Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Preparation of Isolated Mitochondria from Cortical and Hippocampal Tissues
2.3. Measurement of Mitochondrial Respiration Rates in Cortex and Hippocampus
2.4. Western Blot Analysis
2.5. Statistical Analysis
3. Results
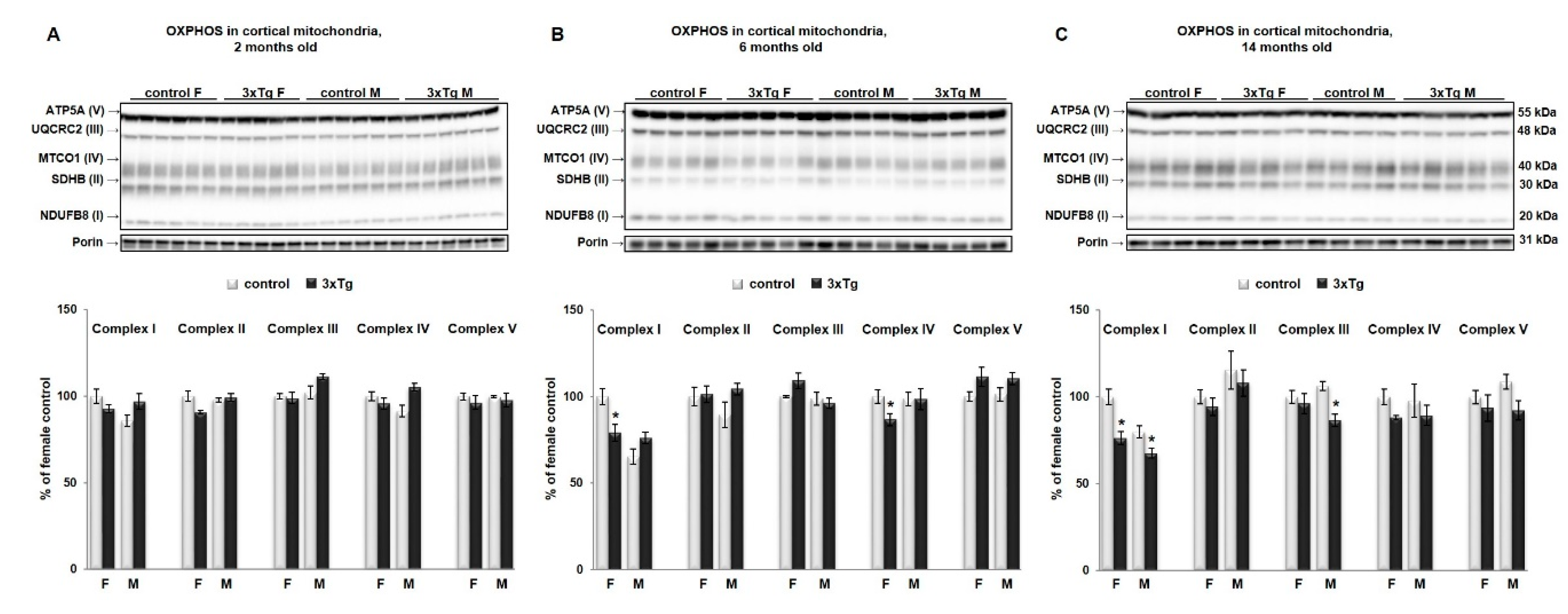
3.1. Bioenergetic Profiles in Isolated Mitochondria from Cortex and Hippocampus of 3xTg Mice at Different Ages
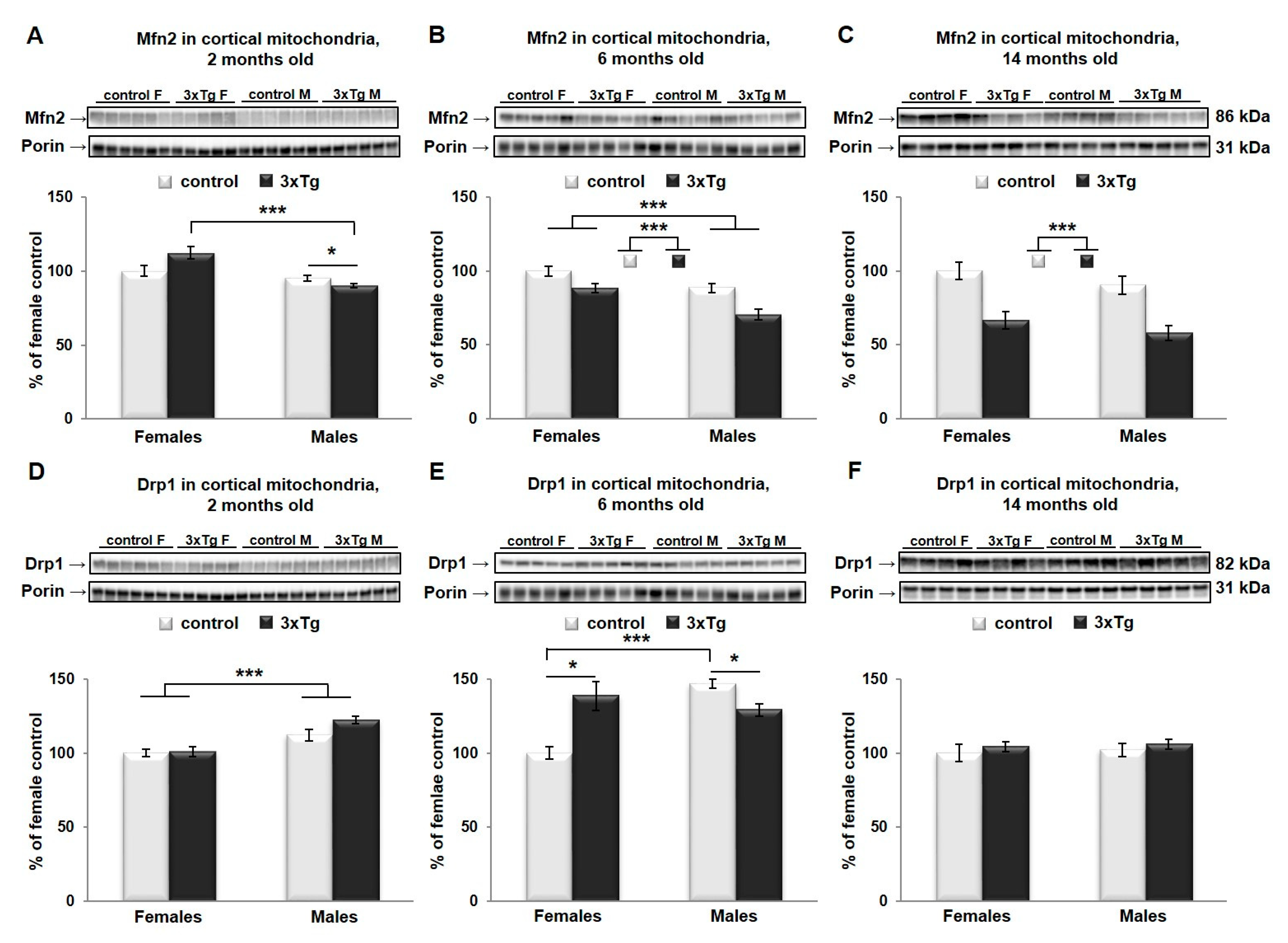
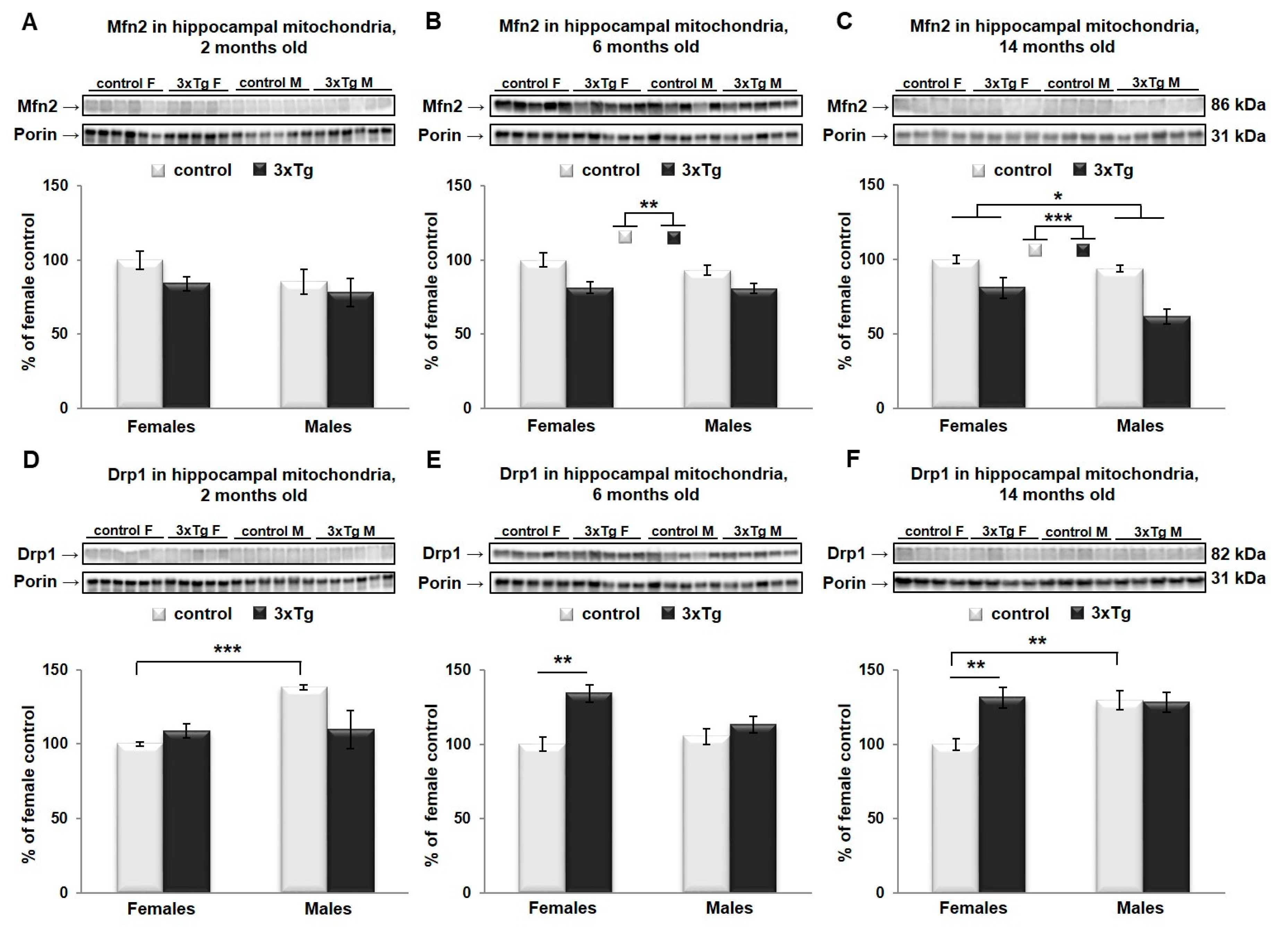
3.2. Changes in Mitochondrial Morphology in 3xTg Mouse Brain
3.3. Uncoupling Protein 4 in Mitochondria of 3xTg Mouse Brain
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Snyder, H.M.; Asthana, S.; Bain, L.; Brinton, R.; Craft, S.; Dubal, D.B.; Espeland, M.A.; Gatz, M.; Mielke, M.M.; Raber, J.; et al. Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the Women’s Alzheimer’s Research Initiative. Alzheimers Dement. 2016, 12, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Podcasy, J.L.; Epperson, C.N. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 2016, 18, 437–446. [Google Scholar] [PubMed]
- Arnold, S.E. Part III. Neuropathology of Alzheimer’s disease. Dis. Mon. 2000, 46, 687–705. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef]
- Swerdlow, R.; Marcus, D.L.; Landman, J.; Kooby, D.; Frey, W., 2nd; Freedman, M.L. Brain glucose metabolism in Alzheimer’s disease. Am. J. Med. Sci. 1994, 308, 141–144. [Google Scholar] [CrossRef]
- Kanfer, J.N.; Singh, I.N.; Pettegrew, J.W.; McCartney, D.G.; Sorrentino, G. Phospholipid metabolism in Alzheimer’s disease and in a human cholinergic cell. J. Lipid Mediat. Cell Signal. 1996, 14, 361–363. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Djordjevic, J.; Albensi, B.C.; Fernyhough, P. Simultaneous evaluation of substrate-dependent oxygen consumption rates and mitochondrial membrane potential by TMRM and safranin in cortical mitochondria. Biosci. Rep. 2015, 36, e00286. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional hypometabolism in the 3xTg mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Galeffi, F.; Gerich, F.J.; Turner, D.A.; Muller, M. Optical and pharmacological tools to investigate the role of mitochondria during oxidative stress and neurodegeneration. Prog. Neurobiol. 2006, 79, 136–171. [Google Scholar] [CrossRef]
