Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
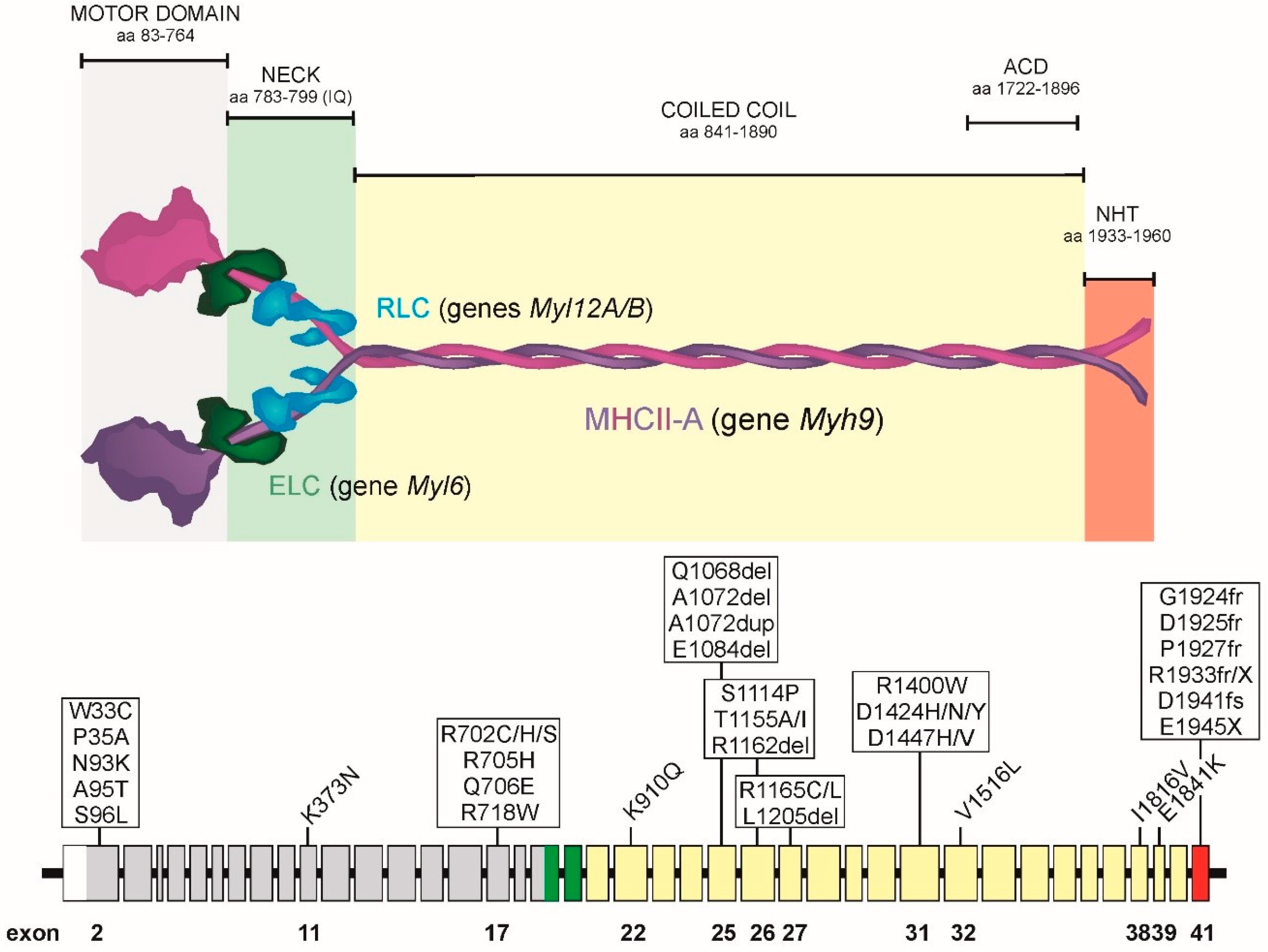
2. Organization of the MYH9 Gene, MHCII-A Protein and NMII-A Hexamer
3. Clinical Features of MYH9-RD Patients
3.1. Macrothrombocytopenia
3.2. Non-Syndromic Sensorineural Deafness
3.3. Nephritis
3.4. Presenile Cataracts
3.5. Other Manifestations of MYH-RD
4. Prevalent MYH9 Genotypes
5. Regulation of NMII-A Activity
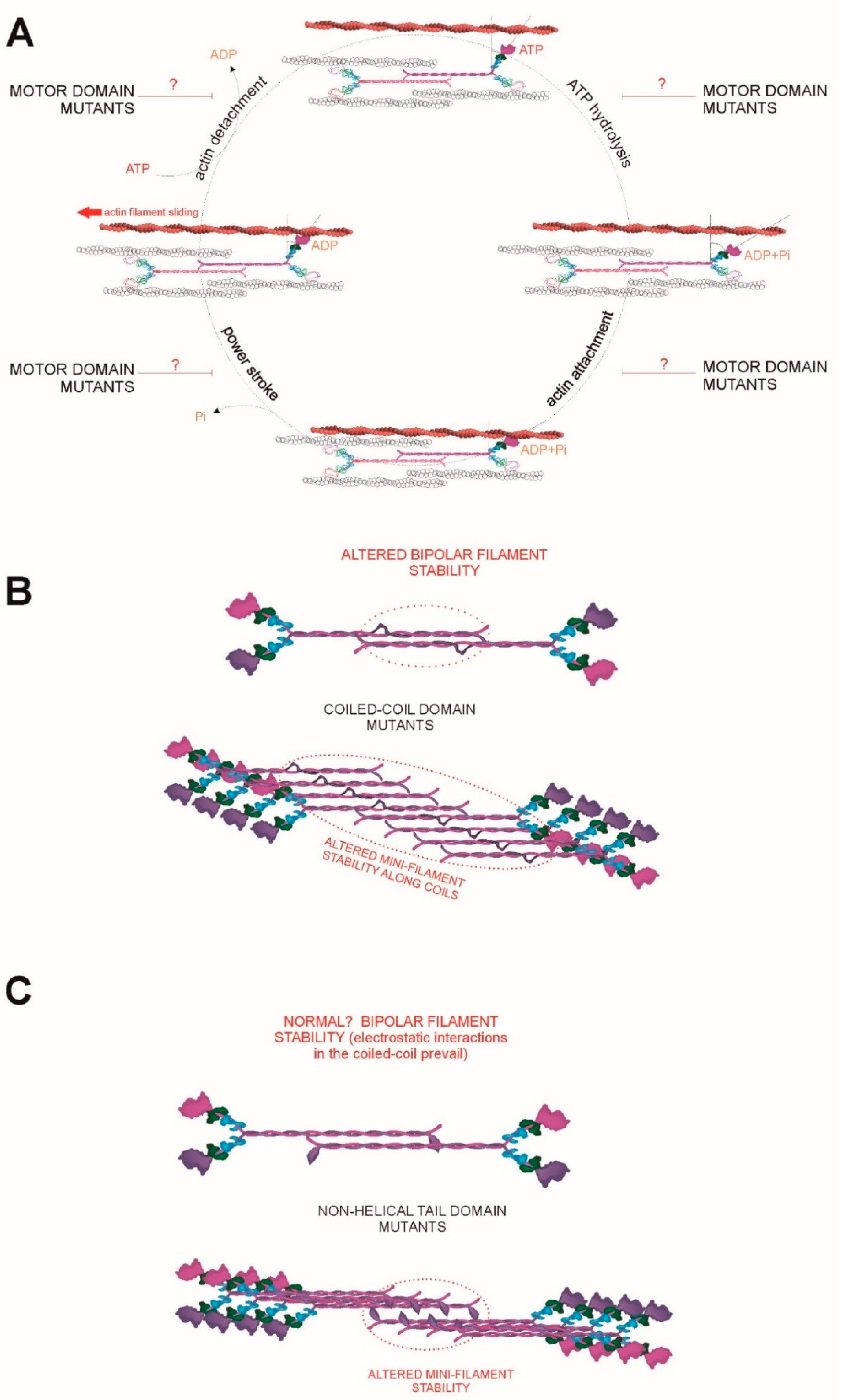
5.1. Regulation of the Conforzmation of NMII-A and Its ATPase Activity
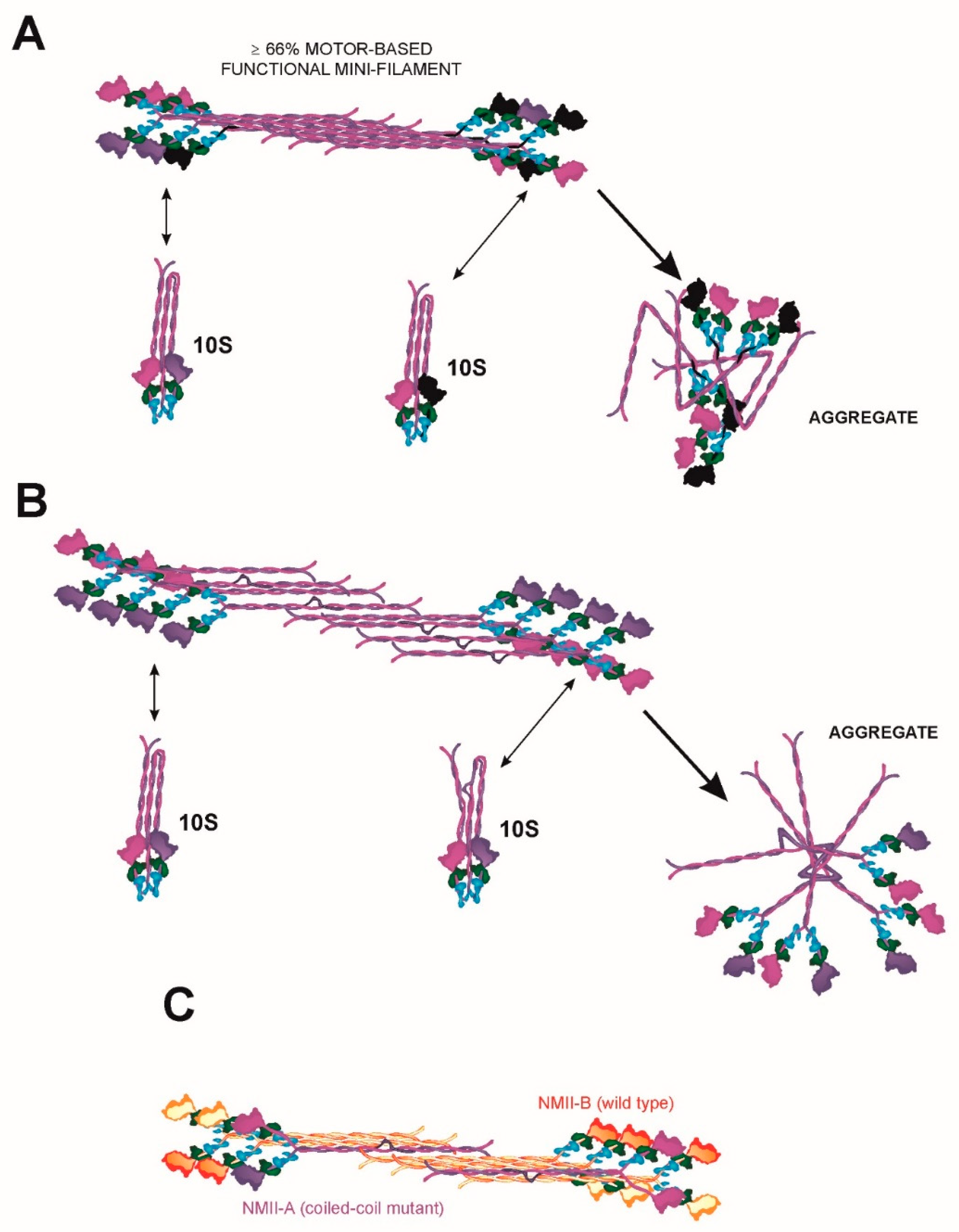
5.2. Regulation of Bipolar Filament Formation and Higher Order Mini-Filament Assembly and Disassembly
6. Three Hypotheses to Explain the Correlation between Genotype and Molecular Phenotype
6.1. A Continuum of MYH9 Mutations that Cause Graded Molecular Defects
6.2. A Continuum of MYH9 Mutations that Causes Graded Protein Accumulation in Aggregates
6.3. Heterodimerization and Copolymerization with Different Myosin Isoforms
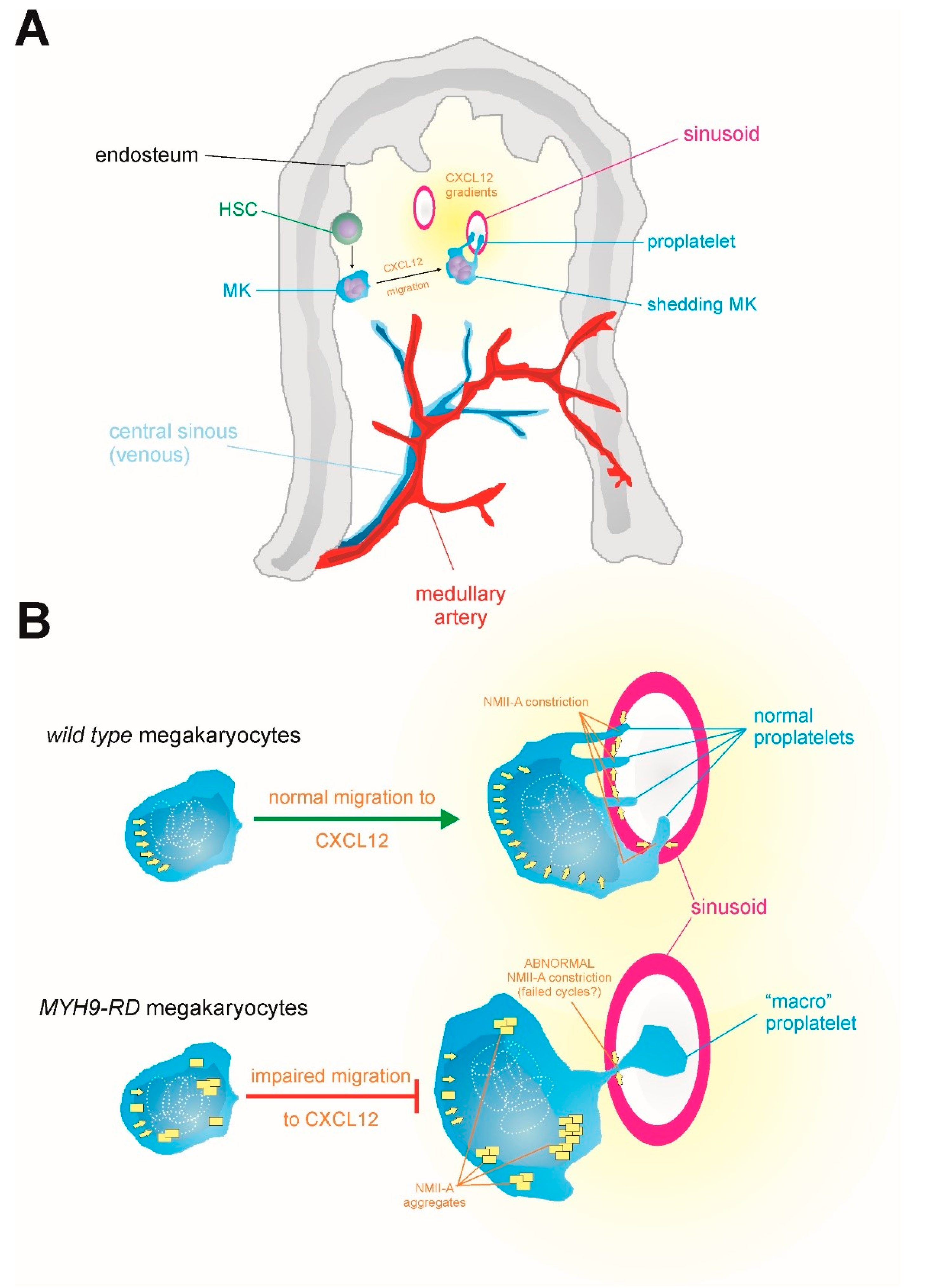
7. Why do MYH9-RD Patients Display Few, but Giant, Platelets?
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| MYH9-RD | MYH9-gene Related Diseases |
| MHCII | myosin heavy chain |
| NMII | non-muscle myosin II |
| SMM | smooth muscle myosin |
| ELC | essential light chain |
| RLC | regulatory light chain |
| NHT | non-helical tailpiece |
| ACD | assembly-competent domain |
| CXCL12 | chemokine CXCL12, also known as stromal cell-derived factor-1 (SDF-1) |
