Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy


Abstract
1. Introduction
2. Targeting Tumors with TCR-Based ACT
2.1. Antigen Recognition by TCRs
2.2. Target Antigen Selection for TCR Gene Therapy
2.3. TCR Affinity
2.4. TCR Gene Therapy Clinical Results
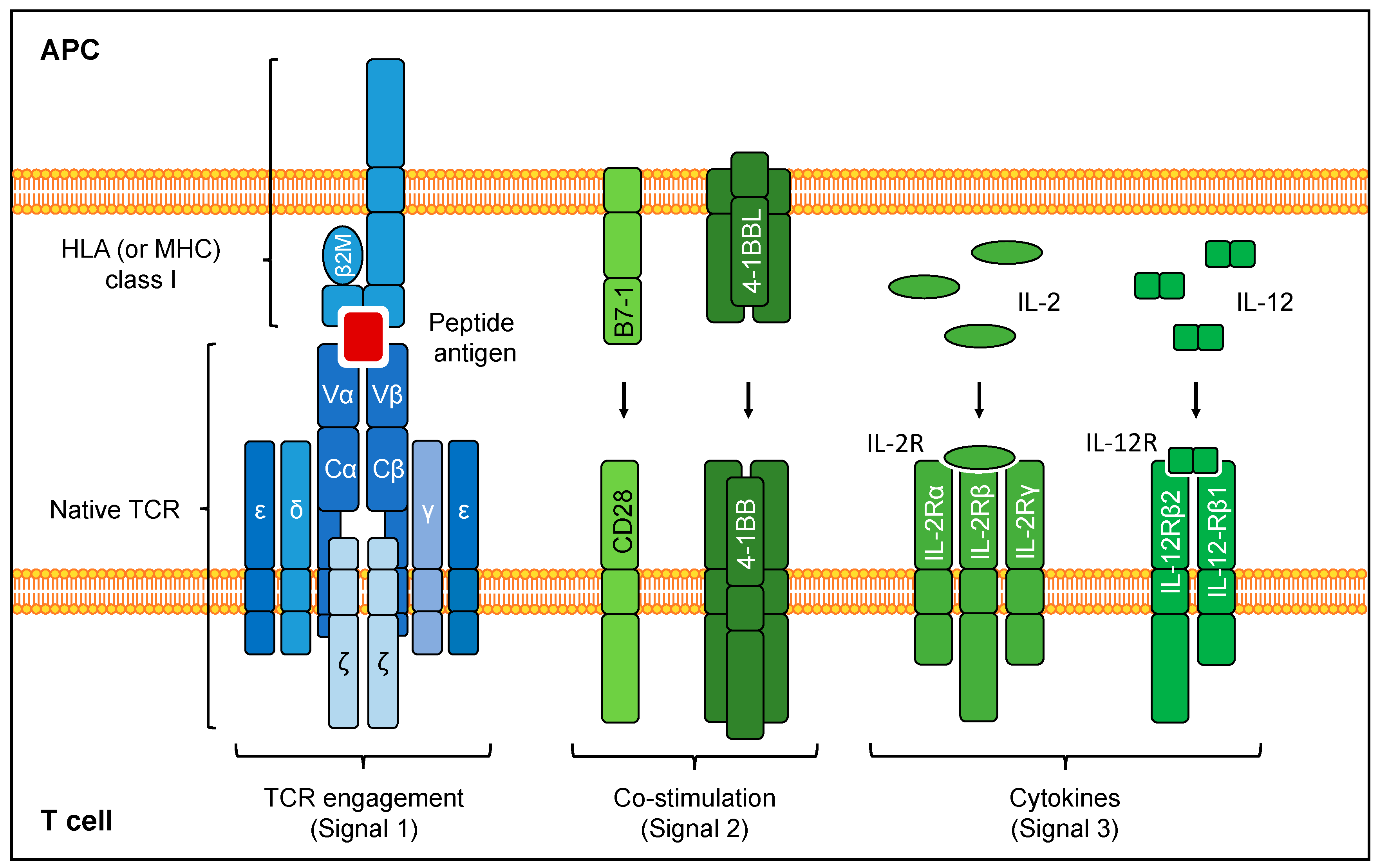
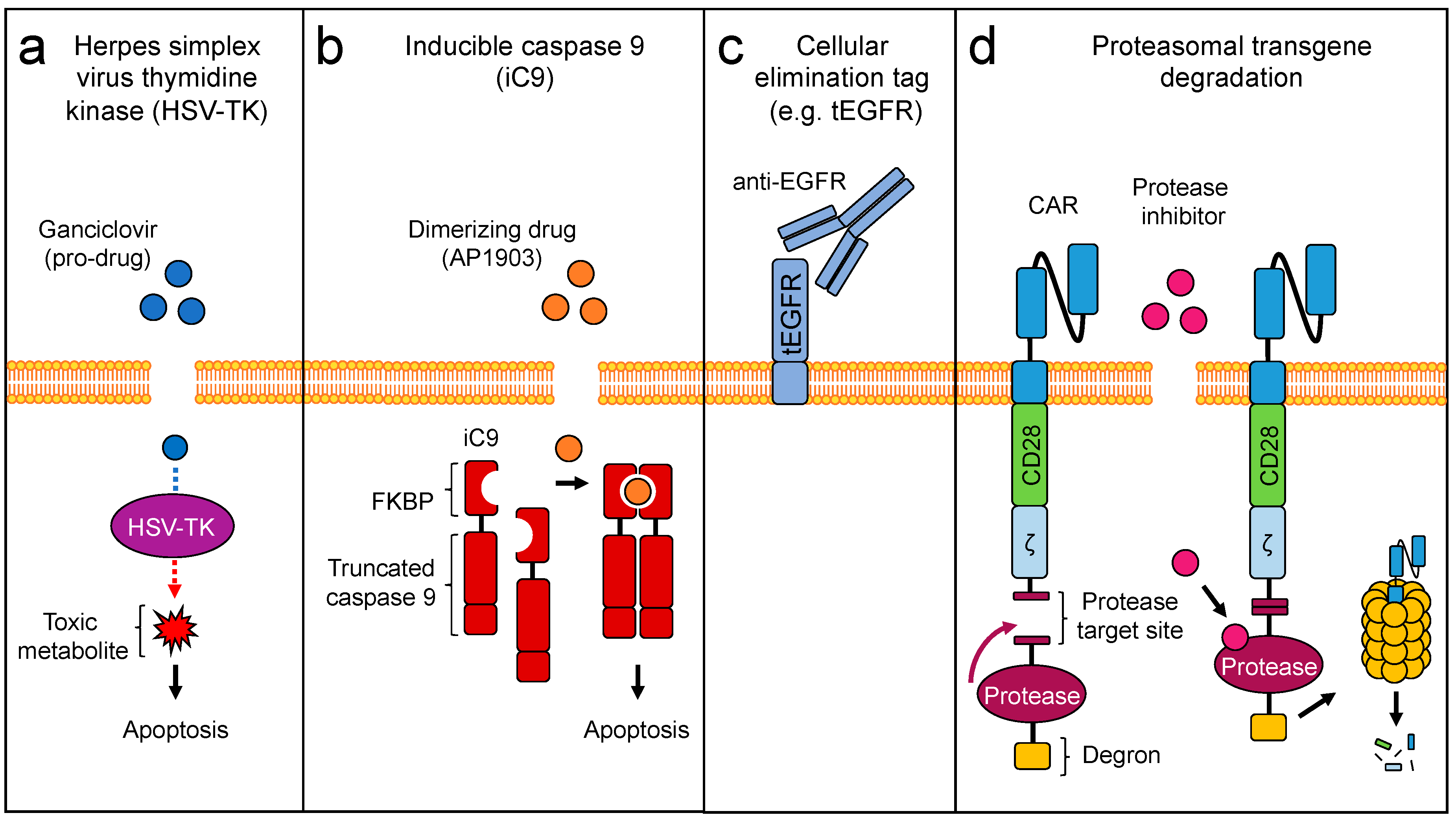
3. Engineering Safety
4. T Cell Engineering Strategies to Enhance Anti-Tumor Activity of TCR-Transgenic T Cells
4.1. Enhancing Functional Avidity
4.2. Engineering MHC-Independent Antigen Specificity through TCR
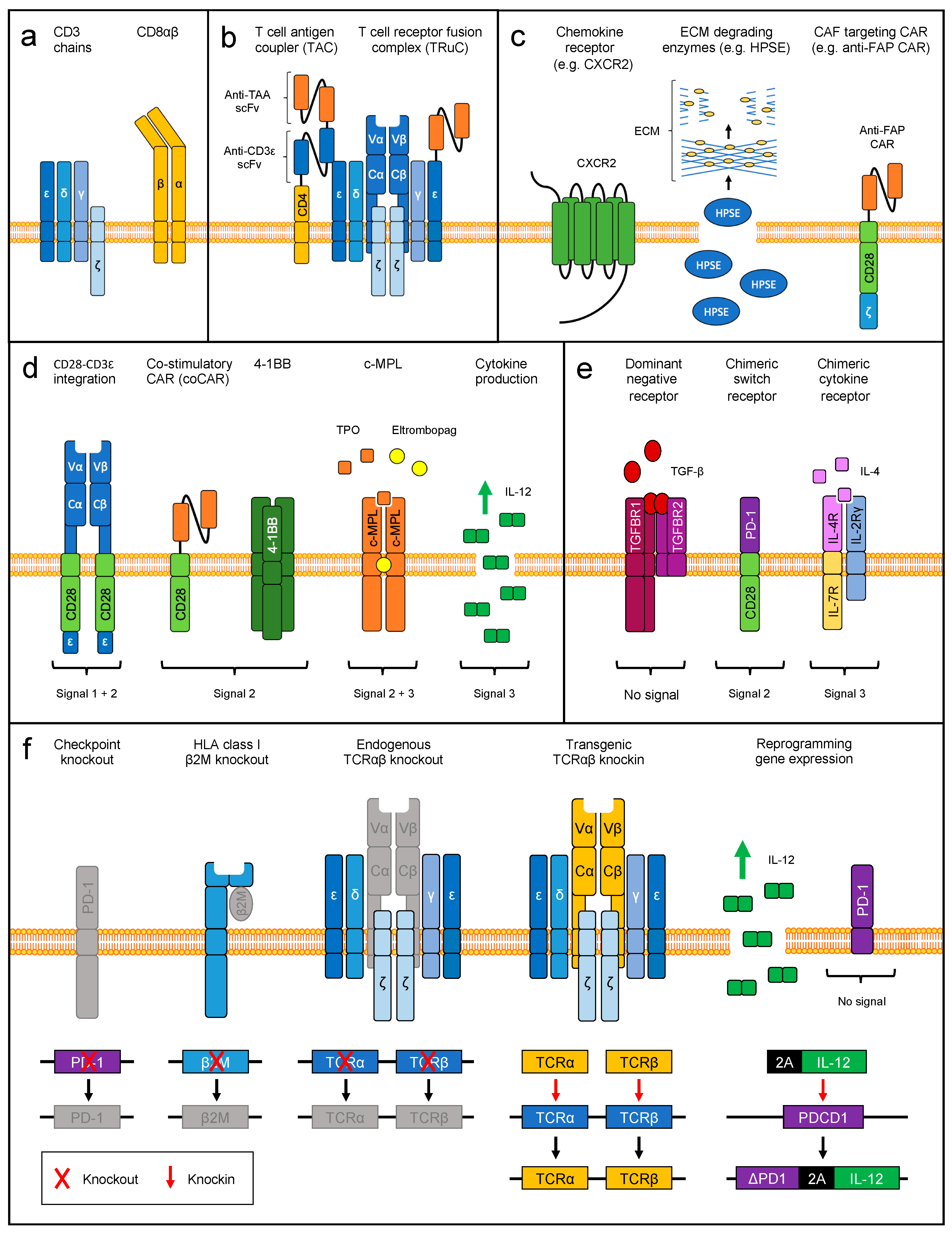
4.3. Engineering Strategies to Target the Tumor Microenvironment
4.3.1. Engineering T Cell Homing and Tumor Infiltration
4.3.2. Delivery of Co-Stimulation
4.3.3. Delivery of Cytokine Signaling
4.3.4. Reverting Immune Inhibitory Signals
5. Therapeutic Genome Editing
5.1. Elimination of Endogenous TCR Specificities
5.2. Multiplexed CRISPR/Cas9-Based Genome Editing
5.3. Targeted Transgene Delivery into Defined Genetic Loci
5.4. Discovery of Novel Immune Engineering Targets
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef]
- Leung, W.; Heslop, H.E. Adoptive Immunotherapy with Antigen-Specific T Cells Expressing a Native TCR. Cancer Immunol. Res. 2019, 7, 528–533. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Faitg, T.H.; Lowther, D.E.; Badros, A.Z.; Chagin, K.; Dengel, K.; Iyengar, M.; Melchiori, L.; Navenot, J.M.; Norry, E.; et al. Long-term safety and activity of NY-ESO-1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. 2019, 3, 2022–2034. [Google Scholar] [CrossRef]
- Doran, S.L.; Stevanovic, S.; Adhikary, S.; Gartner, J.J.; Jia, L.; Kwong, M.L.M.; Faquin, W.C.; Hewitt, S.M.; Sherry, R.M.; Yang, J.C.; et al. T-Cell Receptor Gene Therapy for Human Papillomavirus-Associated Epithelial Cancers: A First-in-Human, Phase I/II Study. J. Clin. Oncol. 2019, 37, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Bonini, C.; Mondino, A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur. J. Immunol. 2015, 45, 2457–2469. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Banchereau, J.; Bhardwaj, N.; Cockett, M.; Disis, M.L.; Dranoff, G.; Gilboa, E.; Hammond, S.A.; Hershberg, R.; Korman, A.J.; et al. The Human Vaccines Project: A roadmap for cancer vaccine development. Sci. Transl. Med. 2016, 8, 334ps339. [Google Scholar] [CrossRef] [PubMed]
- Chabannon, C.; Kuball, J.; Bondanza, A.; Dazzi, F.; Pedrazzoli, P.; Toubert, A.; Ruggeri, A.; Fleischhauer, K.; Bonini, C. Hematopoietic stem cell transplantation in its 60s: A platform for cellular therapies. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Fuca, G.; Reppel, L.; Landoni, E.; Savoldo, B.; Dotti, G. Enhancing Chimeric Antigen Receptor T-Cell Efficacy in Solid Tumors. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Lanitis, E.; Coukos, G.; Irving, M. All systems go: Converging synthetic biology and combinatorial treatment for CAR-T cell therapy. Curr. Opin. Biotechnol. 2020, 65, 75–87. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Raeber, M.E.; Zurbuchen, Y.; Impellizzieri, D.; Boyman, O. The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 2018, 283, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Clin. Invest. 2015, 125, 3413–3421. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Crawford, J.C.; Allen, E.K.; Guo, X.J.; Bakke, J.; Carter, R.A.; Abdelsamed, H.A.; Moustaki, A.; Li, Y.; Chang, T.C.; et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8(+) T cell responses. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, C.; Mezzadra, R.; Schumacher, T.N. TCR repertoires of intratumoral T-cell subsets. Immunol. Rev. 2014, 257, 72–82. [Google Scholar] [CrossRef]
- Lu, Y.C.; Zheng, Z.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, Y.F.; Ray, S.; Franco, Z.; Bliskovsky, V.; et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol. Ther. 2018, 26, 379–389. [Google Scholar] [CrossRef]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef]
- Guillaume, P.; Picaud, S.; Baumgaertner, P.; Montandon, N.; Schmidt, J.; Speiser, D.E.; Coukos, G.; Bassani-Sternberg, M.; Filippakopoulos, P.; Gfeller, D. The C-terminal extension landscape of naturally presented HLA-I ligands. Proc. Natl. Acad. Sci. USA 2018, 115, 5083–5088. [Google Scholar] [CrossRef]
- Ogishi, M.; Yotsuyanagi, H. Quantitative Prediction of the Landscape of T Cell Epitope Immunogenicity in Sequence Space. Front. Immunol. 2019, 10, 827. [Google Scholar] [CrossRef]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 2019, 37, 1283–1286. [Google Scholar] [CrossRef]
- Abelin, J.G.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M. Mass Spectrometry Based Immunopeptidomics for the Discovery of Cancer Neoantigens. Methods Mol. Biol. 2018, 1719, 209–221. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.; McGranahan, N.; Rosenthal, R.; et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zaretsky, J.M.; Ng, A.H.C.; Chour, W.; Bethune, M.T.; Choi, J.; Hsu, A.; Holman, E.; Ding, X.; Guo, K.; et al. Sensitive Detection and Analysis of Neoantigen-Specific T Cell Populations from Tumors and Blood. Cell Rep. 2019, 28, 2728–2738. [Google Scholar] [CrossRef]
- Hammerl, D.; Rieder, D.; Martens, J.W.M.; Trajanoski, Z.; Debets, R. Adoptive T Cell Therapy: New Avenues Leading to Safe Targets and Powerful Allies. Trends Immunol. 2018, 39, 921–936. [Google Scholar] [CrossRef]
- Arnaud, M.; Duchamp, M.; Bobisse, S.; Renaud, P.; Coukos, G.; Harari, A. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol. 2020, 65, 52–59. [Google Scholar] [CrossRef]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Wolchok, J.D. Shared cancer neoantigens: Making private matters public. J. Exp. Med. 2018, 215, 5–7. [Google Scholar] [CrossRef]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef]