- Castellani, R.; Hirai, K.; Aliev, G.; Drew, K.L.; Nunomura, A.; Takeda, A.; Cash, A.D.; Obrenovich, M.E.; Perry, G.; Smith, M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 357–360. [Google Scholar] [CrossRef]
- McBride, H.; Scorrano, L. Mitochondrial dynamics and physiology. Biochim. Biophys. Acta 2013, 1833, 148–149. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, J.K.; Kang, W.S.; Kim, S.K.; Han, C.; Na, H.R.; Park, H.J.; Kim, J.W.; Kim, Y.Y.; Park, M.H.; et al. Association between Mitofusin 2 Gene Polymorphisms and Late-Onset Alzheimer’s Disease in the Korean Population. Psychiatry Investig. 2017, 14, 81–85. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 2016, 1862, 814–828. [Google Scholar] [CrossRef]
- Zhu, X.; Perry, G.; Smith, M.A.; Wang, X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S253–S262. [Google Scholar] [CrossRef]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, Z.H.; Chan, K.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial neuronal uncoupling proteins: A target for potential disease-modification in Parkinson’s disease. Transl. Neurodegener. 2012, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Rose, G.; Crocco, P.; De Rango, F.; Montesanto, A.; Passarino, G. Further support to the uncoupling-to-survive theory: The genetic variation of human UCP genes is associated with longevity. PLoS ONE 2011, 6, e29650. [Google Scholar] [CrossRef] [PubMed]
- Montesanto, A.; Crocco, P.; Dato, S.; Geracitano, S.; Frangipane, F.; Colao, R.; Maletta, R.; Passarino, G.; Bruni, A.C.; Rose, G. Uncoupling protein 4 (UCP4) gene variability in neurodegenerative disorders: Further evidence of association in Frontotemporal dementia. Aging (Albany NY) 2018, 10, 3283–3293. [Google Scholar] [CrossRef]
- Smorodchenko, A.; Rupprecht, A.; Sarilova, I.; Ninnemann, O.; Brauer, A.U.; Franke, K.; Schumacher, S.; Techritz, S.; Nitsch, R.; Schuelke, M.; et al. Comparative analysis of uncoupling protein 4 distribution in various tissues under physiological conditions and during development. Biochim. Biophys. Acta 2009, 1788, 2309–2319. [Google Scholar] [CrossRef]