| MK | megakaryocyte |
| DFNA | Deafness, Autosomal Dominant |
| ROCK | RhoA- Coiled Coil Kinase |
| PKC | protein kinase C |
| TRPM7 | transient receptor potential melastatin 7 |
| CKII | casein kinase II |
References
- Althaus, K.; Greinacher, A. MYH-9 Related Platelet Disorders: Strategies for Management and Diagnosis. Transfus. Med. Hemotherapy 2010, 37, 260–267. [Google Scholar] [CrossRef]
- Pecci, A.; Klersy, C.; Gresele, P.; Lee, K.J.; De Rocco, D.; Bozzi, V.; Russo, G.; Heller, P.G.; Loffredo, G.; Ballmaier, M.; et al. MYH9-related disease: A novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum. Mutat. 2014, 35, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Pecci, A.; Panza, E.; Pujol-Moix, N.; Klersy, C.; Di Bari, F.; Bozzi, V.; Gresele, P.; Lethagen, S.; Fabris, F.; Dufour, C.; et al. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related disease. Hum. Mutat. 2008, 29, 409–417. [Google Scholar] [CrossRef]
- Savoia, A.; De Rocco, D.; Pecci, A. MYH9 gene mutations associated with bleeding. Platelets 2017, 28, 312–315. [Google Scholar] [CrossRef]
- Sellers, J.R. Myosins: A diverse superfamily. Biochim. Biophys. Acta 2000, 1496, 3–22. [Google Scholar] [CrossRef]
- Juanes-Garcia, A.; Llorente-Gonzalez, C.; Vicente-Manzanares, M. Non Muscle Myosin II. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Maupin, P.; Phillips, C.L.; Adelstein, R.S.; Pollard, T.D. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J. Cell Sci. 1994, 107, 3077–3090. [Google Scholar]
