Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Determination of ROS Generation
2.3. MTT Assay
2.4. Cell Proliferation Based on 5-Bromodeoxyuridine (BrdU) Incorporation
2.5. Western Blotting and Antibodies
2.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.7. In Vitro Wound Healing Assay
2.8. Invasion Assay
2.9. siRNA Transfection
2.10. Electric Cell Substrate Impedance Sensing Assay
2.11. Immunofluorescence
2.12. Statistical Analysis
3. Results
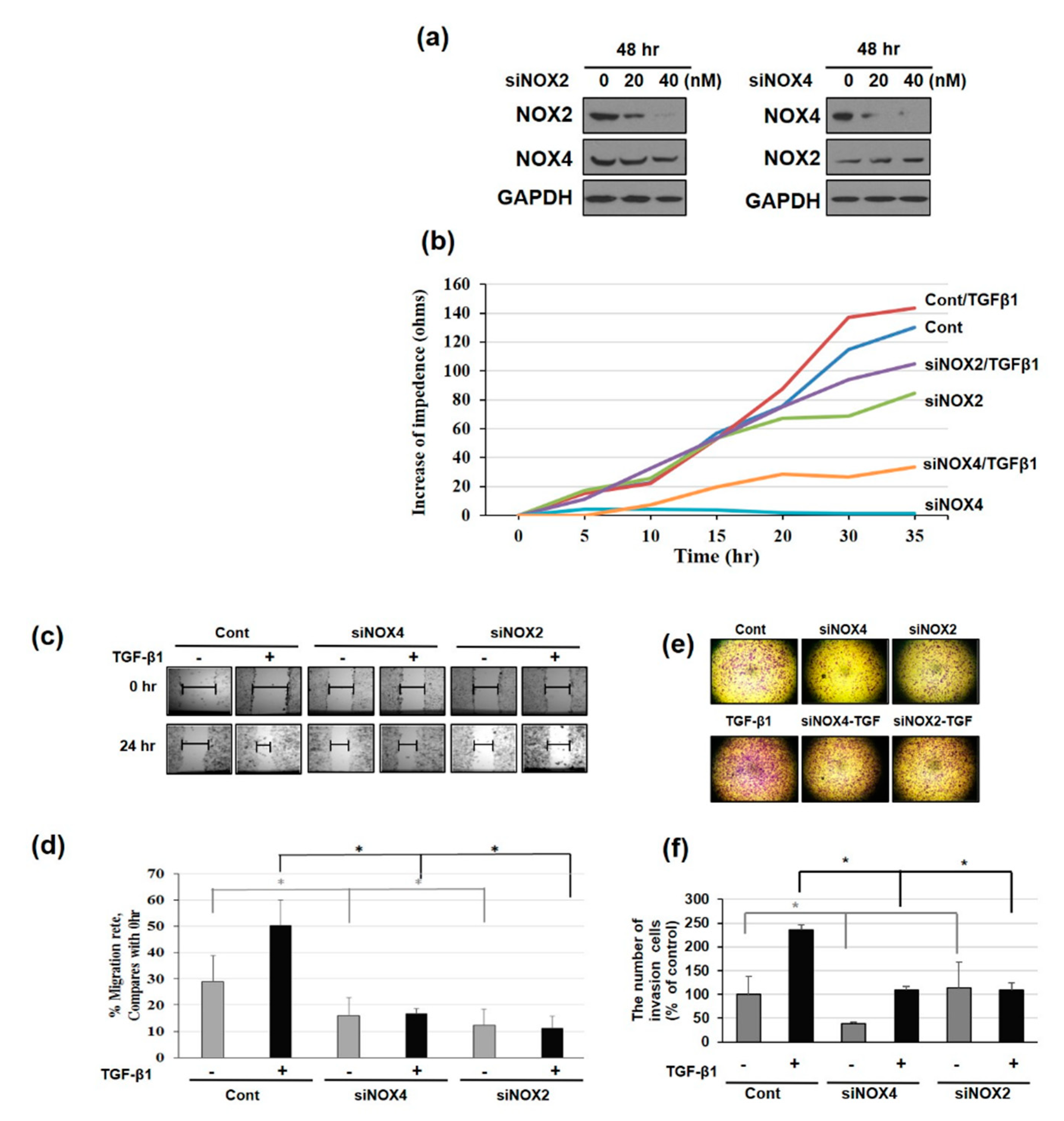
3.1. NAPDH Oxidase Isoforms—NOX2 and NOX4—Regulates EMT and Cell Migration in TGF-β1-Treated HeLa Cells
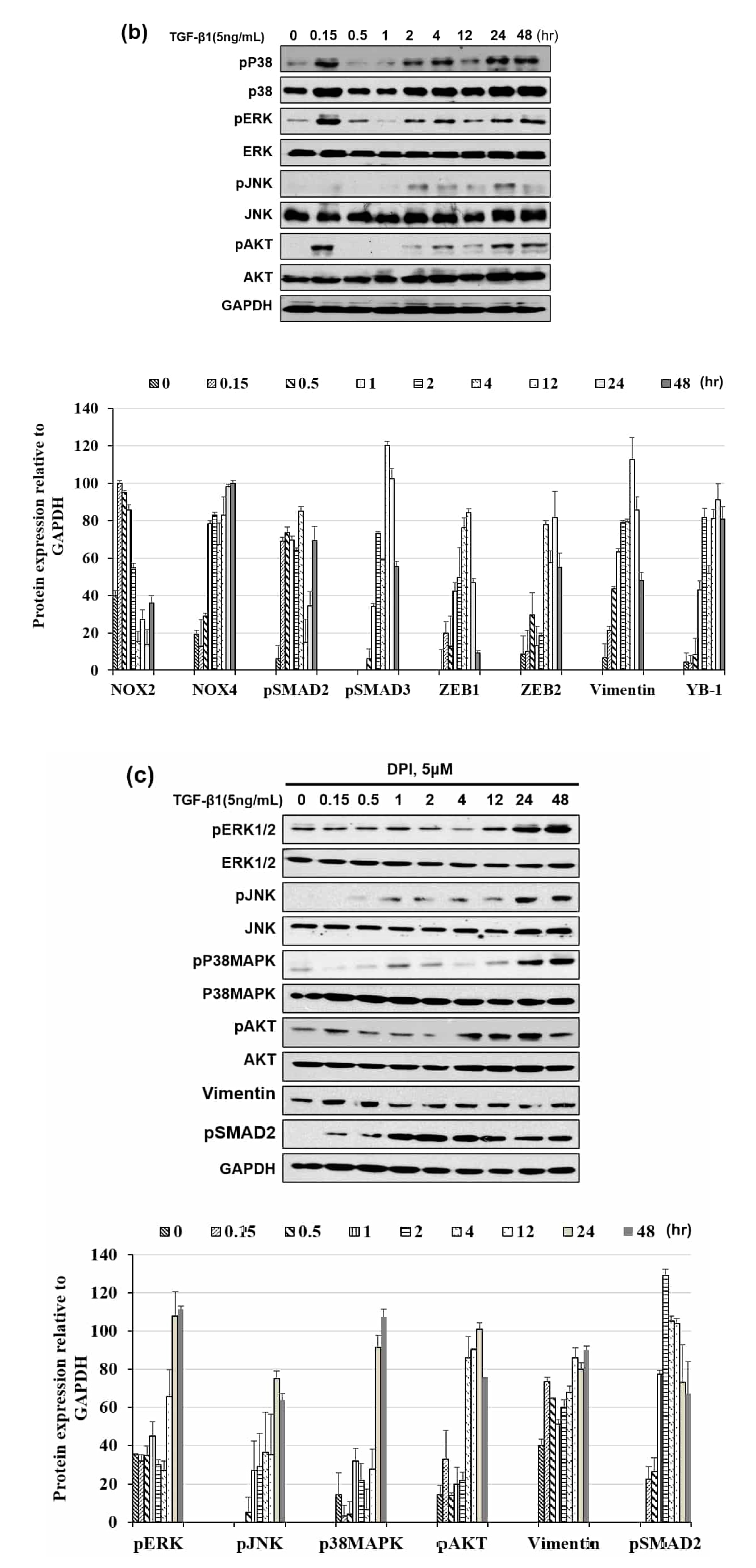
3.2. Distinct Pattern of NOX Isoforms’ Spatial-Temporal Expression in TGF-β1 Treated HeLa Cells
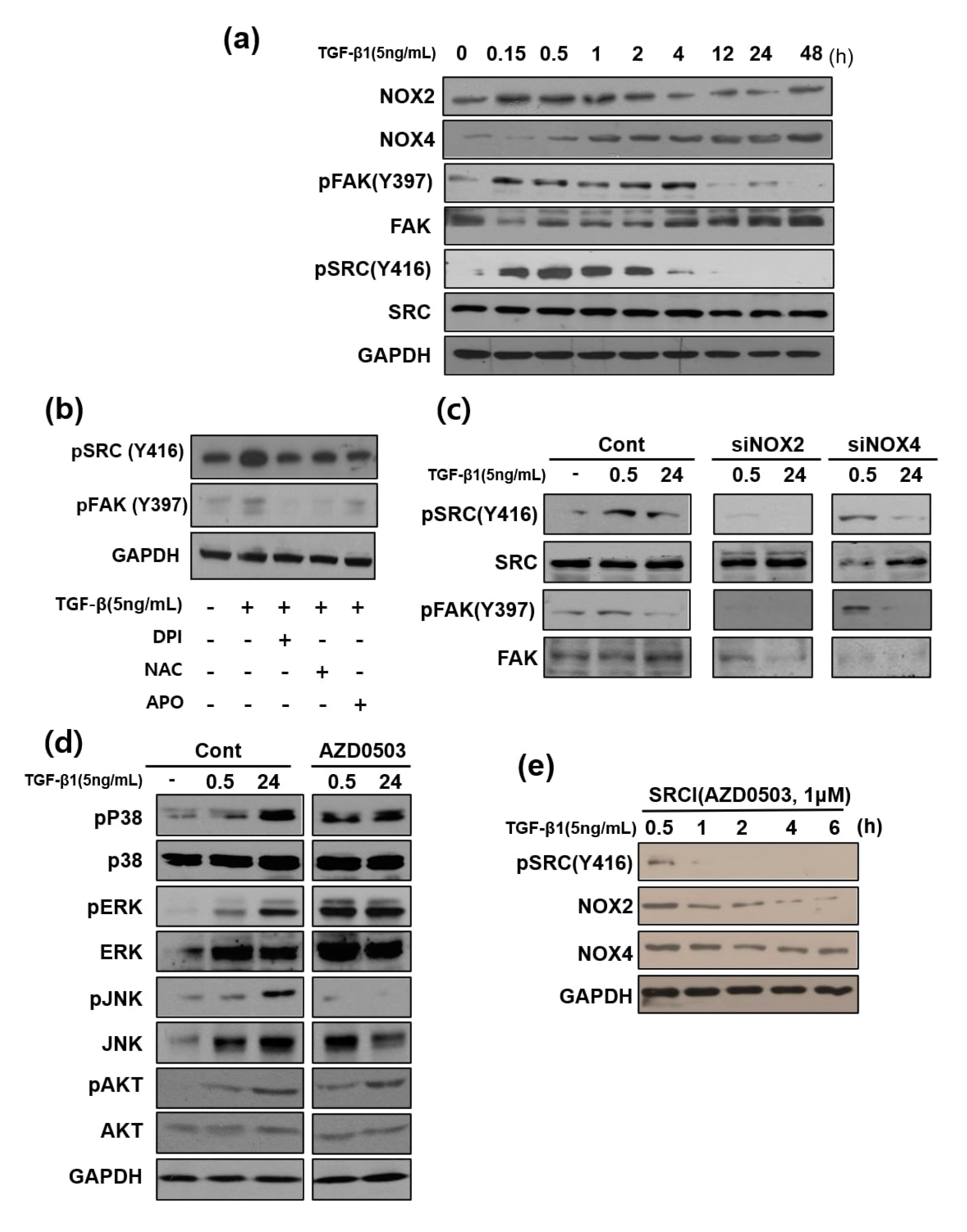
3.3. NOX Regulated Redox Signaling Mechanisms
3.4. Downregulating NOX2/4 Manifests Inhibition on Cell Motility and Proliferation
3.5. NOX4 Promotes Cell Proliferation Through Cyclin D and E Induction
3.6. Focal Adhesion Kinase and SRC Phosphorylation are Involved in TGF-β1-Mediated Cell Migration Via NOX2 Activation
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BrdU | 5-bromodeoxyuridine |
| DCFDA | 2′, 7′-dichlorfluorescein-diacetate |
| DMEM | Dulbecco’s modified Eagle medium |
| DPI | diphenyleneiodonium chloride |
| ECIS | electric cell substrate impedance sensing |
| ECM | extracellular matrix |
| EMT | epithelial–mesenchymal transition |
| FAK | focal adhesion kinase |
| FBS | fetal bovine serum |
| K14 | keratin 14 |
| MTT | 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| NOX | NADPH oxidase |
| PBS | phosphate-buffered saline |
| ROS | reactive oxygen species |
| RT-PCR | reverse transcription-polymerase chain reaction |
| NAC | N-acetyl-l-cysteine |
References
- Adams, E.K.; Chien, L.N.; Florence, C.S.; Raskind-Hood, C. The breast and cervical cancer prevention and treatment act in georgia: Effects on time to medicaid enrollment. Cancer 2009, 115, 1300–1309. [Google Scholar] [CrossRef]