- van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.; Kester, M.G.; van der Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M.; et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Invest. 2019, 129, 774–785. [Google Scholar] [CrossRef]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Mori, L.; Lepore, M.; De Libero, G. The Immunology of CD1- and MR1-Restricted T Cells. Annu. Rev. Immunol. 2016, 34, 479–510. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Invest. 2016, 126, 2341–2355. [Google Scholar] [CrossRef]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef]
- Aleksic, M.; Liddy, N.; Molloy, P.E.; Pumphrey, N.; Vuidepot, A.; Chang, K.M.; Jakobsen, B.K. Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol. 2012, 42, 3174–3179. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Stone, J.D.; Kranz, D.M. Role of T cell receptor affinity in the efficacy and specificity of adoptive T cell therapies. Front. Immunol. 2013, 4, 244. [Google Scholar] [CrossRef]
- Arber, C.; Feng, X.; Abhyankar, H.; Romero, E.; Wu, M.F.; Heslop, H.E.; Barth, P.; Dotti, G.; Savoldo, B. Survivin-specific T cell receptor targets tumor but not T cells. J. Clin. Invest. 2015, 125, 157–168. [Google Scholar] [CrossRef]
- Stone, J.D.; Harris, D.T.; Kranz, D.M. TCR affinity for p/MHC formed by tumor antigens that are self-proteins: Impact on efficacy and toxicity. Curr. Opin. Immunol. 2015, 33, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P.; Dolton, G.M.; Gerry, A.B.; Brewer, J.E.; Bennett, A.D.; Pumphrey, N.J.; Jakobsen, B.K.; Sewell, A.K. Human leucocyte antigen class I-redirected anti-tumour CD4(+) T cells require a higher T cell receptor binding affinity for optimal activity than CD8(+) T cells. Clin. Exp. Immunol. 2017, 187, 124–137. [Google Scholar] [CrossRef]
- Zhao, Y.; Bennett, A.D.; Zheng, Z.; Wang, Q.J.; Robbins, P.F.; Yu, L.Y.; Li, Y.; Molloy, P.E.; Dunn, S.M.; Jakobsen, B.K.; et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J. Immunol. 2007, 179, 5845–5854. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Chervin, A.S.; Sant, A.J.; Kranz, D.M.; Schreiber, H. Long-term persistence of CD4(+) but rapid disappearance of CD8(+) T cells expressing an MHC class I-restricted TCR of nanomolar affinity. Mol. Ther. 2012, 20, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.M.; Stone, J.D.; Chervin, A.S.; Engels, B.; Schreiber, H.; Roy, E.J.; Kranz, D.M. MHC-class I-restricted CD4 T cells: A nanomolar affinity TCR has improved anti-tumor efficacy in vivo compared to the micromolar wild-type TCR. Cancer Immunol. Immunother. 2013, 62, 359–369. [Google Scholar] [CrossRef]
- Holler, P.D.; Holman, P.O.; Shusta, E.V.; O'Herrin, S.; Wittrup, K.D.; Kranz, D.M. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. USA 2000, 97, 5387–5392. [Google Scholar] [CrossRef]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef]
- Dunn, S.M.; Rizkallah, P.J.; Baston, E.; Mahon, T.; Cameron, B.; Moysey, R.; Gao, F.; Sami, M.; Boulter, J.; Li, Y.; et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006, 15, 710–721. [Google Scholar] [CrossRef]
- Richman, S.A.; Kranz, D.M. Display, engineering, and applications of antigen-specific T cell receptors. Biomol. Eng. 2007, 24, 361–373. [Google Scholar] [CrossRef]
- Zoete, V.; Irving, M.; Ferber, M.; Cuendet, M.A.; Michielin, O. Structure-Based, Rational Design of T Cell Receptors. Front. Immunol. 2013, 4, 268. [Google Scholar] [CrossRef]
- Pierce, B.G.; Hellman, L.M.; Hossain, M.; Singh, N.K.; Vander Kooi, C.W.; Weng, Z.; Baker, B.M. Computational design of the affinity and specificity of a therapeutic T cell receptor. PLoS Comput. Biol. 2014, 10, e1003478. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Wang, N.; Riley, T.P.; Anderson, S.D.; Singh, N.K.; Procko, E.; Baker, B.M.; Kranz, D.M. Deep Mutational Scans as a Guide to Engineering High Affinity T Cell Receptor Interactions with Peptide-bound Major Histocompatibility Complex. J. Biol. Chem. 2016, 291, 24566–24578. [Google Scholar] [CrossRef]
- Robbins, P.F.; Li, Y.F.; El-Gamil, M.; Zhao, Y.; Wargo, J.A.; Zheng, Z.; Xu, H.; Morgan, R.A.; Feldman, S.A.; Johnson, L.A.; et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008, 180, 6116–6131. [Google Scholar] [CrossRef]
- Hebeisen, M.; Baitsch, L.; Presotto, D.; Baumgaertner, P.; Romero, P.; Michielin, O.; Speiser, D.E.; Rufer, N. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J. Clin. Invest. 2013, 123, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P.; Gerry, A.B.; Brewer, J.E.; Melchiori, L.; Bridgeman, J.S.; Bennett, A.D.; Pumphrey, N.J.; Jakobsen, B.K.; Price, D.A.; Ladell, K.; et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin. Exp. Immunol. 2015, 180, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.N.; Erdes, E.; Hebeisen, M.; Rufer, N. Chronic TCR-MHC (self)-interactions limit the functional potential of TCR affinity-increased CD8 T lymphocytes. J. Immunother. Cancer 2019, 7, 284. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 2020, 9, 1682381. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Kageyama, S.; Ikeda, H.; Miyahara, Y.; Imai, N.; Ishihara, M.; Saito, K.; Sugino, S.; Ueda, S.; Ishikawa, T.; Kokura, S.; et al. Adoptive Transfer of MAGE-A4 T-cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2268–2277. [Google Scholar] [CrossRef]
- Lu, Y.C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- D'Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef]
- Tawara, I.; Kageyama, S.; Miyahara, Y.; Fujiwara, H.; Nishida, T.; Akatsuka, Y.; Ikeda, H.; Tanimoto, K.; Terakura, S.; Murata, M.; et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood 2017, 130, 1985–1994. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Joo, J.; Riley, J.P.; Yu, Z.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin. Cancer Res. 2009, 15, 169–180. [Google Scholar] [CrossRef]
