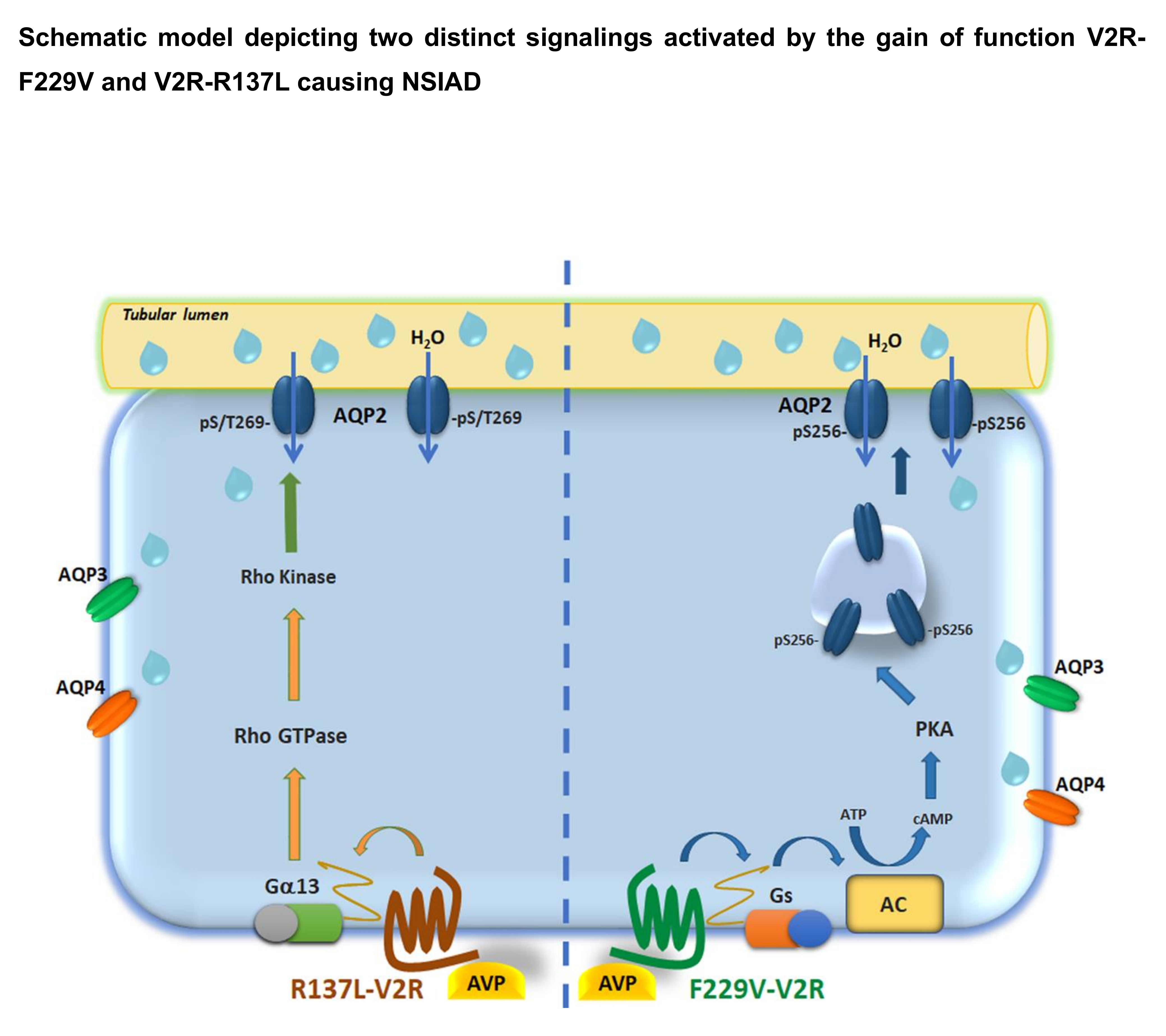{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
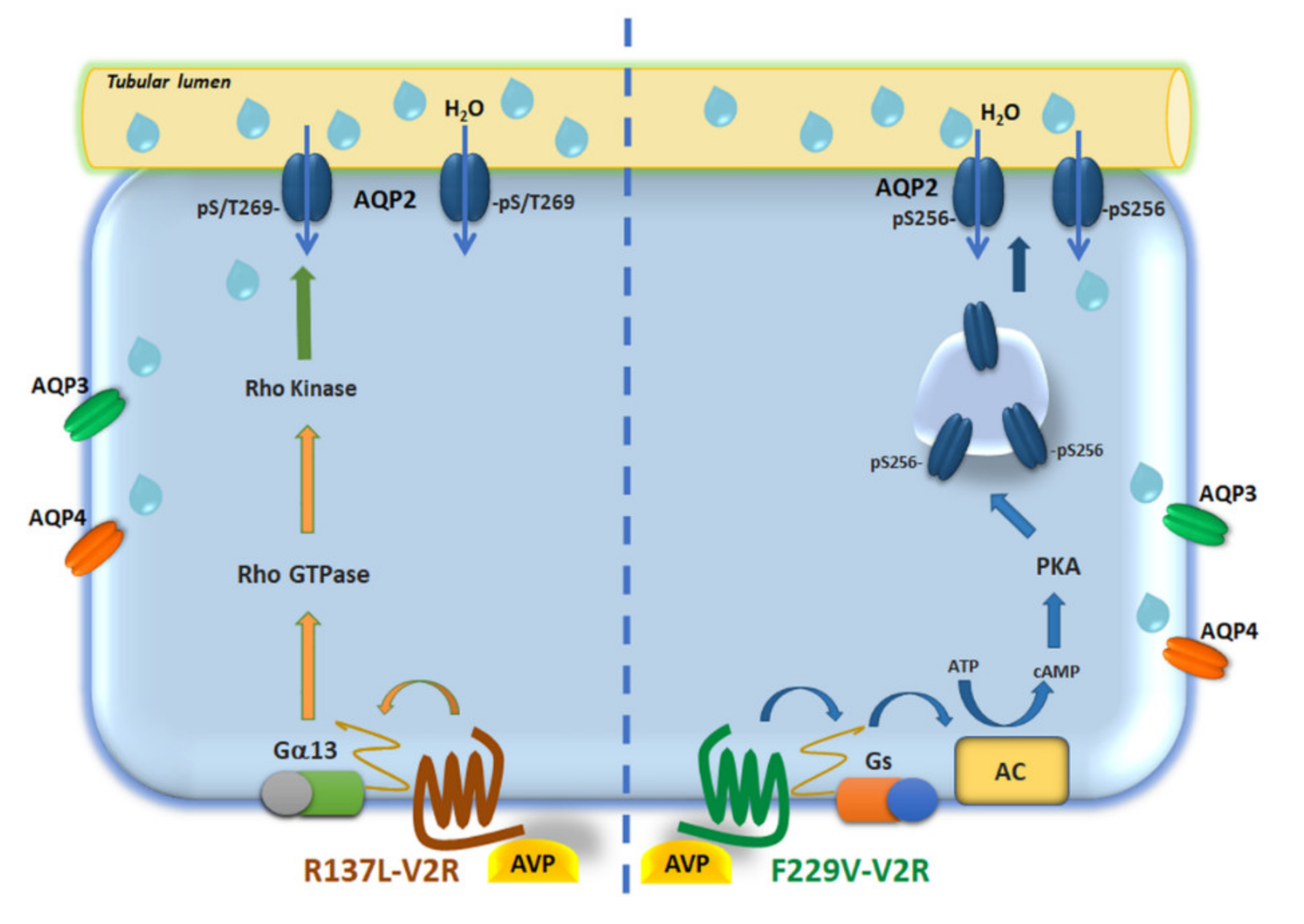{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Antibodies
