Huntington’s Disease—An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components

 and
and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Cytoskeleton Plays an Important Role in the Neuronal Morphology, Plasticity, and Neurodegenerative Processes
2. Microtubules (MTs) and Their Properties Necessary for the Neuron’s Development and Functioning
3. The Role of Endogenous MT Modulators and Tubulin Post-Translational Modifications in the Development of the Nervous System Diseases
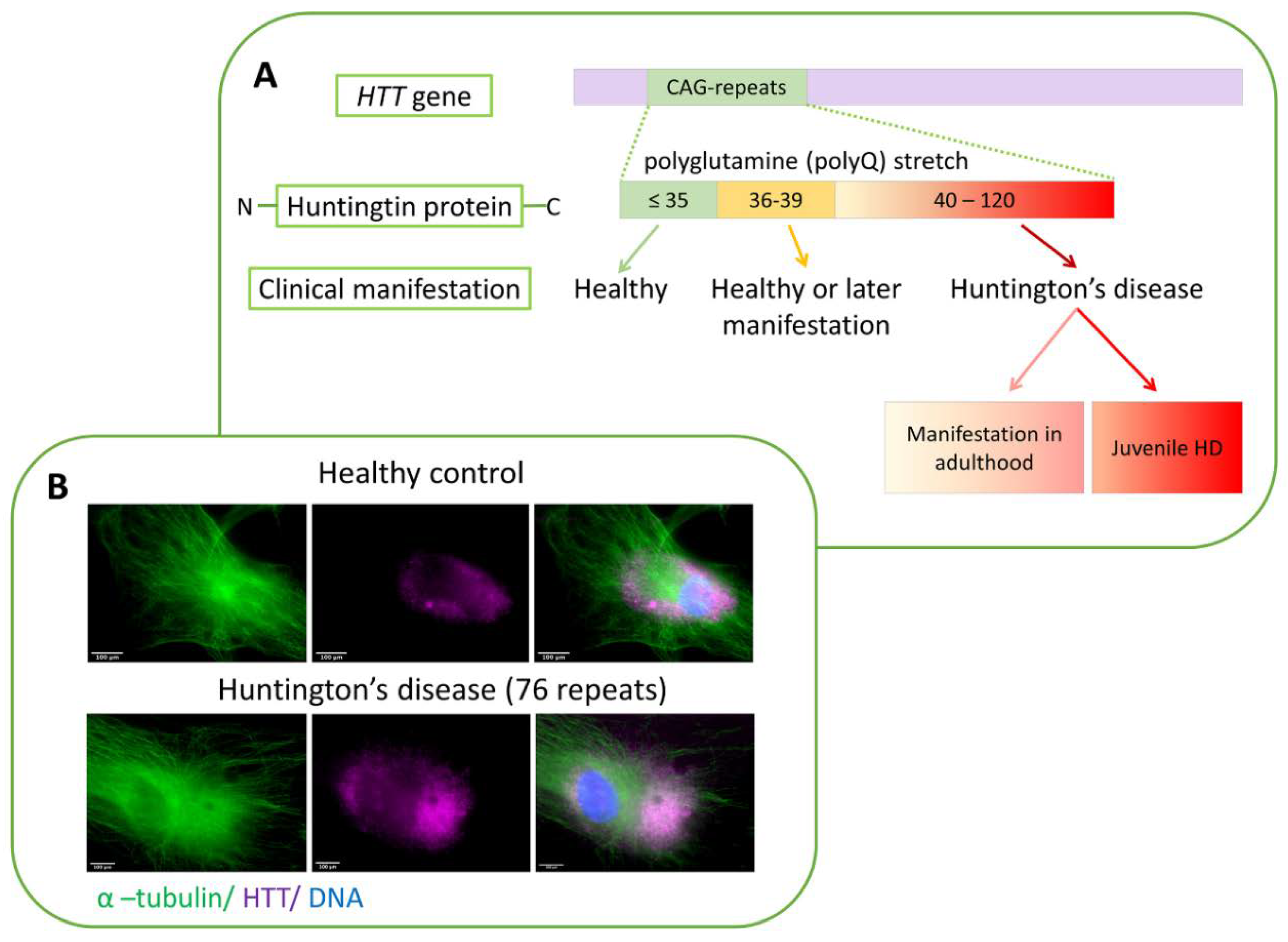
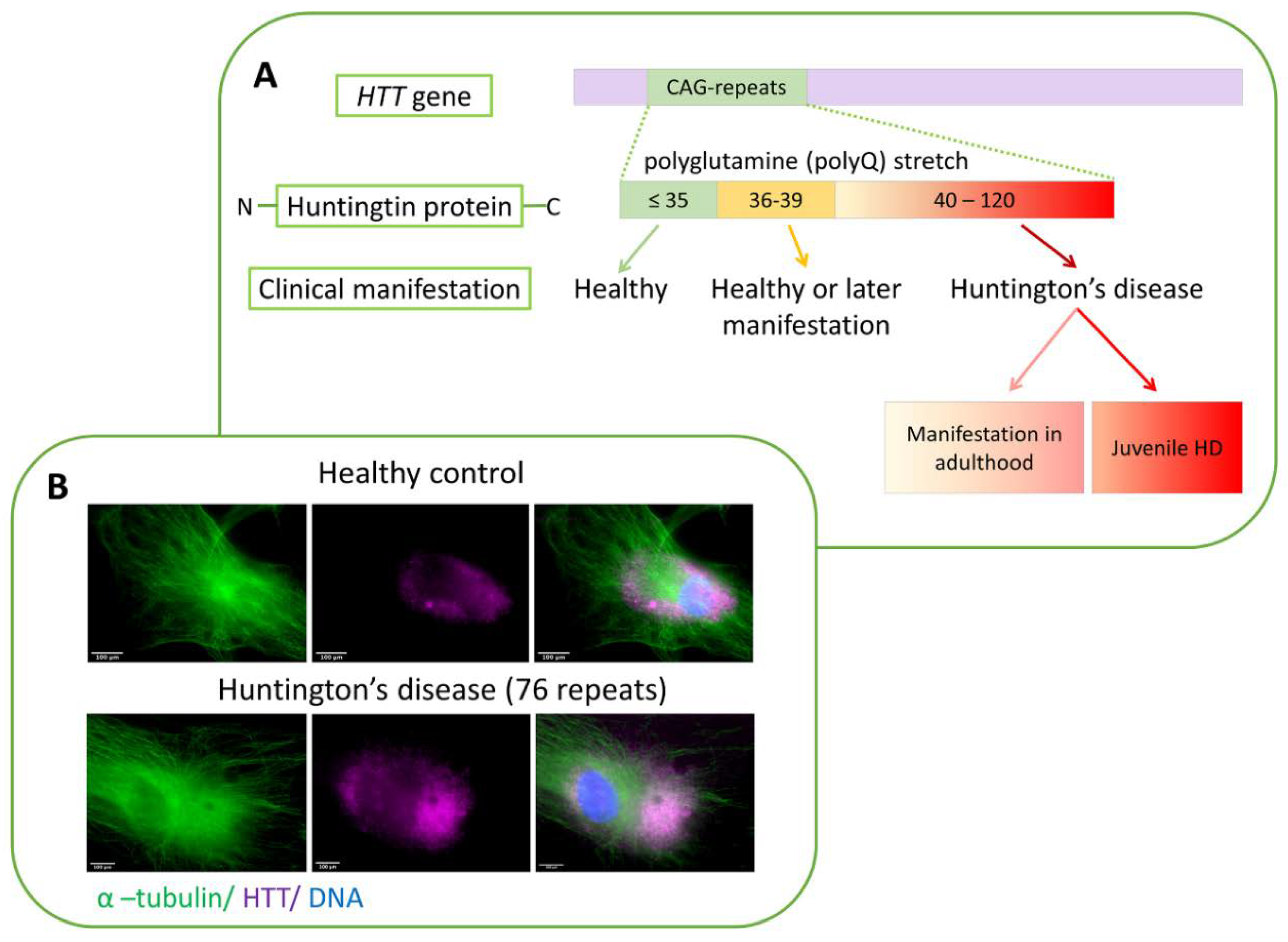
4. Huntington’s Disease—A History of Studies and Unanswered Questions
5. Intracellular Localization of Huntingtin and Its Association with the Cell Cytoskeleton
6. HTT Involvement in Cell Life Processes: Normal Protein Functions and Toxicity of the Mutant Form of HTT
7. The Involvement of HTT in Vesicular Transport and Endocytosis
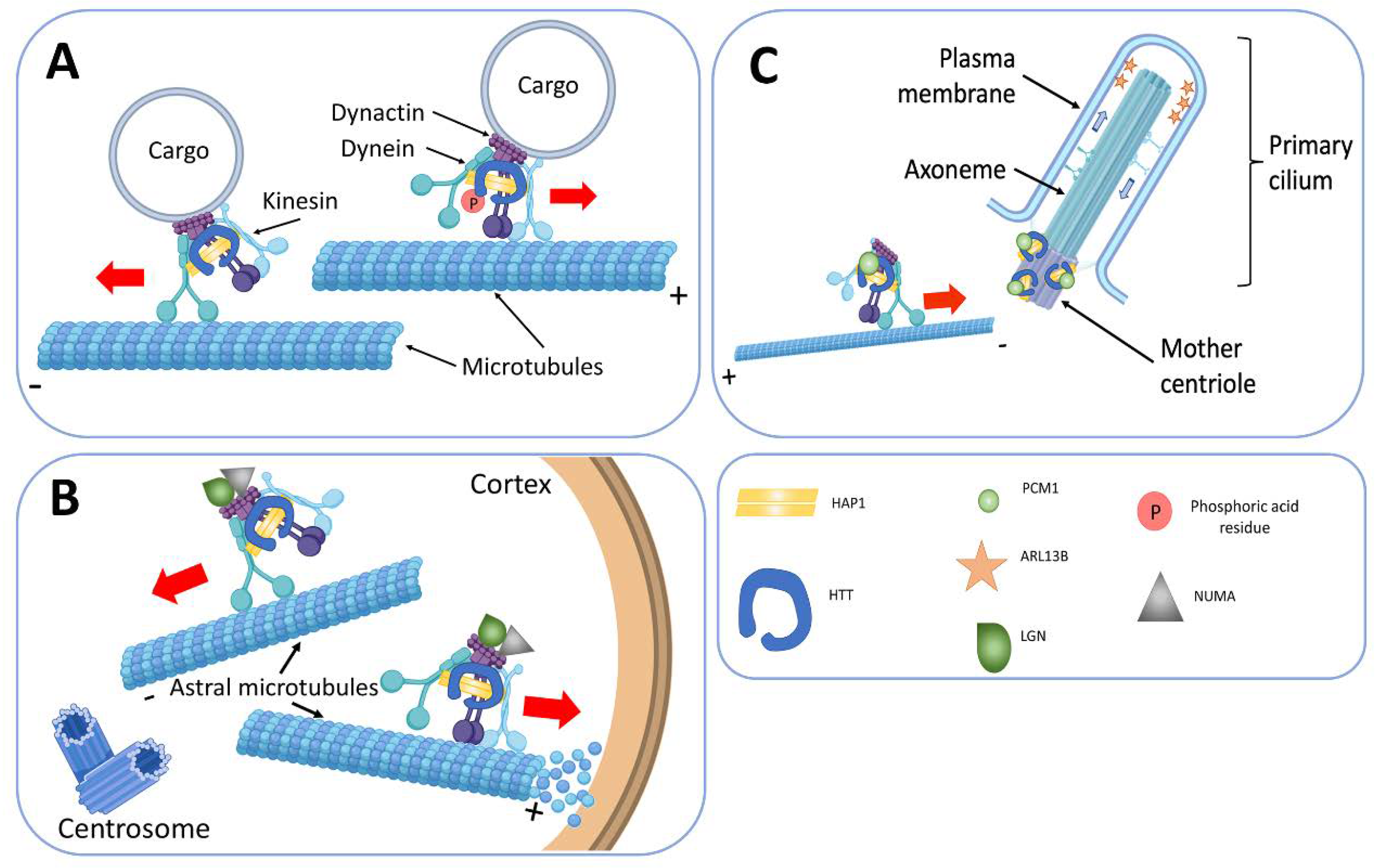
8. HTT and Regulation of the Primary Cilia Formation
9. HTT in the Process of Cell Division
10. Conclusions and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Penazzi, L.; Bakota, L.; Brandt, R. Microtubule dynamics in neuronal development, plasticity, and neurodegeneration. Int. Rev. Cell Mol. Biol. 2016, 321, 89–169. [Google Scholar] [CrossRef]
- Meiring, J.C.; Akhmanova, A. Microtubules keep large cells in shape. J. Cell Biol. 2020, 219, 202004031. [Google Scholar] [CrossRef]
- Alieva, I.B. Role of microtubule cytoskeleton in regulation of endothelial barrier function. Biochemistry 2014, 79, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.; Alieva, I.; Khromova, N.; Kireev, I.I.; Gunning, P.W.; Kopnin, P.B. Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and γ-actin in epithelial cells. Oncotarget 2016, 7, 72699–72715. [Google Scholar] [CrossRef] [Green Version]
- Kopf, A.; Renkawitz, J.; Hauschild, R.; Girkontaite, I.; Tedford, K.; Merrin, J.; Thorn-Seshold, O.; Trauner, D.; Häcker, H.; Fischer, K.-D.; et al. Microtubules control cellular shape and coherence in amoeboid migrating cells. J. Cell Biol. 2020, 219, 219. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C.; Broisy, G.G.; Taylor, E.W. Colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 1968, 7, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Amos, L.; Klug, A. Arrangement of subunits in flagellar microtubules. J. Cell Sci. 1974, 14, 523–549. [Google Scholar] [PubMed]
- Allen, C.; Borisy, G.G. Structural polarity and directional growth of microtubules of Chlamydomonas flagella. J. Mol. Biol. 1974, 90, 381–402. [Google Scholar] [CrossRef]
- Mitchison, T.; Kirschner, M. Dynamic instability of microtubule growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef]
- Walker, R.A.; O’Brien, E.T.; Pryer, N.K.; Soboeiro, M.F.; A Voter, W.; Erickson, H.P.; Salmon, E.D. Dynamic instability of individual microtubules analyzed by video light microscopy: Rate constants and transition frequencies. J. Cell Biol. 1988, 107, 1437–1448. [Google Scholar] [CrossRef]
- Desai, A.; Mitchison, T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelden, E.; Wadsworth, P. Observation and quantification of individual microtubule behavior in vivo: Microtubule dynamics are cell-type specific. J. Cell Biol. 1993, 120, 935–945. [Google Scholar] [CrossRef]
- O’Brien, E.T.; Salmon, E.D.; Walker, R.A.; Erickson, H.P. Effects of magnesium on the dynamic instability of individual microtubules. Biochemistry 1990, 29, 6648–6656. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gildersleeve, R.F.; Cross, A.R.; E Cullen, K.; Fagen, A.P.; Williams, R.C. Microtubules grow and shorten at intrinsically variable rates. J. Biol. Chem. 1992, 267, 7995–8006. [Google Scholar] [PubMed]
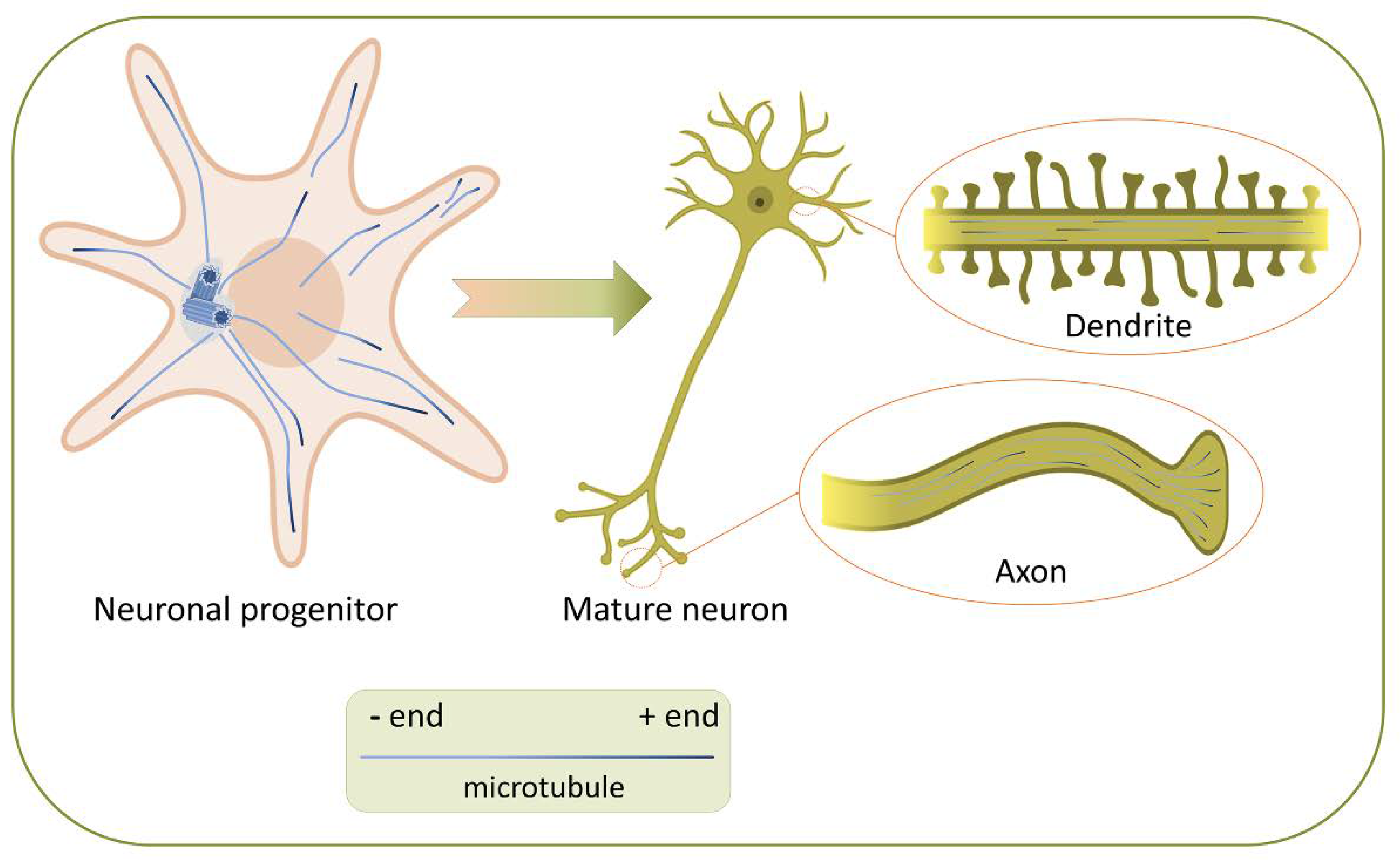