- Chinnasamy, N.; Wargo, J.A.; Yu, Z.; Rao, M.; Frankel, T.L.; Riley, J.P.; Hong, J.J.; Parkhurst, M.R.; Feldman, S.A.; Schrump, D.S.; et al. A TCR targeting the HLA-A*0201-restricted epitope of MAGE-A3 recognizes multiple epitopes of the MAGE-A antigen superfamily in several types of cancer. J. Immunol. 2011, 186, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Pasquini, M.C.; Logan, B.; Stadtmauer, E.A.; Vesole, D.H.; Alyea, E., 3rd; Antin, J.H.; Comenzo, R.; Goodman, S.; Hari, P.; et al. Autologous haemopoietic stem-cell transplantation followed by allogeneic or autologous haemopoietic stem-cell transplantation in patients with multiple myeloma (BMT CTN 0102): A phase 3 biological assignment trial. Lancet Oncol. 2011, 12, 1195–1203. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Johnson, L.A.; Heemskerk, B.; Powell, D.J., Jr.; Cohen, C.J.; Morgan, R.A.; Dudley, M.E.; Robbins, P.F.; Rosenberg, S.A. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 2006, 177, 6548–6559. [Google Scholar] [CrossRef] [PubMed]
- Bownds, S.; Tong-On, P.; Rosenberg, S.A.; Parkhurst, M. Induction of Tumor-Reactive Cytotoxic T-Lymphocytes Using a Peptide from NY-ESO-1 Modified at the Carboxy-terminus to Enhance HLA-A2.1 Binding Affinity and Stability in Solution. J. Immunother. 2001, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Naota, H.; Wang, L.; Hiasa, A.; Goto, M.; Watanabe, M.; Kitano, S.; Okumura, S.; Takemitsu, T.; Yuta, A.; et al. Determination of cellularly processed HLA-A2402-restricted novel CTL epitopes derived from two cancer germ line genes, MAGE-A4 and SAGE. Clin. Cancer Res. 2005, 11, 5581–5589. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Ragnarsson, G.B.; Nguyen, H.N.; Chaney, C.N.; Pufnock, J.S.; Schmitt, T.M.; Duerkopp, N.; Roberts, I.M.; Pogosov, G.L.; Ho, W.Y.; et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 2013, 5, 174ra127. [Google Scholar] [CrossRef]
- Ohminami, H.; Yasukawa, M.; Fujita, S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood 2000, 95, 286–293. [Google Scholar] [CrossRef]
- Yao, X.; Lu, Y.C.; Parker, L.L.; Li, Y.F.; El-Gamil, M.; Black, M.A.; Xu, H.; Feldman, S.A.; van der Bruggen, P.; Rosenberg, S.A.; et al. Isolation and Characterization of an HLA-DPB1*04: 01-restricted MAGE-A3 T-Cell Receptor for Cancer Immunotherapy. J. Immunother. 2016, 39, 191–201. [Google Scholar] [CrossRef]
- Draper, L.M.; Kwong, M.L.; Gros, A.; Stevanovic, S.; Tran, E.; Kerkar, S.; Raffeld, M.; Rosenberg, S.A.; Hinrichs, C.S. Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin. Cancer Res. 2015, 21, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.; Zimmermann, S.; Arber, C.; Irving, M.; Trueb, L.; Coukos, G. Safety and Tolerability of Adoptive Cell Therapy in Cancer. Drug Saf. 2019, 42, 315–334. [Google Scholar] [CrossRef]
- Bonini, C.; Ferrari, G.; Verzeletti, S.; Servida, P.; Zappone, E.; Ruggieri, L.; Ponzoni, M.; Rossini, S.; Mavilio, F.; Traversari, C.; et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997, 276, 1719–1724. [Google Scholar] [CrossRef] [PubMed]
- Verzeletti, S.; Bonini, C.; Marktel, S.; Nobili, N.; Ciceri, F.; Traversari, C.; Bordignon, C. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: Clinical follow-up and improved new vectors. Hum. Gene. Ther. 1998, 9, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.; Vago, L.; Bondanza, A.; Peccatori, J.; Cieri, N.; Marktel, S.; Mastaglio, S.; Bordignon, C.; et al. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015, 6, 95. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pule, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]
- Zhou, X.; Di Stasi, A.; Tey, S.K.; Krance, R.A.; Martinez, C.; Leung, K.S.; Durett, A.G.; Wu, M.F.; Liu, H.; Leen, A.M.; et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014, 123, 3895–3905. [Google Scholar] [CrossRef]
- Zhou, X.; Dotti, G.; Krance, R.A.; Martinez, C.A.; Naik, S.; Kamble, R.T.; Durett, A.G.; Dakhova, O.; Savoldo, B.; Di Stasi, A.; et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood 2015, 125, 4103–4113. [Google Scholar] [CrossRef]
- Zhou, X.; Brenner, M.K. Improving the safety of T-Cell therapies using an inducible caspase-9 gene. Exp. Hematol. 2016, 44, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Vogler, I.; Newrzela, S.; Hartmann, S.; Schneider, N.; von Laer, D.; Koehl, U.; Grez, M. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol. Ther. 2010, 18, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019, 19, 44. [Google Scholar] [CrossRef]
- Vigano, S.; Utzschneider, D.T.; Perreau, M.; Pantaleo, G.; Zehn, D.; Harari, A. Functional avidity: A measure to predict the efficacy of effector T cells? Clin. Dev. Immunol. 2012, 2012, 153863. [Google Scholar] [CrossRef]
- Zhong, S.; Malecek, K.; Johnson, L.A.; Yu, Z.; Vega-Saenz de Miera, E.; Darvishian, F.; McGary, K.; Huang, K.; Boyer, J.; Corse, E.; et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc. Natl. Acad. Sci. USA 2013, 110, 6973–6978. [Google Scholar] [CrossRef]
- Hebeisen, M.; Allard, M.; Gannon, P.O.; Schmidt, J.; Speiser, D.E.; Rufer, N. Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Front. Immunol. 2015, 6, 582. [Google Scholar] [CrossRef]
- Viola, A.; Lanzavecchia, A. T cell activation determined by T cell receptor number and tunable thresholds. Science 1996, 273, 104–106. [Google Scholar] [CrossRef]
- Valitutti, S.; Muller, S.; Dessing, M.; Lanzavecchia, A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J. Exp. Med. 1996, 183, 1917–1921. [Google Scholar] [CrossRef]