2.3. Constructs
2.4. Cell Culture
2.5. Gel Electrophoresis and Immunoblotting
2.6. Water Permeability Assay
2.7. Fluorescence Resonance Energy Transfer Measurements
2.8. F-actin co-Sedimentation Assay
2.9. Rho-Associated kinase (ROCK) Activity Assay
2.10. In Vitro Phosphorylation of Synthetic Peptides
2.11. Statistical Analysis
3. Results
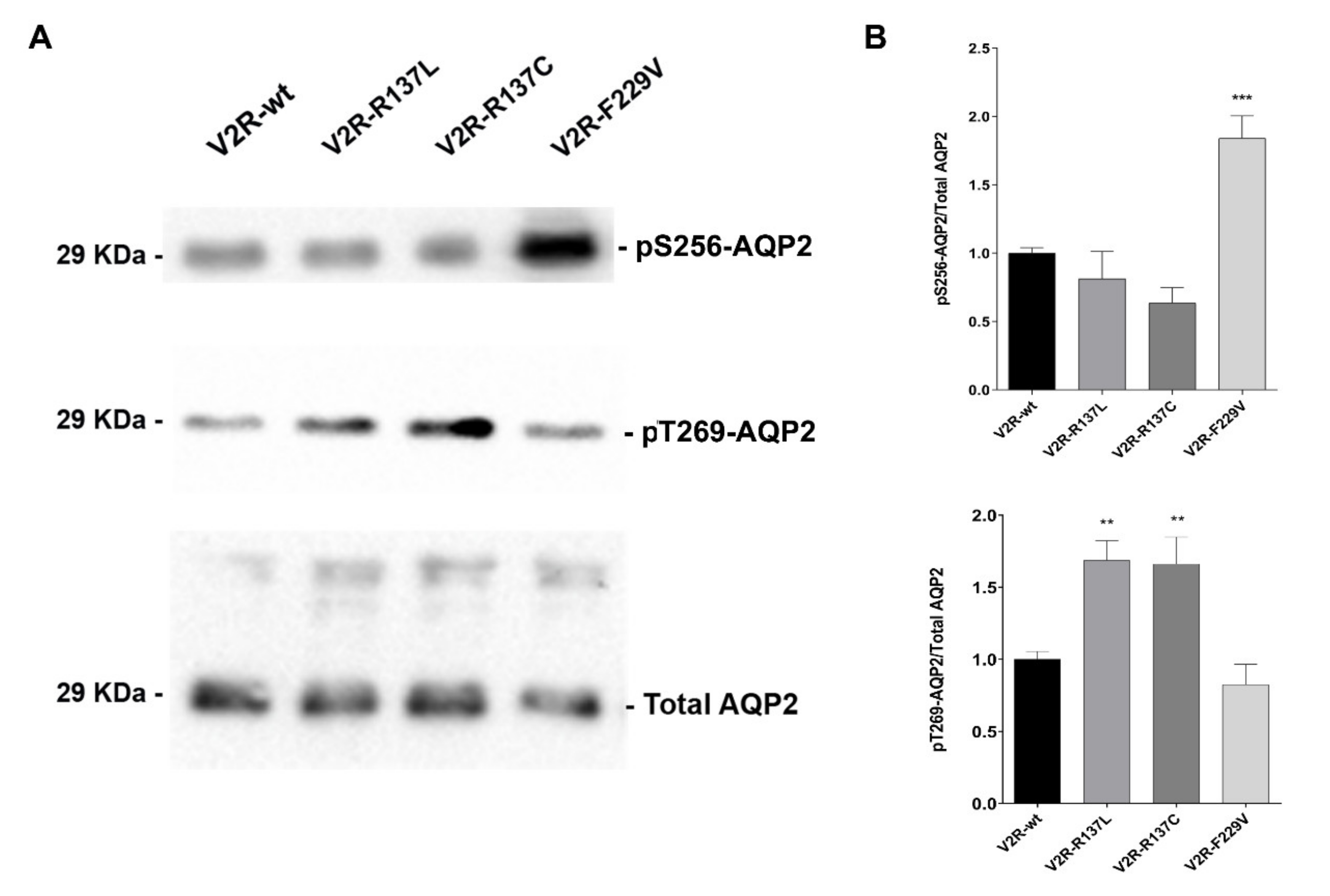
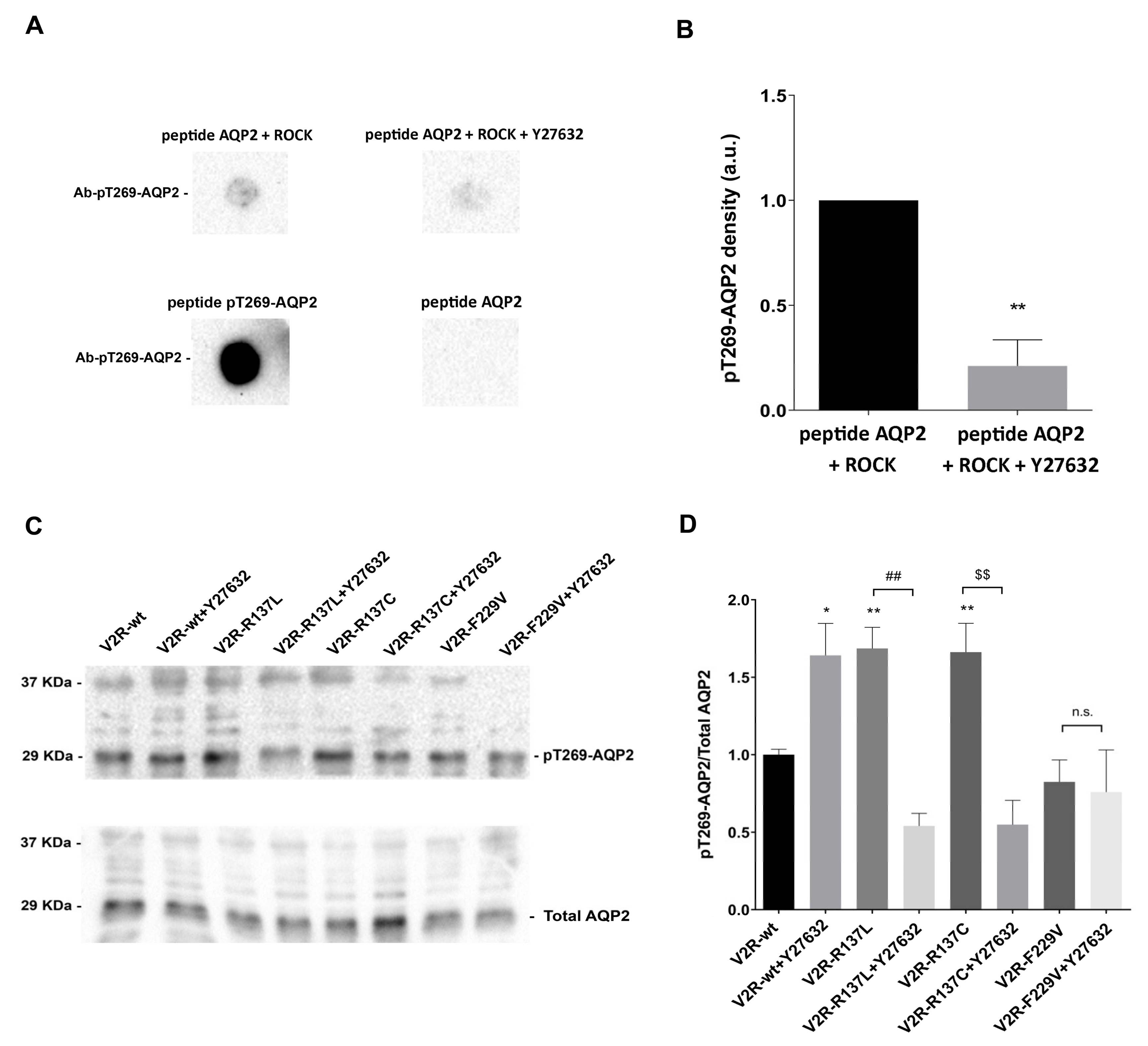
3.1. V2R R137L and R137C expressing cells have significantly higher basal levels of pT269-AQP2
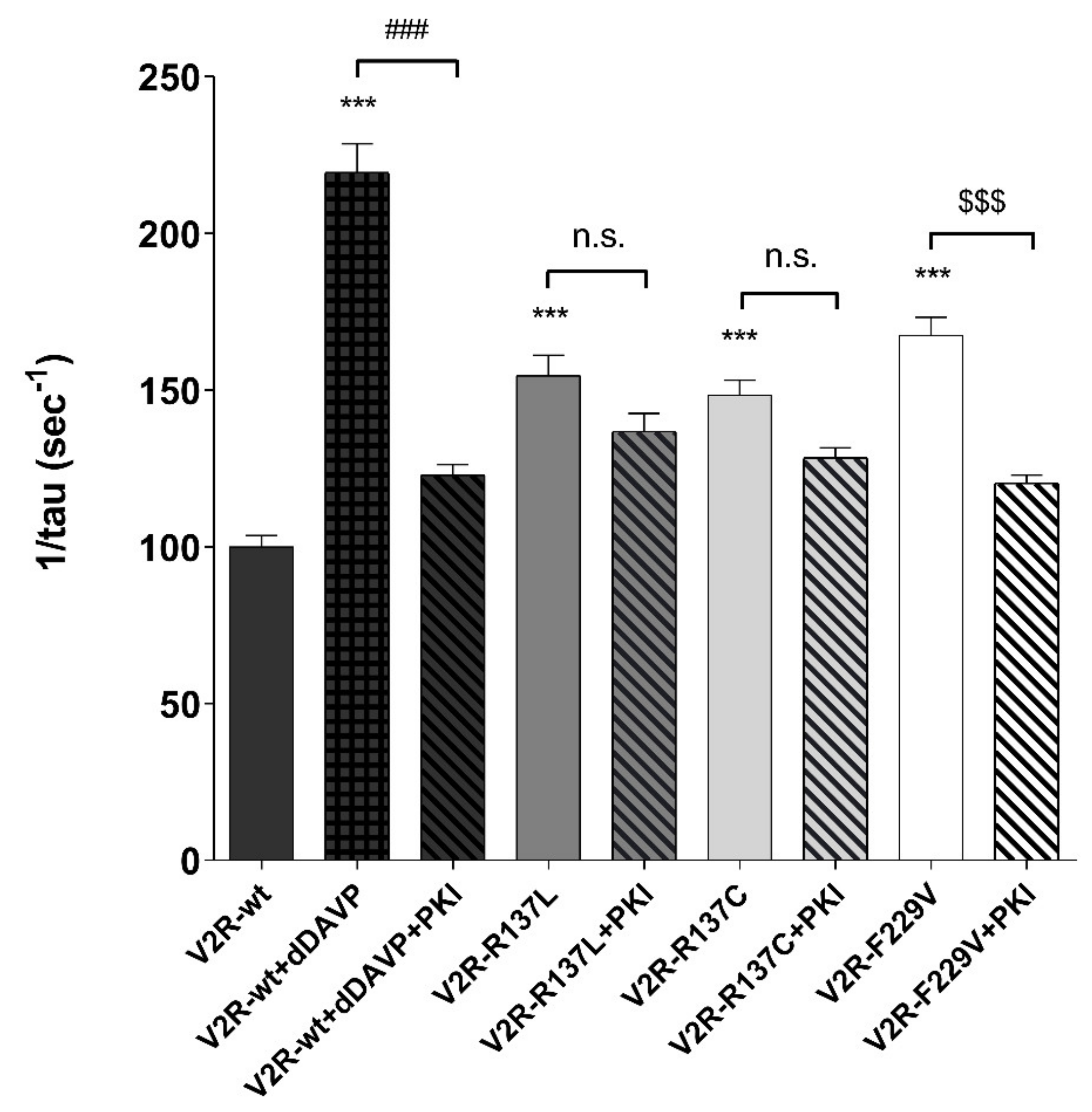
3.2. Sensitivity of Osmotic Water Permeability to PKA Inhibitor.
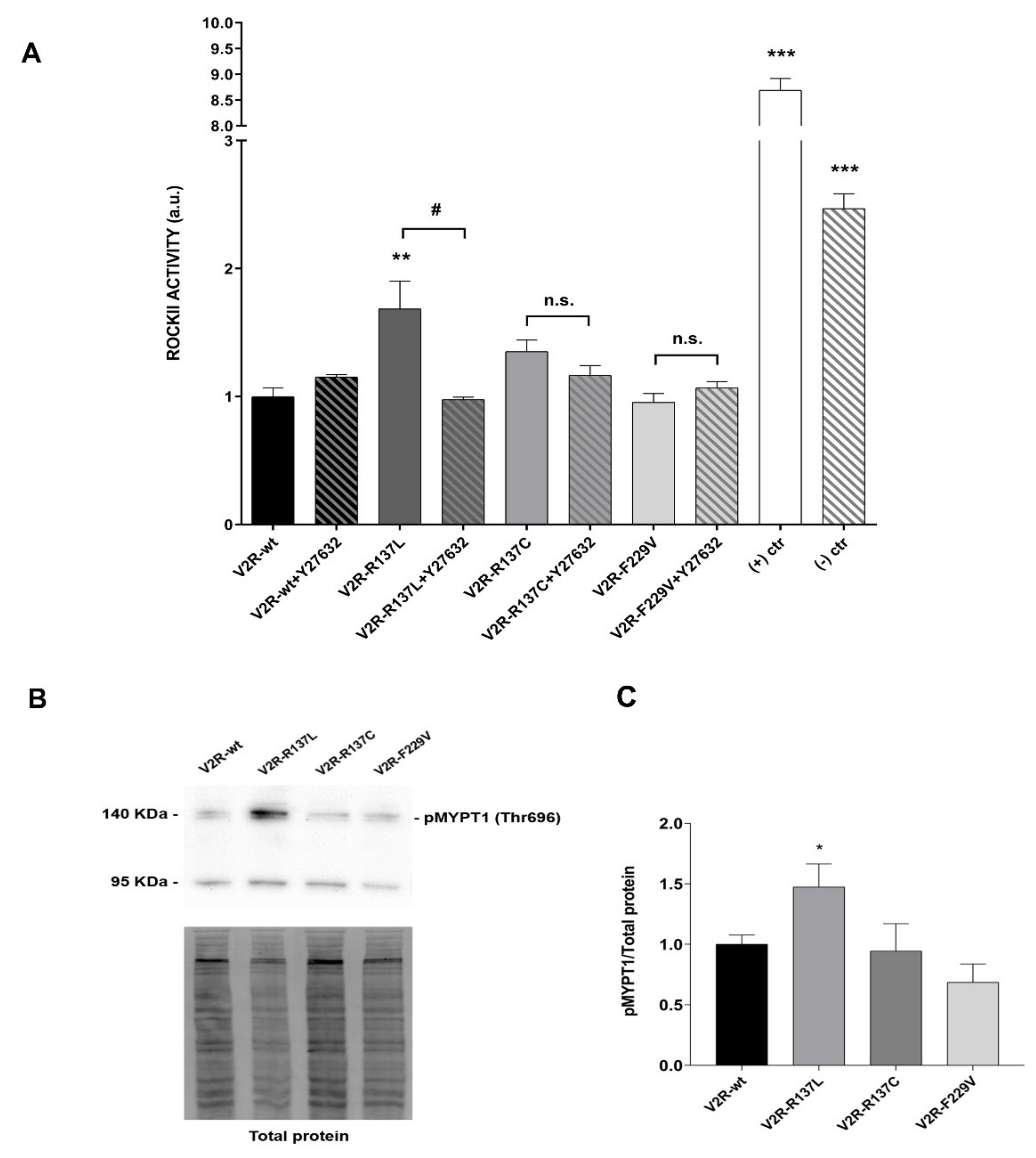
3.3. R137L and R137C Mutants Activate a Signaling Pathway Involving Rho-Associated Kinase (ROCK)
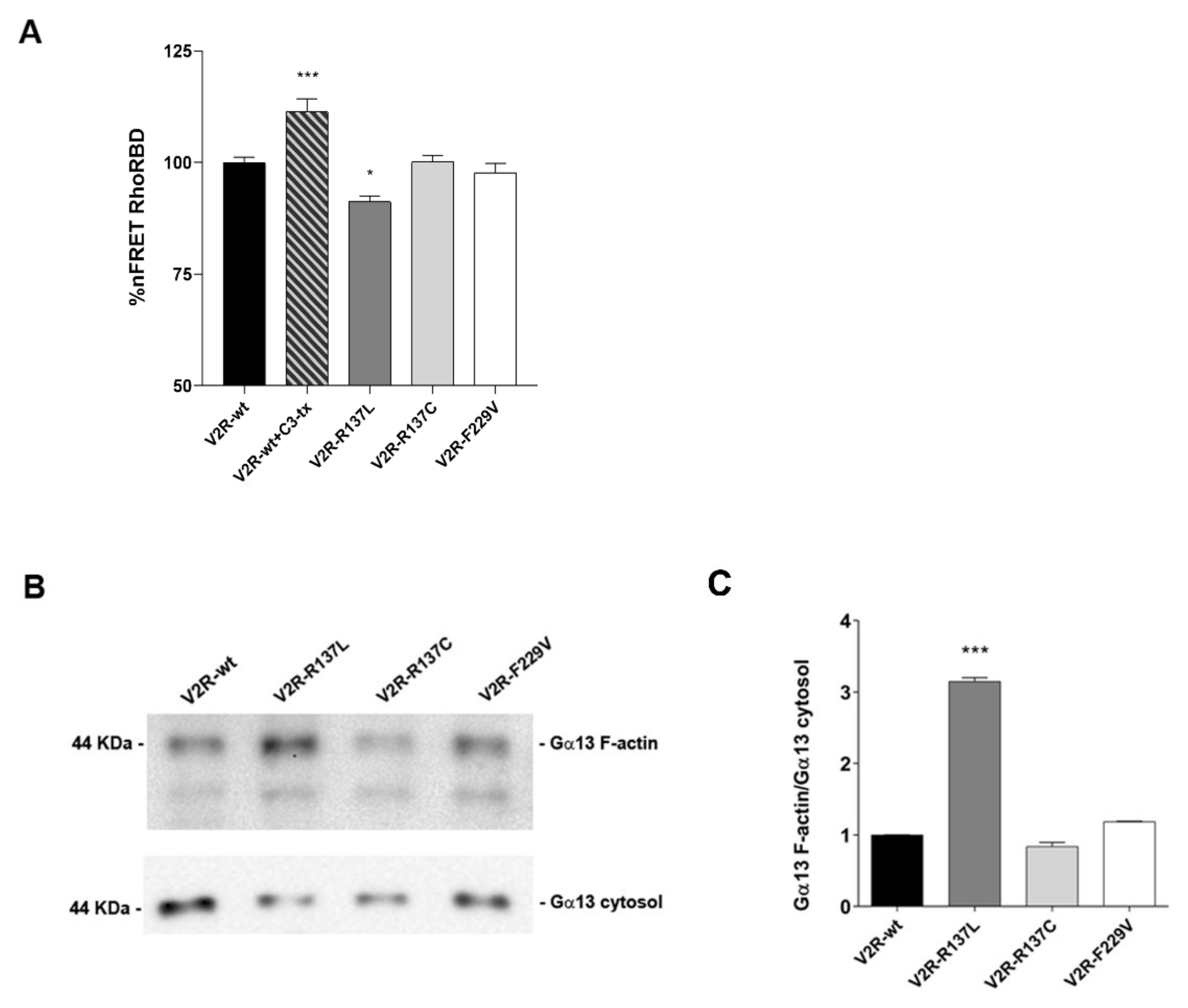
3.4. Gα12/13–Rho–ROCK Signaling is Activated by the V2R-R137L Mutant
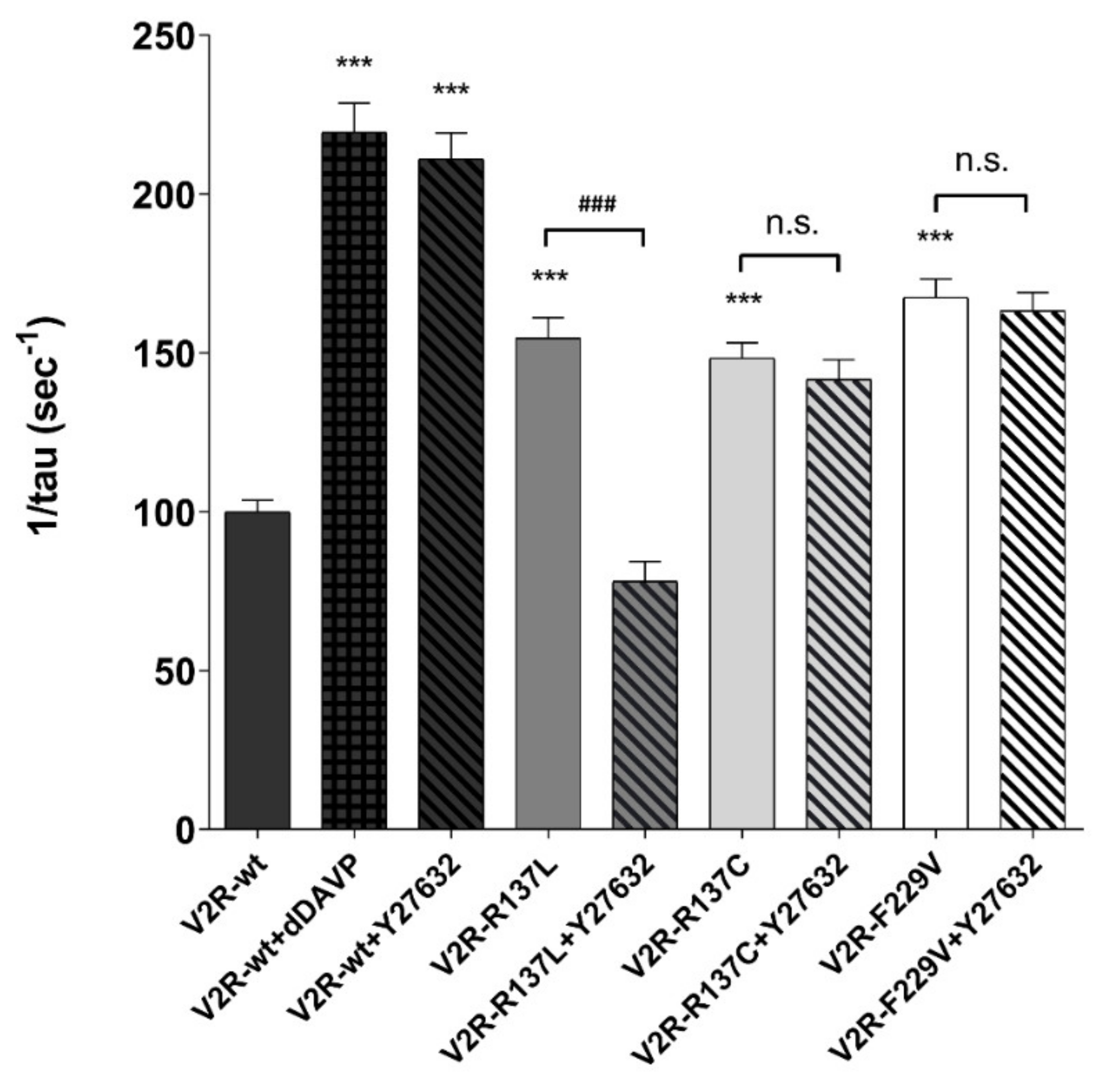
3.5. Inhibition of ROCK Reduces the Basal Osmotic Water Permeability in V2R-R137L Expressing Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldman, B.J.; Rosenthal, S.M.; Vargas, G.A.; Fenwick, R.G.; Huang, E.A.; Matsuda-Abedini, M.; Lustig, R.H.; Mathias, R.S.; Portale, A.A.; Miller, W.L.; et al. Nephrogenic syndrome of inappropriate antidiuresis. N. Engl. J. Med. 2005, 352, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, E.; Greenbaum, L.A.; Rochdi, D.; Abrol, R.; Goddard, W.A., 3rd; Bichet, D.G.; Bouvier, M. Identification and characterization of an activating F229V substitution in the V2 vasopressin receptor in an infant with NSIAD. J. Am. Soc. Nephrol. 2012, 23, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Erdelyi, L.S.; Mann, W.A.; Morris-Rosendahl, D.J.; Gross, U.; Nagel, M.; Varnai, P.; Balla, A.; Hunyady, L. Mutation in the V2 vasopressin receptor gene, AVPR2, causes nephrogenic syndrome of inappropriate diuresis. Kidney Int. 2015, 88, 1070–1078. [Google Scholar] [CrossRef] [PubMed]
- Scheer, A.; Costa, T.; Fanelli, F.; De Benedetti, P.G.; Mhaouty-Kodja, S.; Abuin, L.; Nenniger-Tosato, M.; Cotecchia, S. Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr motif of the alpha(1b)-adrenergic receptor: Effects on receptor isomerization and activation. Mol. Pharmacol. 2000, 57, 219–231. [Google Scholar] [PubMed]