- Vandal, M.; White, P.J.; Chevrier, G.; Tremblay, C.; St-Amour, I.; Planel, E.; Marette, A.; Calon, F. Age-dependent impairment of glucose tolerance in the 3xTg-AD mouse model of Alzheimer’s disease. FASEB J. 2015, 29, 4273–4284. [Google Scholar] [CrossRef]
- Carroll, J.C.; Rosario, E.R.; Kreimer, S.; Villamagna, A.; Gentzschein, E.; Stanczyk, F.Z.; Pike, C.J. Sex differences in beta-amyloid accumulation in 3xTg-AD mice: Role of neonatal sex steroid hormone exposure. Brain Res. 2010, 1366, 233–245. [Google Scholar] [CrossRef]
- Clinton, L.K.; Billings, L.M.; Green, K.N.; Caccamo, A.; Ngo, J.; Oddo, S.; McGaugh, J.L.; LaFerla, F.M. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol. Dis. 2007, 28, 76–82. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Fukumoto, N.; Fujii, T.; Combarros, O.; Kamboh, M.I.; Tsai, S.J.; Matsushita, S.; Nacmias, B.; Comings, D.E.; Arboleda, H.; Ingelsson, M.; et al. Sexually dimorphic effect of the Val66Met polymorphism of BDNF on susceptibility to Alzheimer’s disease: New data and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 235–242. [Google Scholar] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Dranka, B.P.; Zou, L.; Chatham, J.C.; Darley-Usmar, V.M. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem. J. 2009, 424, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Nickens, K.P.; Wikstrom, J.D.; Shirihai, O.S.; Patierno, S.R.; Ceryak, S. A bioenergetic profile of non-transformed fibroblasts uncovers a link between death-resistance and enhanced spare respiratory capacity. Mitochondrion 2013, 13, 662–667. [Google Scholar] [CrossRef]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann. Neurol. 2015. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Drose, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Muller, W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Park, B.S.; Jung, Y.; Reddy, P.H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromol. Med. 2004, 5, 147–162. [Google Scholar] [CrossRef]