- Goff, S.L.; Johnson, L.A.; Black, M.A.; Xu, H.; Zheng, Z.; Cohen, C.J.; Morgan, R.A.; Rosenberg, S.A.; Feldman, S.A. Enhanced receptor expression and in vitro effector function of a murine-human hybrid MART-1-reactive T cell receptor following a rapid expansion. Cancer Immunol. Immunother. 2010, 59, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Boniface, J.J.; Rabinowitz, J.D.; Wulfing, C.; Hampl, J.; Reich, Z.; Altman, J.D.; Kantor, R.M.; Beeson, C.; McConnell, H.M.; Davis, M.M. Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands [corrected]. Immunity 1998, 9, 459–466. [Google Scholar] [CrossRef]
- Cochran, J.R.; Cameron, T.O.; Stern, L.J. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity 2000, 12, 241–250. [Google Scholar] [CrossRef]
- Stone, J.D.; Stern, L.J. CD8 T cells, like CD4 T cells, are triggered by multivalent engagement of TCRs by MHC-peptide ligands but not by monovalent engagement. J. Immunol. 2006, 176, 1498–1505. [Google Scholar] [CrossRef]
- Minguet, S.; Swamy, M.; Alarcon, B.; Luescher, I.F.; Schamel, W.W. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity 2007, 26, 43–54. [Google Scholar] [CrossRef]
- Cameron, T.O.; Cochran, J.R.; Yassine-Diab, B.; Sekaly, R.P.; Stern, L.J. Cutting edge: Detection of antigen-specific CD4+ T cells by HLA-DR1 oligomers is dependent on the T cell activation state. J. Immunol. 2001, 166, 741–745. [Google Scholar] [CrossRef]
- Fahmy, T.M.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Increased TCR avidity after T cell activation: A mechanism for sensing low-density antigen. Immunity 2001, 14, 135–143. [Google Scholar] [CrossRef]
- Kao, C.; Daniels, M.A.; Jameson, S.C. Loss of CD8 and TCR binding to Class I MHC ligands following T cell activation. Int. Immunol. 2005, 17, 1607–1617. [Google Scholar] [CrossRef][Green Version]
- Curtsinger, J.M.; Lins, D.C.; Mescher, M.F. CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells to (CD44low, Ly-6C-) to TCR/CD8 signaling in response to antigen. J. Immunol. 1998, 160, 3236–3243. [Google Scholar]
- Demetriou, M.; Granovsky, M.; Quaggin, S.; Dennis, J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature 2001, 409, 733–739. [Google Scholar] [CrossRef]
- Daniels, M.A.; Devine, L.; Miller, J.D.; Moser, J.M.; Lukacher, A.E.; Altman, J.D.; Kavathas, P.; Hogquist, K.A.; Jameson, S.C. CD8 binding to MHC class I molecules is influenced by T cell maturation and glycosylation. Immunity 2001, 15, 1051–1061. [Google Scholar] [CrossRef]
- Morgan, R.; Gao, G.; Pawling, J.; Dennis, J.W.; Demetriou, M.; Li, B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J. Immunol. 2004, 173, 7200–7208. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Hodge, J.W.; Ahlers, J.D.; Burke, D.S.; Schlom, J.; Berzofsky, J.A. Selective induction of high avidity CTL by altering the balance of signals from APC. J. Immunol. 2003, 170, 2523–2530. [Google Scholar] [CrossRef]
- Hodge, J.W.; Chakraborty, M.; Kudo-Saito, C.; Garnett, C.T.; Schlom, J. Multiple costimulatory modalities enhance CTL avidity. J. Immunol. 2005, 174, 5994–6004. [Google Scholar] [CrossRef]
- Yang, S.; Hodge, J.W.; Grosenbach, D.W.; Schlom, J. Vaccines with enhanced costimulation maintain high avidity memory CTL. J. Immunol. 2005, 175, 3715–3723. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef]
- van Gisbergen, K.P.; Klarenbeek, P.L.; Kragten, N.A.; Unger, P.P.; Nieuwenhuis, M.B.; Wensveen, F.M.; ten Brinke, A.; Tak, P.P.; Eldering, E.; Nolte, M.A.; et al. The costimulatory molecule CD27 maintains clonally diverse CD8(+) T cell responses of low antigen affinity to protect against viral variants. Immunity 2011, 35, 97–108. [Google Scholar] [CrossRef]
- Shibuya, K.; Shirakawa, J.; Kameyama, T.; Honda, S.; Tahara-Hanaoka, S.; Miyamoto, A.; Onodera, M.; Sumida, T.; Nakauchi, H.; Miyoshi, H.; et al. CD226 (DNAM-1) is involved in lymphocyte function-associated antigen 1 costimulatory signal for naive T cell differentiation and proliferation. J. Exp. Med. 2003, 198, 1829–1839. [Google Scholar] [CrossRef]
- Perez, O.D.; Mitchell, D.; Jager, G.C.; South, S.; Murriel, C.; McBride, J.; Herzenberg, L.A.; Kinoshita, S.; Nolan, G.P. Leukocyte functional antigen 1 lowers T cell activation thresholds and signaling through cytohesin-1 and Jun-activating binding protein 1. Nat. Immunol. 2003, 4, 1083–1092. [Google Scholar] [CrossRef]
- Wang, Y.; Shibuya, K.; Yamashita, Y.; Shirakawa, J.; Shibata, K.; Kai, H.; Yokosuka, T.; Saito, T.; Honda, S.; Tahara-Hanaoka, S.; et al. LFA-1 decreases the antigen dose for T cell activation in vivo. Int. Immunol. 2008, 20, 1119–1127. [Google Scholar] [CrossRef]
- Ahmadi, M.; King, J.W.; Xue, S.A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef]
- Wooldridge, L.; van den Berg, H.A.; Glick, M.; Gostick, E.; Laugel, B.; Hutchinson, S.L.; Milicic, A.; Brenchley, J.M.; Douek, D.C.; Price, D.A.; et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J. Biol. Chem. 2005, 280, 27491–27501. [Google Scholar] [CrossRef]
- Holler, P.D.; Kranz, D.M. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 2003, 18, 255–264. [Google Scholar] [CrossRef]
- Wooldridge, L.; Laugel, B.; Ekeruche, J.; Clement, M.; van den Berg, H.A.; Price, D.A.; Sewell, A.K. CD8 controls T cell cross-reactivity. J. Immunol. 2010, 185, 4625–4632. [Google Scholar] [CrossRef]
- Norment, A.M.; Salter, R.D.; Parham, P.; Engelhard, V.H.; Littman, D.R. Cell-cell adhesion mediated by CD8 and MHC class I molecules. Nature 1988, 336, 79–81. [Google Scholar] [CrossRef]
- Luescher, I.F.; Vivier, E.; Layer, A.; Mahiou, J.; Godeau, F.; Malissen, B.; Romero, P. CD8 modulation of T-cell antigen receptor-ligand interactions on living cytotoxic T lymphocytes. Nature 1995, 373, 353–356. [Google Scholar] [CrossRef]
- Veillette, A.; Bookman, M.A.; Horak, E.M.; Bolen, J.B. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell 1988, 55, 301–308. [Google Scholar] [CrossRef]