- Audet, M.; Bouvier, M. Restructuring G-protein- coupled receptor activation. Cell 2012, 151, 14–23. [Google Scholar] [CrossRef]
- Barak, L.S.; Oakley, R.H.; Laporte, S.A.; Caron, M.G. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. USA 2001, 98, 93–98. [Google Scholar] [CrossRef]
- Kocan, M.; Pfleger, K.D. Detection of GPCR/beta-arrestin interactions in live cells using bioluminescence resonance energy transfer technology. Methods Mol. Biol. 2009, 552, 305–317. [Google Scholar] [CrossRef]
- Rochdi, M.D.; Vargas, G.A.; Carpentier, E.; Oligny-Longpre, G.; Chen, S.; Kovoor, A.; Gitelman, S.E.; Rosenthal, S.M.; von Zastrow, M.; Bouvier, M. Functional characterization of vasopressin type 2 receptor substitutions (R137H/C/L) leading to nephrogenic diabetes insipidus and nephrogenic syndrome of inappropriate antidiuresis: Implications for treatments. Mol. Pharmacol. 2010, 77, 836–845. [Google Scholar] [CrossRef]
- Takahashi, K.; Makita, N.; Manaka, K.; Hisano, M.; Akioka, Y.; Miura, K.; Takubo, N.; Iida, A.; Ueda, N.; Hashimoto, M.; et al. V2 vasopressin receptor (V2R) mutations in partial nephrogenic diabetes insipidus highlight protean agonism of V2R antagonists. J. Biol. Chem. 2012, 287, 2099–2106. [Google Scholar] [CrossRef]
- Tiulpakov, A.; White, C.W.; Abhayawardana, R.S.; See, H.B.; Chan, A.S.; Seeber, R.M.; Heng, J.I.; Dedov, I.; Pavlos, N.J.; Pfleger, K.D. Mutations of Vasopressin Receptor 2 Including Novel L312S Have Differential Effects on Trafficking. Mol. Endocrinol. 2016, 30, 889–904. [Google Scholar] [CrossRef]
- Miyado, M.; Fukami, M.; Takada, S.; Terao, M.; Nakabayashi, K.; Hata, K.; Matsubara, Y.; Tanaka, Y.; Sasaki, G.; Nagasaki, K.; et al. Germline-Derived Gain-of-Function Variants of Gsalpha-Coding GNAS Gene Identified in Nephrogenic Syndrome of Inappropriate Antidiuresis. J. Am. Soc. Nephrol. 