- Duborjal, H.; Beugnot, R.; Mousson de Camaret, B.; Issartel, J.P. Large functional range of steady-state levels of nuclear and mitochondrial transcripts coding for the subunits of the human mitochondrial OXPHOS system. Genome Res. 2002, 12, 1901–1909. [Google Scholar] [CrossRef][Green Version]
- Garbian, Y.; Ovadia, O.; Dadon, S.; Mishmar, D. Gene expression patterns of oxidative phosphorylation complex I subunits are organized in clusters. PLoS ONE 2010, 5, e9985. [Google Scholar] [CrossRef]
- van Waveren, C.; Moraes, C.T. Transcriptional co-expression and co-regulation of genes coding for components of the oxidative phosphorylation system. BMC Genom. 2008, 9, 18. [Google Scholar] [CrossRef]
- Telford, J.E.; Kilbride, S.M.; Davey, G.P. Complex I is rate-limiting for oxygen consumption in the nerve terminal. J. Biol. Chem. 2009, 284, 9109–9114. [Google Scholar] [CrossRef]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef]
- Grimm, A.; Lim, Y.A.; Mensah-Nyagan, A.G.; Gotz, J.; Eckert, A. Alzheimer’s disease, oestrogen and mitochondria: An ambiguous relationship. Mol. Neurobiol. 2012, 46, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Han, S.H.; Son, S.M.; Hong, H.S.; Choi, Y.J.; Byun, J.; Mook-Jung, I. Mitochondria-specific accumulation of amyloid beta induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE 2012, 7, e34929. [Google Scholar] [CrossRef] [PubMed]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Bokde, A.L.; Pietrini, P.; Ibanez, V.; Furey, M.L.; Alexander, G.E.; Graff-Radford, N.R.; Rapoport, S.I.; Schapiro, M.B.; Horwitz, B. The effect of brain atrophy on cerebral hypometabolism in the visual variant of Alzheimer disease. Arch. Neurol. 2001, 58, 480–486. [Google Scholar] [CrossRef]
- Mielke, M.M.; Vemuri, P.; Rocca, W.A. Clinical epidemiology of Alzheimer’s disease: Assessing sex and gender differences. Clin. Epidemiol. 2014, 6, 37–48. [Google Scholar] [CrossRef]
- Yu, W.; Bonnet, M.; Farso, M.; Ma, K.; Chabot, J.G.; Martin, E.; Torriglia, A.; Guan, Z.; McLaurin, J.; Quirion, R.; et al. The expression of apoptosis inducing factor (AIF) is associated with aging-related cell death in the cortex but not in the hippocampus in the TgCRND8 mouse model of Alzheimer’s disease. BMC Neurosci 2014, 15, 73. [Google Scholar] [CrossRef]