- Hoogenraad, C.C.; Bradke, F. Control of neuronal polarity and plasticity – a renaissance for microtubules? Trends Cell Biol. 2009, 19, 669–676. [Google Scholar] [CrossRef]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Kapitein, L.C.; Hoogenraad, C.C. Building the neuronal microtubule cytoskeleton. Neuron 2015, 87, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Kuijpers, M.; Hoogenraad, C.C. Centrosomes, microtubules and neuronal development. Mol. Cell. Neurosci. 2011, 48, 349–358. [Google Scholar] [CrossRef]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Dehmelt, L.; Nalbant, P.; Steffen, W.; Halpain, S. A microtubule-based, dynein-dependent force induces local cell protrusions: Implications for neurite initiation. Brain Cell Biol. 2007, 35, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Witte, H.; Neukirchen, D.; Bradke, F. Microtubule stabilization specifies initial neuronal polarization. J. Cell Biol. 2008, 180, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraldo, S.; Gordon-Weeks, P. Cytoskeletal dynamics in growth-cone steering. J. Cell Sci. 2009, 122, 3595–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, K.W.; Schätzle, P.; Tortosa, E.; Pagés, S.; Holtmaat, A.; Kapitein, L.C.; Hoogenraad, C.C. Dendrites in vitro and in vivo contain microtubules of opposite polarity and axon formation correlates with uniform plus-end-out microtubule orientation. J. Neurosci. 2016, 36, 1071–1085. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Viesselmann, C.; Nam, S.; Merriam, E.; Dent, E.W. Activity-dependent dynamic microtubule invasion of dendritic spines. J. Neurosci. 2008, 28, 13094–13105. [Google Scholar] [CrossRef]
- Merriam, E.B.; Millette, M.; Lumbard, D.C.; Saengsawang, W.; Fothergill, T.; Hu, X.; Ferhat, L.; Dent, E.W. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J. Neurosci. 2013, 33, 16471–16482. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Black, M.M.; A Banker, G. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 1989, 109, 3085–3094. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Deitch, J.S.; Black, M.M.; Banker, G.A. Polarity orientation of microtubules in hippocampal neurons: Uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA 1988, 85, 8335–8339. [Google Scholar] [CrossRef] [Green Version]
- Baas, P.W.; Slaughter, T.; Brown, A.; Black, M.M. Microtubule dynamics in axons and dendrites. J. Neurosci. Res. 1991, 30, 134–153. [Google Scholar] [CrossRef]
- Brown, A.; Li, Y.; Slaughter, T.; Black, M.M. Composite microtubules of the axon: Quantitative analysis of tyrosinated and acetylated tubulin along individual axonal microtubules. J. Cell Sci. 1993, 104, 339–352. [Google Scholar]
- Stepanova, T.; Slemmer, J.; Hoogenraad, C.C.; Lansbergen, G.; Dortland, B.; De Zeeuw, C.I.; Grosveld, F.; Van Cappellen, G.; Akhmanova, A.; Galjart, N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J. Neurosci. 2003, 23, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Marinković, P.; Williams, P.R.; Stern, S.; Weigand, E.E.; Engerer, P.; Naumann, R.; Hartmann, J.; Karl, R.M.; Bradke, F.; et al. An assay to image neuronal microtubule dynamics in mice. Nat. Commun. 2014, 5, 4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dent, E.W.; Gertler, F.B. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 2003, 40, 209–227. [Google Scholar] [CrossRef] [Green Version]
- Dent, E.W.; Kalil, K. Axon branching requires interactions between dynamic microtubules and actin filaments. J. Neurosci. 2001, 21, 9757–9769. [Google Scholar] [CrossRef]
- Van de Willige, D.; Hummel, J.J.; Alkemade, C.; Kahn, O.I.; Au, F.K.; Qi, R.Z.; Dogterom, M.; Koenderink, G.H.; Hoogenraad, C.C.; Akhmanova, A. Cytolinker Gas2L1 regulates axon morphology through microtubule-modulated actin stabilization. EMBO Rep. 2019, 20, e47732. [Google Scholar] [CrossRef]