2019, 30, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Decaux, G.; Vandergheynst, F.; Bouko, Y.; Parma, J.; Vassart, G.; Vilain, C. Nephrogenic syndrome of inappropriate antidiuresis in adults: High phenotypic variability in men and women from a large pedigree. J. Am. Soc. Nephrol. 2007, 18, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Tamma, G.; Pellegrino, T.; Vezzi, V.; Ambrosio, C.; Gro, C.; Di Mise, A.; Costa, T.; Valenti, G.; Cotecchia, S. Gain-of-function mutations of the V2 vasopressin receptor in nephrogenic syndrome of inappropriate antidiuresis (NSIAD): A cell-based assay to assess constitutive water reabsorption. Pflug. Arch. 2019, 471, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Di Mise, A.; Tamma, G.; Valenti, G. Vasopressin-aquaporin-2 pathway: Recent advances in understanding water balance disorders. F1000Res 2019, 8. [Google Scholar] [CrossRef]
- Valenti, G.T.; Tamma, G. Vasopressin and the Regulation of Aquaporin-2 in Health and Disease. In Aquaporins in Health and Disease New Molecular Targets for Drug Discovery; Soveral, G.N.S., Casini, A., Eds.; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar] [CrossRef]
- Nedvetsky, P.I.; Tamma, G.; Beulshausen, S.; Valenti, G.; Rosenthal, W.; Klussmann, E. Regulation of aquaporin-2 trafficking. Handb. Exp. Pharmacol. 2009, 133–157. [Google Scholar] [CrossRef]
- Jung, H.J.; Kwon, T.H. Molecular mechanisms regulating aquaporin-2 in kidney collecting duct. Am. J. Physiol. Renal Physiol. 2016, 311, F1318–F1328. [Google Scholar] [CrossRef]
- Wilson, J.L.; Miranda, C.A.; Knepper, M.A. Vasopressin and the regulation of aquaporin-2. Clin. Exp. Nephrol. 2013, 17, 751–764. [Google Scholar] [CrossRef]
- Tamma, G.; Lasorsa, D.; Trimpert, C.; Ranieri, M.; Di Mise, A.; Mola, M.G.; Mastrofrancesco, L.; Devuyst, O.; Svelto, M.; Deen, P.M.; et al. A protein kinase A-independent pathway controlling aquaporin 2 trafficking as a possible cause for the syndrome of inappropriate antidiuresis associated with polycystic kidney disease 1 haploinsufficiency. J. Am. Soc. Nephrol. 2014, 25, 2241–2253. [Google Scholar] [CrossRef][Green Version]