- Kolega, J. Cytoplasmic dynamics of myosin IIA and IIB: Spatial ‘sorting’ of isoforms in locomoting cells. J. Cell Sci. 1998, 111, 2085–2095. [Google Scholar] [PubMed]
- Conti, M.A.; Kawamoto, S.; Adelstein, R.S. Myosins: A Superfamily of Molecular Motors; Coluccio, L.M., Ed.; Springer: Watertown, MA, USA, 2007; pp. 223–264. [Google Scholar]
- Heissler, S.M.; Manstein, D.J. Nonmuscle myosin-2: Mix and match. Cell Mol. Life Sci. 2013, 70, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Smith, R.; Kendrick-Jones, J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature 1983, 302, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Dasbiswas, K.; Hu, S.; Schnorrer, F.; Safran, S.A.; Bershadsky, A.D. Ordering of myosin II filaments driven by mechanical forces: Experiments and theory. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2018, 373, 20170114. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Woodhead, J.L. Structure and function of myosin filaments. Curr. Opin. Struct. Biol. 2006, 16, 204–212. [Google Scholar] [CrossRef] [PubMed]
- El-Mezgueldi, M.; Bagshaw, C.R. Myosins: A Superfamily of Molecular Motors; Coluccio, L.M., Ed.; Springer: Watertown, MA, USA, 2007; pp. 55–93. [Google Scholar]
- Vicente-Manzanares, M.; Newell-Litwa, K.; Bachir, A.I.; Whitmore, L.A.; Horwitz, A.R. Myosin IIA/IIB restrict adhesive and protrusive signaling to generate front-back polarity in migrating cells. J. Cell Biol. 2011, 193, 381–396. [Google Scholar] [CrossRef]