- Arcaro, A.; Gregoire, C.; Bakker, T.R.; Baldi, L.; Jordan, M.; Goffin, L.; Boucheron, N.; Wurm, F.; van der Merwe, P.A.; Malissen, B.; et al. CD8beta endows CD8 with efficient coreceptor function by coupling T cell receptor/CD3 to raft-associated CD8/p56(lck) complexes. J. Exp. Med. 2001, 194, 1485–1495. [Google Scholar] [CrossRef]
- Arcaro, A.; Gregoire, C.; Boucheron, N.; Stotz, S.; Palmer, E.; Malissen, B.; Luescher, I.F. Essential role of CD8 palmitoylation in CD8 coreceptor function. J. Immunol. 2000, 165, 2068–2076. [Google Scholar] [CrossRef]
- Pang, D.J.; Hayday, A.C.; Bijlmakers, M.J. CD8 Raft localization is induced by its assembly into CD8alpha beta heterodimers, Not CD8alpha alpha homodimers. J. Biol. Chem. 2007, 282, 13884–13894. [Google Scholar] [CrossRef]
- Govers, C.; Sebestyen, Z.; Coccoris, M.; Willemsen, R.A.; Debets, R. T cell receptor gene therapy: Strategies for optimizing transgenic TCR pairing. Trends Mol. Med. 2010, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Uckert, W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef]
- Scholten, K.B.; Kramer, D.; Kueter, E.W.; Graf, M.; Schoedl, T.; Meijer, C.J.; Schreurs, M.W.; Hooijberg, E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin. Immunol. 2006, 119, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Jorritsma, A.; Gomez-Eerland, R.; Dokter, M.; van de Kasteele, W.; Zoet, Y.M.; Doxiadis, I.I.; Rufer, N.; Romero, P.; Morgan, R.A.; Schumacher, T.N.; et al. Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood 2007, 110, 3564–3572. [Google Scholar] [CrossRef]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.H.; Fowler, C.; Greenberg, P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef]
- Haga-Friedman, A.; Horovitz-Fried, M.; Cohen, C.J. Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J. Immunol. 2012, 188, 5538–5546. [Google Scholar] [CrossRef]
- Kuball, J.; Hauptrock, B.; Malina, V.; Antunes, E.; Voss, R.H.; Wolfl, M.; Strong, R.; Theobald, M.; Greenberg, P.D. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J. Exp. Med. 2009, 206, 463–475. [Google Scholar] [CrossRef]
- Bethune, M.T.; Gee, M.H.; Bunse, M.; Lee, M.S.; Gschweng, E.H.; Pagadala, M.S.; Zhou, J.; Cheng, D.; Heath, J.R.; Kohn, D.B.; et al. Domain-swapped T cell receptors improve the safety of TCR gene therapy. Elife 2016, 5. [Google Scholar] [CrossRef]
- Sebestyen, Z.; Schooten, E.; Sals, T.; Zaldivar, I.; San Jose, E.; Alarcon, B.; Bobisse, S.; Rosato, A.; Szollosi, J.; Gratama, J.W.; et al. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J. Immunol. 2008, 180, 7736–7746. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, R.A.; Weijtens, M.E.; Ronteltap, C.; Eshhar, Z.; Gratama, J.W.; Chames, P.; Bolhuis, R.L. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Ther. 2000, 7, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Harris, D.T.; Soto, C.M.; Chervin, A.S.; Aggen, D.H.; Roy, E.J.; Kranz, D.M. A novel T cell receptor single-chain signaling complex mediates antigen-specific T cell activity and tumor control. Cancer Immunol. Immunother. 2014, 63, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Fujiwara, H.; Okamoto, S.; An, J.; Nagai, K.; Shirakata, T.; Mineno, J.; Kuzushima, K.; Shiku, H.; Yasukawa, M. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood 2011, 118, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Berdien, B.; Mock, U.; Atanackovic, D.; Fehse, B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014, 21, 539–548. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Georgiadis, C.; Preece, R.; Mock, U.; Qasim, W. TCRalphabeta/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat. Commun. 2019, 10, 4451. [Google Scholar] [CrossRef]
- Watson, H.A.; Durairaj, R.R.P.; Ohme, J.; Alatsatianos, M.; Almutairi, H.; Mohammed, R.N.; Vigar, M.; Reed, S.G.; Paisey, S.J.; Marshall, C.; et al. L-Selectin Enhanced T Cells Improve the Efficacy of Cancer Immunotherapy. Front. Immunol. 2019, 10, 1321. [Google Scholar] [CrossRef]
- Morris, E.C.; Tsallios, A.; Bendle, G.M.; Xue, S.A.; Stauss, H.J. A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc. Natl. Acad. Sci. USA 2005, 102, 7934–7939. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Schepers, K.; van den Boom, M.D.; Topham, D.J.; Schumacher, T.N. Generation of T cell help through a MHC class I-restricted TCR. J. Immunol. 2006, 177, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Frankel, T.L.; Burns, W.R.; Peng, P.D.; Yu, Z.; Chinnasamy, D.; Wargo, J.A.; Zheng, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Both CD4 and CD8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid TCR targeting tyrosinase. J. Immunol. 2010, 184, 5988–5998. [Google Scholar] [CrossRef] [PubMed]
- van Loenen, M.M.; Hagedoorn, R.S.; de Boer, R.; Falkenburg, J.H.; Heemskerk, M.H. Extracellular domains of CD8alpha and CD8ss subunits are sufficient for HLA class I restricted helper functions of TCR-engineered CD4(+) T cells. PLoS ONE 2013, 8, e65212. [Google Scholar] [CrossRef]
- Xue, S.A.; Gao, L.; Ahmadi, M.; Ghorashian, S.; Barros, R.D.; Pospori, C.; Holler, A.; Wright, G.; Thomas, S.; Topp, M.; et al. Human MHC Class I-restricted high avidity CD4(+) T cells generated by co-transfer of TCR and CD8 mediate efficient tumor rejection in vivo. Oncoimmunology 2013, 2, e22590. [Google Scholar] [CrossRef]
- Dossa, R.G.; Cunningham, T.; Sommermeyer, D.; Medina-Rodriguez, I.; Biernacki, M.A.; Foster, K.; Bleakley, M. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood 2018, 131, 108–120. [Google Scholar] [CrossRef]