- Fréal, A.; Rai, D.; Tas, R.P.; Pan, X.; Katrukha, E.A.; Van De Willige, D.; Stucchi, R.; Aher, A.; Yang, C.; Altelaar, A.M.; et al. Feedback-driven assembly of the axon initial segment. Neuron 2019, 104, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Chakraborti, S.; Natarajan, K.; Curiel, J.; Janke, C.; Liu, J. The emerging role of the tubulin code: From the tubulin molecule to neuronal function and disease. Cytoskeleton 2016, 73, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Stouffer, M.A.; Golden, J.A.; Francis, F. Neuronal migration disorders: Focus on the cytoskeleton and epilepsy. Neurobiol. Dis. 2015, 92, 18–45. [Google Scholar] [CrossRef] [PubMed]
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Severin, F.; Lommer, B.; Shevchenko, A.; Zerial, M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006, 172, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmanova, A.; Steinmetz, M. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in differentiated mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 619–637. [Google Scholar] [CrossRef]
- Alves-Silva, J.; Sánchez-Soriano, N.; Beaven, R.; Klein, M.; Parkin, J.; Millard, T.H.; Bellen, H.J.; Venken, K.J.T.; Ballestrem, C.; Kammerer, R.A.; et al. Spectraplakins promote microtubule-mediated axonal growth by functioning as structural microtubule-associated proteins and EB1-dependent +TIPs (tip interacting proteins). J. Neurosci. 2012, 32, 9143–9158. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.-H.; Cheng, T.; Jan, L.Y.; Jan, Y.N. APC and GSK-3β are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 2004, 14, 2025–2032. [Google Scholar] [CrossRef] [Green Version]
- Koester, M.P.; Müller, O.; Pollerberg, G.E. Adenomatous polyposis coli is differentially distributed in growth cones and modulates their steering. J. Neurosci. 2007, 27, 12590–12600. [Google Scholar] [CrossRef] [Green Version]
- Eom, T.-Y.; Stanco, A.; Guo, J.; Wilkins, G.; DesLauriers, D.; Yan, J.; Monckton, C.; Blair, J.; Oon, E.; Perez, A.; et al. Differential regulation of microtubule severing by APC underlies distinct patterns of projection neuron and interneuron migration. Dev. Cell 2014, 31, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lipka, J.; Stucchi, R.; Burute, M.; Pan, X.; Portegies, S.; Tas, R.; Willems, J.; Will, L.; MacGillavry, H.; et al. Microtubule minus-end binding protein CAMSAP2 and kinesin-14 motor KIFC3 control dendritic microtubule organization. Curr. Biol. 2020, 30, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Thyagarajan, P.; Shorey, M.; Seebold, D.Y.; Weiner, A.T.; Albertson, R.M.; Rao, K.S.; Sagasti, A.; Goetschius, D.J.; Rolls, M.M. Patronin-mediated minus end growth is required for dendritic microtubule polarity. J. Cell Biol. 2019, 218, 2084–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, R.P. The roles of microtubule-associated proteins in brain morphogenesis: A review. Brain Res. Rev. 1990, 15, 101–120. [Google Scholar] [CrossRef]
- Sánchez, C.; Díaz-Nido, J.; Avila, J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog. Neurobiol. 2000, 61, 133–168. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhou, Z.; Zhang, L.; Wang, Y.; Zhang, Y.-W.; Zhong, M.; Xu, S.-C.; Chen, C.-H.; Li, L.; Yu, Z.-P. Tau protein is involved in morphological plasticity in hippocampal neurons in response to BDNF. Neurochem. Int. 2012, 60, 233–242. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Lucas, J.J. Altered levels and isoforms of Tau and nuclear membrane invaginations in Huntington’s disease. Front. Cell. Neurosci. 2020, 13, 574. [Google Scholar] [CrossRef]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.M.; Ferrer, I.; Rozemuller, A.J.M.; Hernández, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef]