- Olesen, E.T.; Moeller, H.B.; Assentoft, M.; MacAulay, N.; Fenton, R.A. The vasopressin type 2 receptor and prostaglandin receptors EP2 and EP4 can increase aquaporin-2 plasma membrane targeting through a cAMP-independent pathway. Am. J. Physiol. Renal Physiol. 2016, 311, F935–F944. [Google Scholar] [CrossRef]
- Olesen, E.T.; Fenton, R.A. Aquaporin-2 membrane targeting: Still a conundrum. Am. J. Physiol. Renal Physiol. 2017, 312, F744–F747. [Google Scholar] [CrossRef]
- Ando, F.; Sohara, E.; Morimoto, T.; Yui, N.; Nomura, N.; Kikuchi, E.; Takahashi, D.; Mori, T.; Vandewalle, A.; Rai, T.; et al. Wnt5a induces renal AQP2 expression by activating calcineurin signalling pathway. Nat. Commun. 2016, 7, 13636. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.W.; Terlouw, A.; Janssen, S.A.; Brown, D.; Bouley, R. Inhibition of non-receptor tyrosine kinase Src induces phosphoserine 256-independent aquaporin-2 membrane accumulation. J. Physiol. 2019, 597, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Procino, G.; Strafino, A.; Bononi, E.; Meyer, G.; Paulmichl, M.; Formoso, V.; Svelto, M.; Valenti, G. Hypotonicity induces aquaporin-2 internalization and cytosol-to-membrane translocation of ICln in renal cells. Endocrinology 2007, 148, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Mastrofrancesco, L.; Mira, A.; Tamma, G.; Carmosino, M.; Emma, F.; Svelto, M.; Valenti, G. Aquaporin 2 and apical calcium-sensing receptor: New players in polyuric disorders associated with hypercalciuria. Semin. Nephrol. 2008, 28, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, M.; Tamma, G.; Di Mise, A.; Russo, A.; Centrone, M.; Svelto, M.; Calamita, G.; Valenti, G. Negative feedback from CaSR signaling to aquaporin-2 sensitizes vasopressin to extracellular Ca2+. J. Cell Sci. 2015, 128, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Ranieri, M.; Di Mise, A.; Dossena, S.; Pellegrino, T.; Furia, E.; Nofziger, C.; Debellis, L.; Paulmichl, M.; Valenti, G.; et al. Interleukin-13 increases pendrin abundance to the cell surface in bronchial NCI-H292 cells via Rho/actin signaling. Pflug. Arch. 2017, 469, 1163–1176. [Google Scholar] [CrossRef]