- Schiffhauer, E.S.; Luo, T.; Mohan, K.; Srivastava, V.; Qian, X.; Griffis, E.R.; Iglesias, P.A.; Robinson, D.N. Mechanoaccumulative Elements of the Mammalian Actin Cytoskeleton. Curr. Biol. 2016, 26, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Schiffhauer, E.S.; Ren, Y.; Iglesias, V.A.; Kothari, P.; Iglesias, P.A.; Robinson, D.N. Myosin IIB assembly state determines its mechanosensitive dynamics. J. Cell Biol. 2019, 218, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Adelstein, R.S.; Ma, X. Replacing nonmuscle myosin 2A with myosin 2C1 permits gastrulation but not placenta vascular development in mice. Mol. Biol. Cell 2018, 29, 2326–2335. [Google Scholar] [CrossRef]
- Golomb, E.; Ma, X.; Jana, S.S.; Preston, Y.A.; Kawamoto, S.; Shoham, N.G.; Goldin, E.; Conti, M.A.; Sellers, J.R.; Adelstein, R.S. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 2004, 279, 2800–2808. [Google Scholar] [CrossRef]
- Ma, X.; Adelstein, R.S. The role of vertebrate nonmuscle Myosin II in development and human disease. Bioarchitecture 2014, 4, 88–102. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- White, J.; Barro, M.V.; Makarenkova, H.P.; Sanger, J.W.; Sanger, J.M. Localization of sarcomeric proteins during myofibril assembly in cultured mouse primary skeletal myotubes. Anat. Rec. 2014, 297, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Wang, J.; Fan, Y.; Dube, D.K.; Sanger, J.W.; Sanger, J.M. Myofibril Assembly in Cultured Mouse Neonatal Cardiomyocytes. Anat. Rec. 2018, 301, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Adelstein, R.S. In vivo studies on nonmuscle myosin II expression and function in heart development. Front Biosci. (Landmark Ed.) 2012, 17, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Jacobelli, J.; Chmura, S.A.; Buxton, D.B.; Davis, M.M.; Krummel, M.F. A single class II myosin modulates T cell motility and stopping, but not synapse formation. Nat. Immunol. 2004, 5, 531–538. [Google Scholar] [CrossRef]
- Junt, T.; Schulze, H.; Chen, Z.; Massberg, S.; Goerge, T.; Krueger, A.; Wagner, D.D.; Graf, T.; Italiano, J.E., Jr.; Shivdasani, R.A.; et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007, 317, 1767–1770. [Google Scholar] [CrossRef]
- Dunois-Larde, C.; Capron, C.; Fichelson, S.; Bauer, T.; Cramer-Borde, E.; Baruch, D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood 2009, 114, 1875–1883. [Google Scholar] [CrossRef]
- Jiang, J.; Woulfe, D.S.; Papoutsakis, E.T. Shear enhances thrombopoiesis and formation of microparticles that induce megakaryocytic differentiation of stem cells. Blood 2014, 124, 2094–2103. [Google Scholar] [CrossRef]
- Eckly, A.; Strassel, C.; Freund, M.; Cazenave, J.P.; Lanza, F.; Gachet, C.; Leon, C. Abnormal megakaryocyte morphology and proplatelet formation in mice with megakaryocyte-restricted MYH9 inactivation. Blood 2008, 113, 3182–3189. [Google Scholar] [CrossRef]