- Blum, D.; Herrera, F.; Francelle, L.; Mendes, T.; Basquin, M.; Obriot, H.; Demeyer, D.; Sergeant, N.; Gerhardt, E.; Brouillet, E.; et al. Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum. Mol. Genet. 2014, 24, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Morales, M. Distribution of acetylated α-tubulin in brain. Cell Tissue Res. 1991, 265, 415–423. [Google Scholar] [CrossRef]
- Cambray-Deakin, M.A.; Burgoyne, R.D. Posttranslational modifications of alpha-tubulin: Acetylated and detyrosinated forms in axons of rat cerebellum. J. Cell Biol. 1987, 104, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.J.; Burgoyne, R.D. Differential localisation of tyrosinated, detyrosinated, and acetylated α-tubulins in neurites and growth cones of dorsal root ganglion neurons. Cell Motil. Cytoskelet. 1989, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Konishi, Y.; Setou, M. Tubulin tyrosination is required for the maintenance of neuronal polarity. Neurosci. Res. 2009, 65, S38. [Google Scholar] [CrossRef]
- Magiera, M.M.; Bodakuntla, S.; Žiak, J.; Lacomme, S.; Sousa, P.M.; Leboucher, S.; Hausrat, T.J.; Bosc, C.; Andrieux, A.; Kneussel, M.; et al. Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J. 2018, 37, e100440. [Google Scholar] [CrossRef]
- Magiera, M.M.; Singh, P.; Gadadhar, S.; Janke, C. Tubulin posttranslational modifications and emerging links to human disease. Cell 2018, 173, 1323–1327. [Google Scholar] [CrossRef] [Green Version]
- Akhmanova, A.; Hoogenraad, C.C. More is not always better: Hyperglutamylation leads to neurodegeneration. EMBO J. 2018, 37, e101023. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Huntington’s Chorea; Springer: Berlin/Heidelberg, Germany, 1981; p. 192. [Google Scholar]
- Myers, R.H. Huntington’s disease genetics. J. Am. Soc. Exp. Neurother. 2004, 1, 255–262. [Google Scholar] [CrossRef]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef]
- Li, Z.; Karlovich, C.A.; Fish, M.P.; Scott, M.P.; Myers, R.M. A putative drosophila homolog of the Huntington’s disease gene. Hum. Mol. Genet. 1999, 8, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Macmillan, J.; Snell, R.G.; Tyler, A.; Houlihan, G.; Fenton, I.; Cheadle, J.; Lazarou, L.; Shaw, J.; Harper, P. Molecular analysis and clinical correlations of the Huntington’s disease mutation. Lancet 1993, 342, 954–958. [Google Scholar] [CrossRef]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Tautz, D.; Schlötterer, C. Simple sequences. Curr. Opin. Genet. Dev. 1994, 4, 832–837. [Google Scholar] [CrossRef]
- Baig, S.S.; Strong, M.; Quarrell, O. The global prevalence of Huntington’s disease: A systematic review and discussion. Neurodegener. Dis. Manag. 2016, 6, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illarioshkin, S.N.; Klyushnikov, S.A.; Seliverstov, Y.A. Huntington’s Disease; ATMO: Moscow, Russia, 2018; p. 472. (In Russian) [Google Scholar]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M.; et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993, 4, 387–392. [Google Scholar] [CrossRef]
- Holmans, P.; Massey, T.H.; Jones, L. Genetic modifiers of Mendelian disease: Huntington’s disease and the trinucleotide repeat disorders. Hum. Mol. Genet. 2017, 26, R83–R90. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.M.; Humbert, S. Huntingtin: Here, there, everywhere! J. Huntingt. Dis. 2013, 2, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Zeitlin, S.; Liu, J.-P.; Chapman, D.L.; Papaioannou, V.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007, 3, e82. [Google Scholar] [CrossRef] [Green Version]