- Rath, J.A.; Bajwa, G.; Carreres, B.; Hoyer, E.; Gruber, I.; Martínez-Paniagua, M.A.; Yu, Y.R.; Nouraee, N.; Sadeghi, F.; Wu, M.; et al. Single-cell transcriptomics identifies multiple pathways underlying anti-tumor function of TCR and CD8-alpha-beta engineered human CD4+ T cells. Sci. Adv. 2020, in press. [Google Scholar]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- D'Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Riviere, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 2011, 121, 1822–1826. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O'Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldorsdottir, H.R.; Holmen Olofsson, G.; Met, O.; Thor Straten, P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, 7, e1450715. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; van den Broek, M.; Mischo, A.; Soltermann, A.; Jungel, A.; Marroquin Belaunzaran, O.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Govers, C.; Sebestyén, Z.; Roszik, J.; van Brakel, M.; Berrevoets, C.; Szöőr, Á.; Panoutsopoulou, K.; Broertjes, M.; Van, T.; Vereb, G.; et al. TCRs Genetically Linked to CD28 and CD3ε Do Not Mispair with Endogenous TCR Chains and Mediate Enhanced T Cell Persistence and Anti-Melanoma Activity. J. Immunol. 2014, 193, 5315–5326. [Google Scholar] [CrossRef]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, M.G.; Daum, R.; Rooney, C.M.; Arber, C.; Omer, B. Enhancing Tumor Directed T Cells with a Costimulatory CAR. Mol. Ther. 2020, 28, 210–211. [Google Scholar] [CrossRef]
- Katsarou, A.; Sjostrand, M.L.; Nianias, A.; Poels, R.; Zweegman, S.; Mutis, T.; Sadelain, M.; Groen, R.W.J.; Themeli, M. Co-Targeting CD38 with a Chimeric Costimulatory Receptor Enhances Adoptive T Cell Therapy for Hematological Malignancies. Mol. Ther. 2020, 28, 40–41. [Google Scholar] [CrossRef]
- Daniel-Meshulam, I.; Horovitz-Fried, M.; Cohen, C.J. Enhanced antitumor activity mediated by human 4-1BB-engineered T cells. Int. J. Cancer 2013, 133, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, C.D.; Brenner, D.A.; Mukherjee, M.; Hirsch, R.A.; Ott, L.; Wu, M.F.; Liu, H.; Dakhova, O.; Orange, J.S.; Brenner, M.K.; et al. c-MPL provides tumor-targeted T-cell receptor-transgenic T cells with costimulation and cytokine signals. Blood 2017, 130, 2739–2749. [Google Scholar] [CrossRef]
- Wagner, H.J.; Bollard, C.M.; Vigouroux, S.; Huls, M.H.; Anderson, R.; Prentice, H.G.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. A strategy for treatment of Epstein-Barr virus-positive Hodgkin's disease by targeting interleukin 12 to the tumor environment using tumor antigen-specific T cells. Cancer Gene Ther. 2004, 11, 81–91. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Muranski, P.; Kaiser, A.; Boni, A.; Sanchez-Perez, L.; Yu, Z.; Palmer, D.C.; Reger, R.N.; Borman, Z.A.; Zhang, L.; et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010, 70, 6725–6734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kerkar, S.P.; Yu, Z.; Zheng, Z.; Yang, S.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther. 2011, 19, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Alsaieedi, A.; Holler, A.; Velica, P.; Bendle, G.; Stauss, H.J. Safety and efficacy of Tet-regulated IL-12 expression in cancer-specific T cells. Oncoimmunology 2019, 8, 1542917. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef]
- Lacuesta, K.; Buza, E.; Hauser, H.; Granville, L.; Pule, M.; Corboy, G.; Finegold, M.; Weiss, H.; Chen, S.Y.; Brenner, M.K.; et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J. Immunother. 2006, 29, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.E.; Dotti, G.; Lu, A.; Khalil, M.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Bollard, C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J. Immunother. 2008, 31, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther 2013, 20, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Bendle, G.M.; Linnemann, C.; Bies, L.; Song, J.Y.; Schumacher, T.N. Blockade of TGF-beta signaling greatly enhances the efficacy of TCR gene therapy of cancer. J. Immunol. 2013, 191, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Tripic, T.; Cruz, C.R.; Dotti, G.; Gottschalk, S.; Torrano, V.; Dakhova, O.; Carrum, G.; Ramos, C.A.; Liu, H.; et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 1128–1139. [Google Scholar] [CrossRef]
- Shin, J.H.; Park, H.B.; Oh, Y.M.; Lim, D.P.; Lee, J.E.; Seo, H.H.; Lee, S.J.; Eom, H.S.; Kim, I.H.; Lee, S.H.; et al. Positive conversion of negative signaling of CTLA4 potentiates antitumor efficacy of adoptive T-cell therapy in murine tumor models. Blood 2012, 119, 5678–5687. [Google Scholar] [CrossRef]
- Ankri, C.; Shamalov, K.; Horovitz-Fried, M.; Mauer, S.; Cohen, C.J. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J. Immunol. 2013, 191, 4121–4129. [Google Scholar] [CrossRef]
- Schlenker, R.; Olguin-Contreras, L.F.; Leisegang, M.; Schnappinger, J.; Disovic, A.; Ruhland, S.; Nelson, P.J.; Leonhardt, H.; Harz, H.; Wilde, S.; et al. Chimeric PD-1:28 Receptor Upgrades Low-Avidity T cells and Restores Effector Function of Tumor-Infiltrating Lymphocytes for Adoptive Cell Therapy. Cancer Res. 2017, 77, 3577–3590. [Google Scholar] [CrossRef]
- Oda, S.K.; Daman, A.W.; Garcia, N.M.; Wagener, F.; Schmitt, T.M.; Tan, X.; Chapuis, A.G.; Greenberg, P.D. A CD200R-CD28 fusion protein appropriates an inhibitory signal to enhance T-cell function and therapy of murine leukemia. Blood 2017, 130, 2410–2419. [Google Scholar] [CrossRef]
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov. 2018, 8, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Li, P.J.; Blaeschke, F.; Nies, J.F.; Apathy, R.; Mowery, C.; Yu, R.; Nguyen, M.L.T.; Lee, Y.; Truong, A.; et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 2020. [Google Scholar] [CrossRef]