- Yang, J.T.; Wang, Z.J.; Cai, H.Y.; Yuan, L.; Hu, M.M.; Wu, M.N.; Qi, J.S. Sex Differences in Neuropathology and Cognitive Behavior in APP/PS1/tau Triple-Transgenic Mouse Model of Alzheimer’s Disease. Neurosci. Bull. 2018, 34, 736–746. [Google Scholar] [CrossRef]
- Hua, X.; Hibar, D.P.; Lee, S.; Toga, A.W.; Jack, C.R., Jr.; Weiner, M.W.; Thompson, P.M.; Alzheimer’s Disease Neuroimaging, I. Sex and age differences in atrophic rates: An ADNI study with n=1368 MRI scans. Neurobiol. Aging 2010, 31, 1463–1480. [Google Scholar] [CrossRef]
- Skup, M.; Zhu, H.; Wang, Y.; Giovanello, K.S.; Lin, J.A.; Shen, D.; Shi, F.; Gao, W.; Lin, W.; Fan, Y.; et al. Alzheimer’s Disease Neuroimaging, I., Sex differences in grey matter atrophy patterns among AD and aMCI patients: Results from ADNI. Neuroimage 2011, 56, 890–906. [Google Scholar] [CrossRef]
- Djordjevic, J.; Thomson, E.; Chowdhury, S.R.; Snow, W.M.; Perez, C.; Wong, T.P.; Fernyhough, P.; Albensi, B.C. Brain region- and sex-specific alterations in mitochondrial function and NF-kappaB signaling in the TgCRND8 mouse model of Alzheimer’s disease. Neuroscience 2017, 361, 81–92. [Google Scholar] [CrossRef]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.W.; Marciniack, C.; Laferla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 2012, 1820, 553–564. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef]
- Deak, F.; Freeman, W.M.; Ungvari, Z.; Csiszar, A.; Sonntag, W.E. Recent Developments in Understanding Brain Aging: Implications for Alzheimer’s Disease and Vascular Cognitive Impairment. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Guevara, R.; Santandreu, F.M.; Valle, A.; Gianotti, M.; Oliver, J.; Roca, P. Sex-dependent differences in aged rat brain mitochondrial function and oxidative stress. Free Radic Biol. Med. 2009, 46, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, M.; Czapski, G.A.; Wojtowicz, S.; Wieczorek, I.; Wencel, P.L.; Strosznajder, R.P.; Jaber, V.; Lukiw, W.J.; Strosznajder, J.B. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid beta Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2019. [Google Scholar]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Fang, D.; Yan, S.; Yu, Q.; Chen, D.; Yan, S.S. Mfn2 is Required for Mitochondrial Development and Synapse Formation in Human Induced Pluripotent Stem Cells/hiPSC Derived Cortical Neurons. Sci. Rep. 2016, 6, 31462. [Google Scholar] [CrossRef]
- Chan, S.L.; Liu, D.; Kyriazis, G.A.; Bagsiyao, P.; Ouyang, X.; Mattson, M.P. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J. Biol. Chem. 2006, 281, 37391–37403. [Google Scholar] [CrossRef]
- Pfeiffer, M.; Kayzer, E.B.; Yang, X.; Abramson, E.; Kenaston, M.A.; Lago, C.U.; Lo, H.H.; Sedensky, M.M.; Lunceford, A.; Clarke, C.F.; et al. Caenorhabditis elegans UCP4 protein controls complex II-mediated oxidative phosphorylation through succinate transport. J. Biol. Chem. 2011, 286, 37712–37720. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, R.; Kempuraj, D.; Zaheer, S.; Raikwar, S.; Ahmed, M.E.; Selvakumar, G.P.; Iyer, S.S.; Zaheer, A. Glia Maturation Factor and Mitochondrial Uncoupling Proteins 2 and 4 Expression in the Temporal Cortex of Alzheimer’s Disease Brain. Front. Aging Neurosci. 2017, 9, 150. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djordjevic, J.; Roy Chowdhury, S.; Snow, W.M.; Perez, C.; Cadonic, C.; Fernyhough, P.; Albensi, B.C. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells 2020, 9, 1541. https://doi.org/10.3390/cells9061541
Djordjevic J, Roy Chowdhury S, Snow WM, Perez C, Cadonic C, Fernyhough P, Albensi BC. Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells. 2020; 9(6):1541. https://doi.org/10.3390/cells9061541
Chicago/Turabian StyleDjordjevic, Jelena, Subir Roy Chowdhury, Wanda M. Snow, Claudia Perez, Chris Cadonic, Paul Fernyhough, and Benedict C. Albensi. 2020. "Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice" Cells 9, no. 6: 1541. https://doi.org/10.3390/cells9061541
APA StyleDjordjevic, J., Roy Chowdhury, S., Snow, W. M., Perez, C., Cadonic, C., Fernyhough, P., & Albensi, B. C. (2020). Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3xTg Alzheimer’s Mice. Cells, 9(6), 1541. https://doi.org/10.3390/cells9061541