- Culver, B.P.; Savas, J.N.; Park, S.K.; Choi, J.H.; Zheng, S.; Zeitlin, S.O.; Yates, J.R.; Tanese, N. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 2012, 287, 21599–21614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A large scale Huntingtin protein interaction network implicates rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 2014, 289, 6709–6726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Monte, S.M.; Vonsattel, J.-P.; Richardson, E.P. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988, 47, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Vonsattel, J.P.G.; Keller, C.; del Pilar, A.M. Neuropathology of Huntington’s disease. Handb. Clin. Neurol. 2008, 89, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.; Weber, B.H.F.; Hayden, M.R. New insights into the clinical features, pathogenesis and molecular genetics of Huntington disease. Brain Pathol. 1992, 2, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Richardson, E.P. Huntington’s disease: Some recent neuropathological studies. Neuropathol. Appl. Neurobiol. 1990, 16, 451–460. [Google Scholar] [CrossRef]
- Halliday, G.M.; McRitchie, D.; Macdonald, V.; Double, K.L.; Trent, R.; McCusker, E. Regional specificity of brain atrophy in Huntington’s disease. Exp. Neurol. 1998, 154, 663–672. [Google Scholar] [CrossRef]
- Rosas, H.D.; Salat, D.H.; Lee, S.Y.; Zaleta, A.K.; Pappu, V.; Fischl, B.; Greve, U.; Hevelone, N.; Hersch, S.M. Cerebral cortex and the clinical expression of Huntington’s disease: Complexity and heterogeneity. Brain 2008, 131, 1057–1068. [Google Scholar] [CrossRef] [Green Version]
- Douaud, G.; Gaura, V.; Ribeiro, M.-J.; Lethimonnier, F.; Maroy, R.; Verny, C.; Krystkowiak, P.; Damier, P.; Bachoud-Lévi, A.-C.; Hantraye, P.; et al. Distribution of grey matter atrophy in Huntington’s disease patients: A combined ROI-based and voxel-based morphometric study. NeuroImage 2006, 32, 1562–1575. [Google Scholar] [CrossRef]
- Myers, R.H.; Vonsattel, J.P.; Paskevich, P.A.; Kiely, M.P.H.; Stevens, B.A.; Cupples, L.A.; Richardson, E.P.; Bird, E.D. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J. Neuropathol. Exp. Neurol. 1991, 50, 729–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Burg, J.M.; Bjorkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
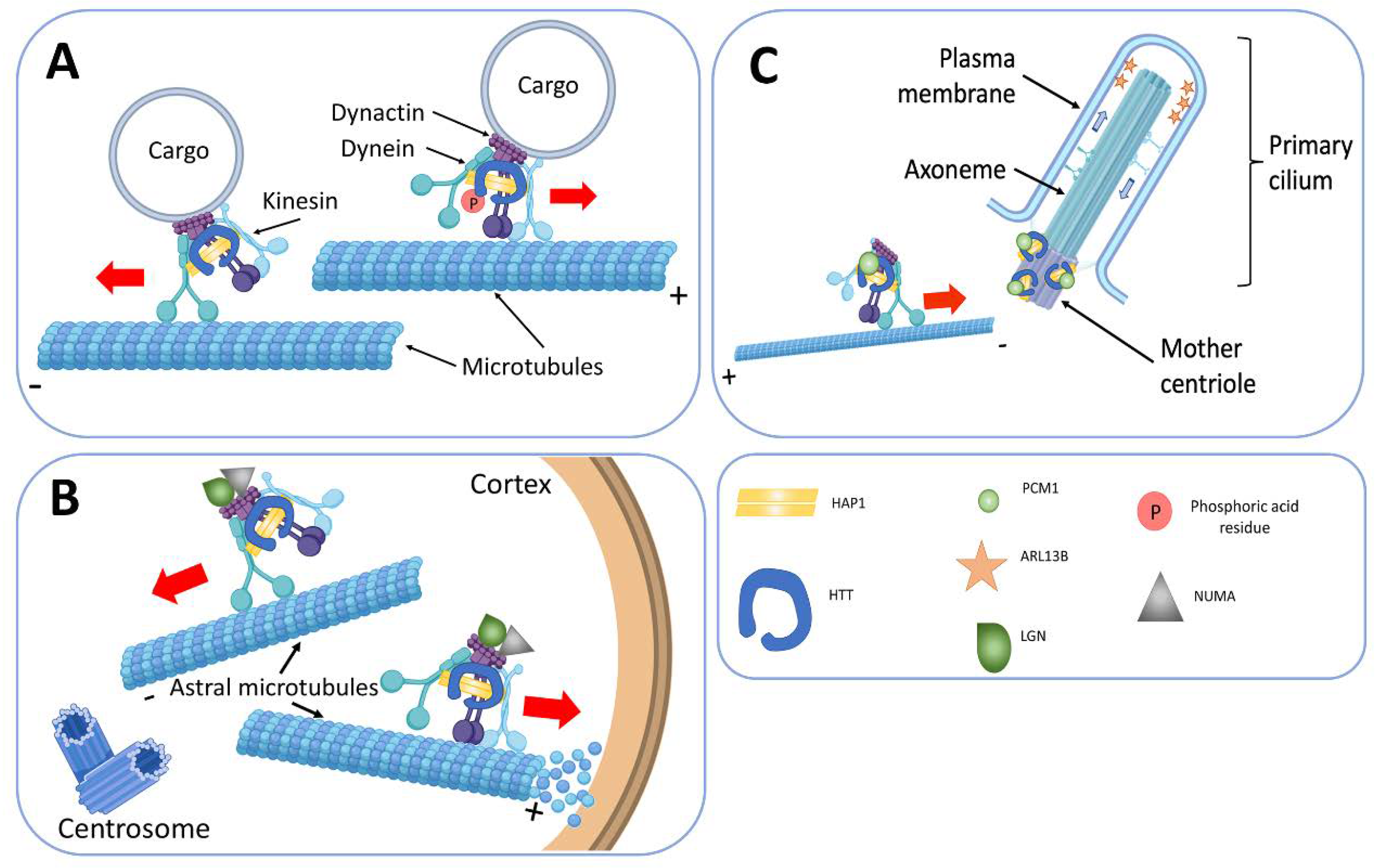
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.; Holzbaur, E.L.; Ross, C.A. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 1997, 6, 2205–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Shirendeb, U.P. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1822, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proskura, A.; Vechkapova, S.O.; Zapara, T.A.; Ratushniak, A.S. Protein–protein interactions of huntingtin in the hippocampus. Mol. Biol. 2017, 51, 647–653. [Google Scholar] [CrossRef]
- Steffan, J.S. Does Huntingtin play a role in selective macroautophagy? Cell Cycle 2010, 9, 3401–3413. [Google Scholar] [CrossRef]
- Martínez-Vicente, M.; Tallóczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.-L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Elias, S.; Thion, M.S.; Yu, H.; Sousa, C.M.; Lasgi, C.; Morin, X.; Humbert, S. Huntingtin regulates mammary stem cell division and differentiation. Stem Cell Rep. 2014, 2, 491–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjes, P.; Wanker, E. The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem. Sci. 2003, 28, 425–433. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.-P.; Carraway, R.; A Reeves, S.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Fusco, F.R.; Chen, Q.; L’Amoreaux, W.; Figueredo-Cardenas, G.; Jiao, Y.; Coffman, J.; Surmeier, D.J.; Honig, M.G.; Carlock, L.R.; Reiner, A. Cellular localization of huntingtin in striatal and cortical neurons in rats: Lack of correlation with neuronal vulnerability in Huntington’s disease. J. Neurosci. 