- Wilkie, S.; Burbridge, S.E.; Chiapero-Stanke, L.; Pereira, A.C.; Cleary, S.; van der Stegen, S.J.; Spicer, J.F.; Davies, D.M.; Maher, J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010, 285, 25538–25544. [Google Scholar] [CrossRef] [PubMed]
- Leen, A.M.; Sukumaran, S.; Watanabe, N.; Mohammed, S.; Keirnan, J.; Yanagisawa, R.; Anurathapan, U.; Rendon, D.; Heslop, H.E.; Rooney, C.M.; et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. 2014, 22, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef]
- Su, S.; Zou, Z.; Chen, F.; Ding, N.; Du, J.; Shao, J.; Li, L.; Fu, Y.; Hu, B.; Yang, Y.; et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology 2017, 6, e1249558. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Cheng, C.; Mu, W.; Liu, X.; Li, N.; Wei, X.; Liu, X.; Xia, C.; Wang, H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017, 11, 554–562. [Google Scholar] [CrossRef]
- Shi, L.; Meng, T.; Zhao, Z.; Han, J.; Zhang, W.; Gao, F.; Cai, J. CRISPR knock out CTLA-4 enhances the anti-tumor activity of cytotoxic T lymphocytes. Gene 2017, 636, 36–41. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef]
- Marotte, L.; Simon, S.; Vignard, V.; Dupre, E.; Gantier, M.; Cruard, J.; Alberge, J.B.; Hussong, M.; Deleine, C.; Heslan, J.M.; et al. Increased antitumor efficacy of PD-1-deficient melanoma-specific human lymphocytes. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Drakes, D.J.; Rafiq, S.; Purdon, T.J.; Lopez, A.V.; Chandran, S.S.; Klebanoff, C.A.; Brentjens, R.J. Optimization of T-cell receptor-modified T cells for cancer therapy. Cancer Immunol. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Lee, P.H.; Vodnala, S.K.; Gurusamy, D.; Kishton, R.J.; Yu, Z.; Eidizadeh, A.; Eil, R.; Fioravanti, J.; Gattinoni, L.; et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest. 2019, 129, 1551–1565. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Young, M.; Barth, P. Reprogramming cellular functions with engineered membrane proteins. Curr. Opin. Biotechnol. 2017, 47, 92–101. [Google Scholar] [CrossRef]
- Singh, N.; Shi, J.; June, C.H.; Ruella, M. Genome-Editing Technologies in Adoptive T Cell Immunotherapy for Cancer. Curr. Hematol. Malig. Rep. 2017, 12, 522–529. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gonen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Schober, K.; Muller, T.R.; Gokmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic replacement of T-cell receptor alpha- and beta-chains with preservation of near-physiological T-cell function. Nat Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Shifrut, E.; Carnevale, J.; Tobin, V.; Roth, T.L.; Woo, J.M.; Bui, C.T.; Li, P.J.; Diolaiti, M.E.; Ashworth, A.; Marson, A. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018, 175, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019, 178, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Certo, M.T.; Morgan, R.A. Salient Features of Endonuclease Platforms for Therapeutic Genome Editing. Mol. Ther. 2016, 24, 422–429. [Google Scholar] [CrossRef][Green Version]
- Wang, D.; Zhang, F.; Gao, G. CRISPR-Based Therapeutic Genome Editing: Strategies and In Vivo Delivery by AAV Vectors. Cell 2020, 181, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Genovese, P.; Magnani, Z.; Ruggiero, E.; Landoni, E.; Camisa, B.; Schiroli, G.; Provasi, E.; Lombardo, A.; Reik, A.; et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood 2017, 130, 606–618. [Google Scholar] [CrossRef]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Potrel, P.; Bas, C.; Lemaire, L.; Galetto, R.; et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.L.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef]
- Knipping, F.; Osborn, M.J.; Petri, K.; Tolar, J.; Glimm, H.; von Kalle, C.; Schmidt, M.; Gabriel, R. Genome-wide Specificity of Highly Efficient TALENs and CRISPR/Cas9 for T Cell Receptor Modification. Mol. Ther. Methods Clin. Dev. 2017, 4, 213–224. [Google Scholar] [CrossRef]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfo, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Sather, B.D.; Romano Ibarra, G.S.; Sommer, K.; Curinga, G.; Hale, M.; Khan, I.F.; Singh, S.; Song, Y.; Gwiazda, K.; Sahni, J.; et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015, 7, 307ra156. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Lee, B.; Honaker, Y.; Leung, W.H.; Grier, A.E.; Jacobs, H.M.; Sommer, K.; Sahni, J.; Jackson, S.W.; Scharenberg, A.M.; et al. Homology-Directed Recombination for Enhanced Engineering of Chimeric Antigen Receptor T Cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Roth, T.L.; Li, P.J.; Chen, P.A.; Apathy, R.; Mamedov, M.R.; Vo, L.T.; Tobin, V.R.; Goodman, D.; Shifrut, E.; et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020, 38, 44–49. [Google Scholar] [CrossRef]
- Sachdeva, M.; Busser, B.W.; Temburni, S.; Jahangiri, B.; Gautron, A.S.; Marechal, A.; Juillerat, A.; Williams, A.; Depil, S.; Duchateau, P.; et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019, 10, 5100. [Google Scholar] [CrossRef]
- Nobles, C.L.; Sherrill-Mix, S.; Everett, J.K.; Reddy, S.; Fraietta, J.A.; Porter, D.L.; Frey, N.; Gill, S.I.; Grupp, S.A.; Maude, S.L.; et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J. Clin. Invest. 2020, 130, 673–685. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O'Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Antigen/ HLA | Cancer | Protocol | TCR Origin | Name | Pts | Response | Toxicities | Reference |
|---|---|---|---|---|---|---|---|---|
| MART1/ A*02:01 | Melanoma | Lymphodepletion (Cy/Flu), ACT, high dose IL-2, peptide vaccine | Human TILs [94] | MART1 | 17 | 2 PR, 15 NR | None | [81] |
| MART1/ A*02:01 gp100/ A*02:01 | Melanoma | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Human TILs, vaccinated mice [95] | DMF5 gp100 | 20 16 | 6 PR, 14 NR 1 CR, 2 PR, 13 NR | AE: 11× grade 2, 8× grade 3 AE: 10× grade 2, 1× grade 3 (skin, eye, ear) | [82] |