- Hoffert, J.D.; Fenton, R.A.; Moeller, H.B.; Simons, B.; Tchapyjnikov, D.; McDill, B.W.; Yu, M.J.; Pisitkun, T.; Chen, F.; Knepper, M.A. Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J. Biol. Chem. 2008, 283, 24617–24627. [Google Scholar] [CrossRef]
- Moeller, H.B.; Knepper, M.A.; Fenton, R.A. Serine 269 phosphorylated aquaporin-2 is targeted to the apical membrane of collecting duct principal cells. Kidney Int. 2009, 75, 295–303. [Google Scholar] [CrossRef]
- Procino, G.; Mastrofrancesco, L.; Tamma, G.; Lasorsa, D.R.; Ranieri, M.; Stringini, G.; Emma, F.; Svelto, M.; Valenti, G. Calcium-sensing receptor and aquaporin 2 interplay in hypercalciuria-associated renal concentrating defect in humans. An in vivo and in vitro study. PLoS ONE 2012, 7, e33145. [Google Scholar] [CrossRef]
- He, Z.; Yang, Y.; Wen, Z.; Chen, C.; Xu, X.; Zhu, Y.; Wang, Y.; Wang, D.W. CYP2J2 metabolites, epoxyeicosatrienoic acids, attenuate Ang II-induced cardiac fibrotic response by targeting Galpha12/13. J. Lipid Res. 2017, 58, 1338–1353. [Google Scholar] [CrossRef]
- Xie, S.; Mason, F.M.; Martin, A.C. Loss of Galpha12/13 exacerbates apical area dependence of actomyosin contractility. Mol. Biol. Cell 2016, 27, 3526–3536. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Carmosino, M.; Svelto, M.; Valenti, G. Bradykinin signaling counteracts cAMP-elicited aquaporin 2 translocation in renal cells. J. Am. Soc. Nephrol. 2005, 16, 2881–2889. [Google Scholar] [CrossRef]
- Vaiskunaite, R.; Adarichev, V.; Furthmayr, H.; Kozasa, T.; Gudkov, A.; Voyno-Yasenetskaya, T.A. Conformational activation of radixin by G13 protein alpha subunit. J. Biol. Chem. 2000, 275, 26206–26212. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Klussmann, E.; Maric, K.; Aktories, K.; Svelto, M.; Rosenthal, W.; Valenti, G. Rho inhibits cAMP-induced translocation of aquaporin-2 into the apical membrane of renal cells. Am. J. Physiol. Renal Physiol. 2001, 281, F1092–F1101. [Google Scholar] [CrossRef] [PubMed]