1999, 19, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClory, H.; Wang, X.; Sapp, E.; Gatune, L.W.; Iuliano, M.; Wu, C.-Y.; Nathwani, G.; Kegel-Gleason, K.B.; DiFiglia, M.; Li, X. The COOH-terminal domain of huntingtin interacts with RhoGEF kalirin and modulates cell survival. Sci. Rep. 2018, 8, 8000. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Martin-Aparicio, E.; Ávila, J.; Lucas, J.J. Nuclear localization of N-terminal mutant huntingtin is cell cycle dependent. Eur. J. Neurosci. 2002, 16, 355–359. [Google Scholar] [CrossRef]
- Hoffner, G.; Kahlem, P.; Djian, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with β-tubulin: Relevance to Huntington’s disease. J. Cell Sci. 2002, 115, 941–948. [Google Scholar]
- Miller, J.P.; Yates, B.E.; Al-Ramahi, I.; Berman, A.E.; Sanhueza, M.; Kim, E.; De Haro, M.; DeGiacomo, F.; Torcassi, C.; Holcomb, J.; et al. A genome-scale RNA–interference screen identifies RRAS signaling as a pathologic feature of Huntington’s disease. PLoS Genet. 2012, 8, e1003042. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Tousley, A.; Iuliano, M.; Weisman, E.; Sapp, E.; Richardson, H.; Vodicka, P.; Alexander, J.; Aronin, N.; DiFiglia, M.; Kegel-Gleason, K.B. Huntingtin associates with the actin cytoskeleton and α-actinin isoforms to influence stimulus dependent morphology changes. PLoS ONE 2019, 14, e0212337. [Google Scholar] [CrossRef] [PubMed]
- Giorgini, F. A flexible polyglutamine hinge opens new doors for understanding huntingtin function. Proc. Natl. Acad. Sci. USA 2013, 110, 14516–14517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, N.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nature 2015, 17, 262–275. [Google Scholar] [CrossRef] [Green Version]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castaño, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal–lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; E Davies, J.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, U.F.; Duden, R.; O’Kane, C.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; A Cozens, B.; DiFiglia, M.; Sharp, A.H.; A Ross, C.; Scherzinger, E.; Wanker, E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.; Arrasate, M.; Shaby, B.A.; Mitra, S.; Masliah, E.; Finkbeiner, S. Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington’s disease molecular pathogenesis. J. Neurosci. 2010, 30, 10541–10550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.; Arrasate, M.; Brooks, E.; Libeu, C.P.; Legleiter, J.; Hatters, D.M.; Curtis, J.; Cheung, K.; Krishnan, P.; Mitra, S.; et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Methods 2011, 7, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Finkbeiner, S.; Devys, D.; E Greenberg, M. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Valor, L.M. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: A genome-wide perspective. Mol. Neurobiol. 2014, 51, 406–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, H.; Gusella, J.F. The predominantly HEAT-like motif structure of huntingtin and its association and coincident nuclear entry with dorsal, an NF-kB/Rel/dorsal family transcription factor. BMC Neurosci. 2002, 3, 15. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.-Z.; Gohler, H.; Wanker, E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early huntington’s disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Marcora, E.; Gowan, K.; Lee, J.E. Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc. Natl. Acad. Sci. USA 2003, 100, 9578–9583. [Google Scholar] [CrossRef] [Green Version]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.; Bouzou, B.; Cha, J.-H.J. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2009, 19, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Zuccato, C.; Cattaneo, E. Huntington’s disease. In Neurotrophic Factors. Handbook of Experimental Pharmacology; Lewin, G., Carter, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 220. [Google Scholar]