- Savagner, P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21, 1–7. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T. Oxidative innate immune defenses by Nox/Duox Family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ackland, M.L.; Newgreen, D.F.; Fridman, M.; Waltham, M.C.; Arvanitis, A.; Minichiello, J.; Price, J.T.; Thompson, E.W. Epidermal growth factor-induced epithelio-mesenchymal transition in human breast carcinoma cells. Lab. Investig. 2003, 83, 435–448. [Google Scholar] [CrossRef]
- Gonzalez-Moreno, O.; Lecanda, J.; Green, J.E.; Segura, V.; Catena, R.; Serrano, D.; Calvo, A. VEGF elicits epithelial-mesenchymal transition (EMT) in prostate intraepithelial neoplasia (PIN)-like cells via an autocrine loop. Exp. Cell Res. 2010, 316, 554–567. [Google Scholar] [CrossRef]
- Wu, Q.; Hou, X.; Xia, J.; Qian, X.; Miele, L.; Sarkar, F.H.; Wang, Z. Emerging roles of PDGF-D in EMT progression during tumorigenesis. Cancer Treat. Rev. 2013, 39, 640–646. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Liu, Y.; De Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the tissue microenvironment: Relevance in fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef]
- Costanza, B.; Umelo, I.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-β in Cancer. J. Clin. Med. 2017, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal. Transduct. Target. Ther. 2017, 2, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Amara, N.; Goven, D.; Prost, F.; Muloway, R.; Crestani, B.; Boczkowski, J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 2010, 65, 733–738. [Google Scholar] [CrossRef]
- Carnesecchi, S.; Deffert, C.; Donati, Y.; Basset, O.; Hinz, B.; Preynat-Seauve, O.; Guichard, C.; Arbiser, J.L.; Banfi, B.; Pache, J.-C.; et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid. Redox Signal. 2011, 15, 607–619. [Google Scholar] [CrossRef]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem. J. 2010, 433, 393–402. [Google Scholar] [CrossRef]