| MART-1/ A*02:01 | Melanoma | Lymphodepletion (Cy/Flu), ACT (D0), high dose IL-2, DC vaccine (D1, D14, D30) | Human TILs [95] | DMF5 | 14 | 7 SD, 6 PD, 1 N/A | Frozen: None Fresh: SAEs: 2× grade 4 (ARDS) | [83] |
| NY-ESO-1/ A*02:01 | Sarcoma, Melanoma | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Human TILs, affinity-enhanced [73] | NY-ESO-1 1G4-a95:LY | 6 11 | Sarcoma: 4 PR, 2 PD Melanoma: 2 CR, 3 PR, 6 PD | None | [4] |
| NY-ESO-1/ A*02:01 | Sarcoma, Melanoma | Lymphodepletion (Cy/Flu), ACT, high dose IL-2, some vaccinated | Human TILs, affinity-enhanced [73] | NY-ESO-1 1G4-a95:LY | 18 20 | Sarcoma: 1 CR, 10 PR, 7 NR Melanoma: 4 CR, 7 PR, 9 NR | None | [5] |
| NY-ESO-1/ A*02:01 | Multiple myeloma | High dose Melphalan + ASCT, ACT (D2), PCV vaccine (D14, 42, 90), low dose lenalidomide (start D100) | Human TILs, affinity-enhanced [73] | NY-ESO-1c259 | 20 | 14 nCR/CR, 2 VGPR, 2 PR, 1 SD, 1 PD | SAE: 7× grade 3–4, AE: 17× grade 1–3 | [6] |
| NY-ESO-1/ A*02:01 | Sarcoma | Lymphodepletion (Cy/Flu), ACT | Human TILs, affinity-enhanced [73] | NY-ESO-1c259 SPEAR T cells | 12 | 1 CR, 5 PR, 6 SD | AE: 11× grade 3–4, CRS in 5 pts (2× grade 1, 1× grade 2, 2× grade 3) | [87] |
| NY-ESO-1/ A*02:01 | Sarcoma | Lymphodepletion (Cy/Flu or Cy), ACT | Human TILs, affinity-enhanced [73] | NY-ESO-1c259 SPEAR T cells | 42 | 1 CR, 14 PR, 24 SD, 3 PD | Not described | [88] |
| NY-ESO-1/ A*02:01 | Sarcoma, Multiple Myeloma | Lymphodepletion (Cy/Flu), ACT | Vaccinated patient [96] | 8F TCR NYCE T cells | 3 | 2 SD, 1 PD | AE: 20× grade 3–4 | [89] |
| MAGE-A3/A12/ A*02:01 | Sarcoma, Melanoma, Esophageal cancer | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Vaccinated mice, affinity-enhanced [92] | MAGE-A3 | 9 | 1 CR, 4 PR, 4 NR | SAE: 2× grade 5 neurotoxicity, 1× grade 4 neurotoxicity attributed to TCR T cells | [79] |
| MAGE-A3/ A*01:01 | Melanoma, Multiple myeloma | Lymphodepletion (Cy), Split ACT D5 (30%) D6 (70%), high dose Melphalan + ASCT, ACT (D2) | Vaccinated patient, affinity-enhanced [78] | MAGE-A3a3a | 2 | Not evaluable | SAE: 2× grade 5 cardiac toxicity attributed to TCR T cells | [77] |
| MAGE-A4/ A*24:02 | Esophageal cancer | ACT (D0), peptide vaccine (D14, D28) | Human healthy donor [97] | MAGE-A4 | 10 | 7 PD, 3 SD | None | [85] |
| CEA/ A*02:01 | Colorectal cancer | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Vaccinated mice, affinity-enhanced [91] | CEA-reactive TCR | 3 | 2 NR, 1 PD | SAE: 2× grade 3 diarrhea, DLT, 3× inflammatory colitis attributed to TCR T cells | [84] |
| WT1/ A*02:01 | AML | Allogeneic HCT, Prophylactic ACT if NED (D47-190), low dose IL-2, second ACT (in 7 pts) | Human healthy donor [98] | TCR-C4 | 12 | 12 CR | SAE: 2× grade 3 CRS, 24× grade 3–4 cytopenias, cGVHD: 6×, aGVHD: 2× grade 2, 1× grade 3 | [9] |
| WT-1/ A*24:02 | AML MDS | ACT (D0 and D28), peptide vaccine (D30 and D44) | Human healthy donor [99] | WT-1 | 8 | 1 SD, 3 blast reduction, 4 PD | None | [90] |
| MAGE-A3/ DPB1*0401 | Metastatic/ refractory cancer | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Vaccinated patient [100] | MAGE-A3 (6F9 TCR) | 17 | 1 CR, 3 PR, 13 NR | AE: 11× grade 2, 5× grade 3, 4× grade 4 | [86] |
| HPV16 -E6/ A*02:01 | Metastatic HPV16+ cancer | Lymphodepletion (Cy/Flu), ACT, high dose IL-2 | Human TILs [101] | E6 TCR | 12 | 2 PR, 4SD, 7 PD (1 pt treated 2x) | AE: 68× grade 3–4, cytopenias and IL-2 side-effects | [8] |
| Goal of Engineering | Modification/Construct | Target of Modification | Mechanism of Action | References |
|---|---|---|---|---|
| Enhance tumor infiltration | Enforce chemokine receptor expression | Chemokines in the TME |
| [189] |
| Enforce expression of ECM degrading enzymes | ECM |
| [193] | |
| CAR targeting the tumor stroma | Tumor stroma |
| [194,195,196] | |
| Provide co-stimulation (signal 2) | CD28-CD3ε integration into TCRαβ chains | No additional recognition |
| [197] |
| Co-stimulatory CAR (coCAR) | Cell surface antigen on the tumor target or bystander cell |
| [200,201] | |
| Co-stimulatory receptors (e.g., 4-1BB) | Cell surface co-stimulatory ligand recognition on tumor target or bystander cell |
| [202] | |
| Provide cytokine signals (signal 3) | Natural receptor targeting a microenvironment factor (e.g., c-MPL) | Soluble factor recognition in the TME (e.g., thrombopoietin, TPO) |
| [203] |
| Enforce secretion of effector cytokines (e.g., IL-12, IL-18) | No additional recognition |
| [204,205,206,207,208,209] | |
| Constitutively active cytokine receptor (e.g., IL-7R) | No additional recognition |
| [210] | |
| Revert immune inhibition | Dominant negative receptors (DNR) | Inhibitory signals in the TME (e.g., TGF-β) |
| [211,212,213,214,215,216] |
| Chimeric switch receptors (CSR) | Inhibitory signals in the TME (e.g., checkpoint ligands) |
| [217,218,219,220,221,222,223] | |
| Chimeric cytokine receptors (CCR) | Inhibitory cytokines in the TME (e.g., IL-4) |
| [224,225] | |
| Knockout of checkpoint receptors | No additional recognition |
| [226,227,228,229,230,231] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rath, J.A.; Arber, C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells 2020, 9, 1485. https://doi.org/10.3390/cells9061485
Rath JA, Arber C. Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells. 2020; 9(6):1485. https://doi.org/10.3390/cells9061485
Chicago/Turabian StyleRath, Jan A., and Caroline Arber. 2020. "Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy" Cells 9, no. 6: 1485. https://doi.org/10.3390/cells9061485
APA StyleRath, J. A., & Arber, C. (2020). Engineering Strategies to Enhance TCR-Based Adoptive T Cell Therapy. Cells, 9(6), 1485. https://doi.org/10.3390/cells9061485