- El-Daher, M.-T.; Hangen, E.; Bruyère, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015, 34, 2255–2271. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, J.M. Towards an understanding of energy impairment in Huntington’s disease brain. J. Huntingt. Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogomazova, A.N.; Eremeev, A.V.; Pozmogova, G.E.; Lagarkova, M.A. The role of mutant RNA in the pathogenesis of Huntington’s disease and other polyglutamine diseases. Mol. Biol. 2019, 53, 954–967. [Google Scholar] [CrossRef]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.C.; et al. Non-ATG–initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2010, 108, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bañez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN translation in Huntington disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pages, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; E MacDonald, M.; Leßmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zala, D.; Hinckelmann, M.-V.; Saudou, F. Huntingtin’s function in axonal transport is conserved in drosophila melanogaster. PLoS ONE 2013, 8, e60162. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Zajac, A.L.; Tokito, M.; Holzbaur, E.L. Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol. Biol. Cell 2011, 22, 478–492. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pages, M.; Li, X.-J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Her, L.-S.; Goldstein, L.S. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant Huntingtin. J. Neurosci. 2008, 28, 13662–13672. [Google Scholar] [CrossRef] [PubMed]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; Macaskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant Huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zala, D.; Hinckelmann, M.-V.; Yu, H.; Da Cunha, M.M.L.; Liot, G.; Cordelières, F.P.; Marco, S.; Saudou, F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 2013, 152, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Engqvist-Goldstein, A.E.; Warren, R.A.; Kessels, M.M.; Keen, J.H.; Heuser, J.; Drubin, D.G. The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J. Cell Biol. 2001, 154, 1209–1224. [Google Scholar] [CrossRef] [Green Version]
- Waelter, S.; Scherzinger, E.; Hasenbank, R.; Nordhoff, E.; Lurz, R.; Goehler, H.; Gauss, C.; Sathasivam, K.; Bates, G.P.; Lehrach, H.; et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum. Mol. Genet. 2001, 10, 1807–1817. [Google Scholar] [CrossRef] [Green Version]
- Legendre-Guillemin, V.; Metzler, M.; Charbonneau, M.; Gan, L.; Chopra, V.; Philie, J.; Hayden, M.R.; McPherson, P.S. HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin. Identification of a novel interaction with clathrin light chain. J. Biol. Chem. 2002, 277, 19897–19904. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Sapp, E.; Valencia, A.; Kegel, K.B.; Qin, Z.-H.; Alexander, J.; Masso, N.; Reeves, P.; Ritch, J.J.; Zeitlin, S.; et al. A function of huntingtin in guanine nucleotide exchange on Rab11. NeuroReport 2008, 19, 1643–1647. [Google Scholar] [CrossRef]