- Hu, T.; Ramachandrarao, S.P.; Siva, S.; Valancius, C.; Zhu, Y.; Mahadev, K.; Toh, I.; Goldstein, B.J.; Woolkalis, M.; Sharma, K. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am. J. Physiology. Ren. Physiol. 2005, 289, F816–F825. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Tang, Y.; Fukai, T.; Dikalov, S.I.; Ma, Y.; Fujimoto, M.; Quinn, M.T.; Pagano, P.J.; Johnson, C.; Alexander, R.W. Novel role of gp91phox-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ. Res. 2002, 91, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Djamali, A.; Reese, S.; Hafez, O.; Vidyasagar, A.; Jacobson, L.; Swain, W.; Kolehmainen, C.; Huang, L.; Wilson, N.A.; Torrealba, J.R. Nox2 is a mediator of chronic CsA nephrotoxicity. Am. J. Transplant. 2012, 12, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; De Marchis, F.; Pusterla, T.; Conti, A.; Alessio, M.; Bianchi, M.E. Src family kinases are necessary for cell migration induced by extracellular HMGB1. J. Leukoc. Biol. 2009, 86, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. β3 Integrin and Src facilitate transforming growth factor-β mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, 1–16. [Google Scholar] [CrossRef]
- Galliher, A.J.; Schiemann, W.P. Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67, 3752–3758. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Fransvea, E.; Mazzocca, A.; Antonaci, S.; Giannelli, G. Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology 2009, 49, 839–850. [Google Scholar] [CrossRef]
- Kim, Y.M.; Cho, M. Activation of NADPH oxidase subunit NCF4 induces ROS-mediated EMT signaling in HeLa cells. Cell. Signal. 2014, 26, 784–796. [Google Scholar] [CrossRef]
- Muthuramalingam, K.; Cho, M.; Kim, Y. Role of NAPDH oxidase and its therapeutic intervention in TGF-β-mediated EMT progression: An in vitro analysis on HeLa cervical cancer cells. Appl. Biol. Chem. 2020, 63. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. A J. Virtual Libr. 2005, 10, 1881–1896. [Google Scholar] [CrossRef]