- Powlson, A.S.; Challis, B.G.; Halsall, D.J.; Schoenmakers, E.; Gurnell, M. Nephrogenic syndrome of inappropriate antidiuresis secondary to an activating mutation in the arginine vasopressin receptor AVPR2. Clin. Endocrinol. (Oxf.) 2016, 85, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Hoffert, J.D.; Pisitkun, T.; Wang, G.; Shen, R.F.; Knepper, M.A. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: Regulation of aquaporin-2 phosphorylation at two sites. Proc. Natl. Acad. Sci. USA 2006, 103, 7159–7164. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Praetorius, J.; Rützler, M.R.; Fenton, R.A. Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 424–429. [Google Scholar] [CrossRef]
- Klussmann, E.; Tamma, G.; Lorenz, D.; Wiesner, B.; Maric, K.; Hofmann, F.; Aktories, K.; Valenti, G.; Rosenthal, W. An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 2001, 276, 20451–20457. [Google Scholar] [CrossRef]
- Bankir, L.; Bouby, N.; Ritz, E. Vasopressin: A novel target for the prevention and retardation of kidney disease? Nat. Rev. Nephrol. 2013, 9, 223–239. [Google Scholar] [CrossRef]
- Natochin, Y.V.; Golosova, D.V. Vasopressin receptor subtypes and renal sodium transport. Vitam. Horm. 2020, 113, 239–258. [Google Scholar] [CrossRef]
- Liu, J.; Wess, J. Different single receptor domains determine the distinct G protein coupling profiles of members of the vasopressin receptor family. J. Biol. Chem. 1996, 271, 8772–8778. [Google Scholar] [CrossRef] [PubMed]
- Devost, D.; Zingg, H.H. Homo- and hetero-dimeric complex formations of the human oxytocin receptor. J. Neuroendocrinol. 2004, 16, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Muratspahic, E.; Monjon, E.; Duerrauer, L.; Rogers, S.M.; Cullen, D.A.; Vanden Broeck, J.; Gruber, C.W. Oxytocin/vasopressin-like neuropeptide signaling in insects. Vitam. Horm. 2020, 113, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Shmygol, A.; Gullam, J.; Blanks, A.; Thornton, S. Multiple mechanisms involved in oxytocin-induced modulation of myometrial contractility. Acta Pharmacol. Sin. 2006, 27, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Moore, F.; Da Silva, C.; Wilde, J.I.; Smarason, A.; Watson, S.P.; Lopez Bernal, A. Up-regulation of p21- and RhoA-activated protein kinases in human pregnant myometrium. Biochem. Biophys. Res. Commun. 