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; E MacDonald, M. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Hattula, K.; Peränen, J. FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr. Biol. 2000, 10, 1603–1606. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, D. Expression of primary cilia in mammalian cells. Cell Biol. Int. 1996, 20, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Alieva, I.B.; A Vorobjev, I. Vertebrate primary cilia: A sensory part of centrosomal complex in tissue cells, but a “sleeping beauty” in cultured cells? Cell Biol. Int. 2004, 28, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Pazour, G.J.; Witman, G.B. The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol. 2003, 15, 105–110. [Google Scholar] [CrossRef]
- Andersen, S.S.L. Molecular characteristics of the centrosome. Nat. Eng. Resist. Plant Viruses 1999, 187, 51–109. [Google Scholar] [CrossRef]
- Keryer, G.; Pineda, J.R.; Liot, G.; Kim, J.; Dietrich, P.; Benstaali, C.; Smith, K.; Cordelières, F.P.; Spassky, N.; Ferrante, R.J.; et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Investig. 2011, 121, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Haremaki, T.; Deglincerti, A.; Brivanlou, A.H. Huntingtin is required for ciliogenesis and neurogenesis during early Xenopus development. Dev. Biol. 2015, 408, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, A.; Tebbe, L.; Weber, C.; Messaddeq, N.; Morlé, L.; Kessler, P.; Wolfrum, U.; Trottier, Y. A novel function of Huntingtin in the cilium and retinal ciliopathy in Huntington’s disease mice. Neurobiol. Dis. 2015, 80, 15–28. [Google Scholar] [CrossRef]
- Sathasivam, K.; Woodman, B.; Mahal, A.; Bertaux, F.; Wanker, E.; Shima, D.T.; Bates, G.P. Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington’s disease (HD) transgenic mice and HD patients. Hum. Mol. Genet. 2001, 10, 2425–2435. [Google Scholar] [CrossRef] [Green Version]
- Rigamonti, D.; Sipione, S.; Goffredo, D.; Zuccato, C.; Fossale, E.; Cattaneo, E. Huntingtin’s neuroprotective activity occurs via inhibition of procaspase-9 processing. J. Biol. Chem. 2001, 276, 14545–14548. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Leavitt, B.R.; Van Raamsdonk, J.M.; Dragatsis, I.; Goldowitz, D.; E MacDonald, M.; Hayden, M.R.; Friedlander, R.M. Huntingtin inhibits caspase-3 activation. EMBO J. 2006, 25, 5896–5906. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Van De Willige, D.; Hoogenraad, C.C.; Akhmanova, A. Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life Sci. 2016, 73, 2053–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csobonyeiova, M.; Polak, S.; Danišovič, L. Recent overview of the use of iPSCs Huntington’s disease modeling and therapy. Int. J. Mol. Sci. 2020, 21, 2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorzhak, A.; Grantyn, R. Single synapse indicators of glutamate release and uptake in acute brain slices from normal and Huntington mice. J. Vis. Exp. 2020, e60113. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Carter, R.L.; Cho, I.K.; Chan, A.W.S. Cell-based therapies for Huntington’s disease. Drug Discov. Today 2014, 19, 980–984. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taran, A.S.; Shuvalova, L.D.; Lagarkova, M.A.; Alieva, I.B. Huntington’s Disease—An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells 2020, 9, 1514. https://doi.org/10.3390/cells9061514
Taran AS, Shuvalova LD, Lagarkova MA, Alieva IB. Huntington’s Disease—An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells. 2020; 9(6):1514. https://doi.org/10.3390/cells9061514
Chicago/Turabian StyleTaran, Aleksandra S., Lilia D. Shuvalova, Maria A. Lagarkova, and Irina B. Alieva. 2020. "Huntington’s Disease—An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components" Cells 9, no. 6: 1514. https://doi.org/10.3390/cells9061514
APA StyleTaran, A. S., Shuvalova, L. D., Lagarkova, M. A., & Alieva, I. B. (2020). Huntington’s Disease—An Outlook on the Interplay of the HTT Protein, Microtubules and Actin Cytoskeletal Components. Cells, 9(6), 1514. https://doi.org/10.3390/cells9061514