- Boudreau, H.E.; Casterline, B.W.; Burke, D.J.; Leto, T.L. Wild-type and mutant p53 differentially regulate NADPH oxidase 4 in TGF-β-mediated migration of human lung and breast epithelial cells. Br. J. Cancer 2014, 110, 2569–2582. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Fujii, T.; Anai, S.; Fujimoto, K.; Konishi, N. ROS generation via NOX4 and its utility in the cytological diagnosis of urothelial carcinoma of the urinary bladder. Bmc Urol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G 2 -M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases (NOX): Insights into selected functions and mechanisms of action in cancer and stem cells. Oxidative Med. Cell. Longev. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Jarman, E.R.; Khambata, V.S.; Cope, C.; Jones, P.; Roger, J.; Ye, L.Y.; Duggan, N.; Head, D.; Pearce, A.; Press, N.J.; et al. An Inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 2014, 50, 158–169. [Google Scholar] [CrossRef]
- Felty, Q.; Singh, K.P.; Roy, D. Estrogen-induced G1/S transition of G0-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene 2005, 24, 4883–4893. [Google Scholar] [CrossRef]
- Menon, S.G.; Coleman, M.C.; Walsh, S.A.; Spitz, D.R.; Goswami, P.C. Differential susceptibility of nonmalignant human breast epithelial cells and breast cancer cells to Thiol antioxidant-induced G 1 -Dela. Antioxid. Redox Signal. 2005, 7, 711–718. [Google Scholar] [CrossRef]
- Alam, H.; Sehgal, L.; Kundu, S.T.; Dalal, S.N.; Vaidya, M.M. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol. Biol. Cell 2011, 22, 4068–4078. [Google Scholar] [CrossRef]
- Somasiri, A.; Howarth, A.; Goswami, D.; Dedhar, S.; Roskelley, C.D. Overexpression of the integrin-linked kinase mesenchymally transforms mammary epithelial cells. J. Cell Sci. 2001, 114, 1125–1136. [Google Scholar]
- Bianchi, A.; Gervasi, M.E.; Bakin, A.V. Role of β5-integrin in epithelial-mesenchymal transition in response to TGFβ. Cell Cycle 2010, 9, 1647–1659. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Wei, W.C.; Wang, Y.K.; Lin, S.C.; Sung, J.M.; Tang, M.J. Transforming growth factor-β1 induces Smad3-dependent β1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am. J. Pathol. 2010, 177, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.Y.; Chiao, C.C.; Kuo, W.Y.; Hsiao, Y.C.; Chen, Y.J.; Wei, Y.Y.; Lai, T.H.; Fong, Y.C.; Tang, C.H. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-κB-dependent pathway in human chondrosarcoma cells. Biochem. Pharmacol. 2008, 75, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Cao, S.; Kang, N.; Huebert, R.C.; Shah, V.H. Fibronectin induces endothelial cell migration through β1 integrin and src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J. Biol. Chem. 2012, 287, 7190–7202. [Google Scholar] [CrossRef]
- Nam, E.H.; Lee, Y.; Park, Y.K.; Lee, J.W.; Kim, S. Zeb2 upregulates integrin α5 expression through cooperation with sp1 to induce invasion during epithelial-mesenchymal transition of human cancer cells. Carcinogenesis 2012, 33, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Radisky, D.C.; Yang, D.; Xu, R.; Radisky, E.S.; Bissell, M.J.; Bishop, J.M. MYC suppresses cancer metastasis by direct transcriptional silencing of α v and β 3 integrin subunits. Nat. Cell Biol. 2012, 14, 567–574. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| NOX2 | CAACCTGGAAGGCTACCACT | CCTCATTCACAGCGCAGT |
| NOX4 | CCAAGCAGGAGAACCAGGAG | GCAACGTCAGCAGCATGTAG |
| Snail | GAGGACAGTGGGAAAGGCTC | TGGCTTCGCATGTGCATCTT |
| Slug | GAACTCACACGGGGGAGAAG | ACACAGCAGCCAGATTCCTC |
| Vimentin | AATGGCTCGTCACCTTCGTGAAT | CAGATTAGTTTTCCCTCAGGTTCAG |
| E-cadherin | CCTGGGACTCCACCTACAGA | GGATGACACAGCGTGAGAGA |
| GAPDH | GAGAAGGCTGGGGCTCATTT | ACTGATGGCATGGACTGTGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.M.; Muthuramalingam, K.; Cho, M. Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells 2020, 9, 1555. https://doi.org/10.3390/cells9061555
Kim YM, Muthuramalingam K, Cho M. Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells. 2020; 9(6):1555. https://doi.org/10.3390/cells9061555
Chicago/Turabian StyleKim, Young Mee, Karthika Muthuramalingam, and Moonjae Cho. 2020. "Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells" Cells 9, no. 6: 1555. https://doi.org/10.3390/cells9061555
APA StyleKim, Y. M., Muthuramalingam, K., & Cho, M. (2020). Redox Regulation of NOX Isoforms on FAK(Y397)/SRC(Y416) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells, 9(6), 1555. https://doi.org/10.3390/cells9061555