2000, 269, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Maggi, M.; Del Carlo, P.; Fantoni, G.; Giannini, S.; Torrisi, C.; Casparis, D.; Massi, G.; Serio, M. Human myometrium during pregnancy contains and responds to V1 vasopressin receptors as well as oxytocin receptors. J. Clin. Endocrinol. Metab. 1990, 70, 1142–1154. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranieri, M.; Venneri, M.; Pellegrino, T.; Centrone, M.; Di Mise, A.; Cotecchia, S.; Tamma, G.; Valenti, G. The Vasopressin Receptor 2 Mutant R137L Linked to the Nephrogenic Syndrome of Inappropriate Antidiuresis (NSIAD) Signals through an Alternative Pathway that Increases AQP2 Membrane Targeting Independently of S256 Phosphorylation. Cells 2020, 9, 1354. https://doi.org/10.3390/cells9061354
Ranieri M, Venneri M, Pellegrino T, Centrone M, Di Mise A, Cotecchia S, Tamma G, Valenti G. The Vasopressin Receptor 2 Mutant R137L Linked to the Nephrogenic Syndrome of Inappropriate Antidiuresis (NSIAD) Signals through an Alternative Pathway that Increases AQP2 Membrane Targeting Independently of S256 Phosphorylation. Cells. 2020; 9(6):1354. https://doi.org/10.3390/cells9061354
Chicago/Turabian StyleRanieri, Marianna, Maria Venneri, Tommaso Pellegrino, Mariangela Centrone, Annarita Di Mise, Susanna Cotecchia, Grazia Tamma, and Giovanna Valenti. 2020. "The Vasopressin Receptor 2 Mutant R137L Linked to the Nephrogenic Syndrome of Inappropriate Antidiuresis (NSIAD) Signals through an Alternative Pathway that Increases AQP2 Membrane Targeting Independently of S256 Phosphorylation" Cells 9, no. 6: 1354. https://doi.org/10.3390/cells9061354
APA StyleRanieri, M., Venneri, M., Pellegrino, T., Centrone, M., Di Mise, A., Cotecchia, S., Tamma, G., & Valenti, G. (2020). The Vasopressin Receptor 2 Mutant R137L Linked to the Nephrogenic Syndrome of Inappropriate Antidiuresis (NSIAD) Signals through an Alternative Pathway that Increases AQP2 Membrane Targeting Independently of S256 Phosphorylation. Cells, 9(6), 1354. https://doi.org/10.3390/cells9061354