- Pal, K.; Nowak, R.; Billington, N.; Liu, R.; Ghosh, A.; Sellers, J.R.; Fowler, V.M. Megakaryocyte migration defects due to nonmuscle myosin IIA mutations underly thrombocytopenia in MYH9-Related Disease. Blood 2020, 135, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Saez, P.J.; Attia, R.; Lennon-Dumenil, A.M.; Lavi, I.; Piel, M.; Vargas, P. Myosin II Activity Is Selectively Needed for Migration in Highly Confined Microenvironments in Mature Dendritic Cells. Front. Immunol. 2019, 10, 747. [Google Scholar] [CrossRef] [PubMed]
- Petrie, R.J.; Koo, H.; Yamada, K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 2014, 345, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. [Google Scholar] [CrossRef] [PubMed]
- Seri, M.; Pecci, A.; Di Bari, F.; Cusano, R.; Savino, M.; Panza, E.; Nigro, A.; Noris, P.; Gangarossa, S.; Rocca, B.; et al. MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Med. (Baltim.) 2003, 82, 203–215. [Google Scholar] [CrossRef]
- Lalwani, A.K.; Goldstein, J.A.; Kelley, M.J.; Luxford, W.; Castelein, C.M.; Mhatre, A.N. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am. J. Hum. Genet. 2000, 67, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Canzi, P.; Pecci, A.; Manfrin, M.; Rebecchi, E.; Zaninetti, C.; Bozzi, V.; Benazzo, M. Severe to profound deafness may be associated with MYH9-related disease: Report of 4 patients. Acta Otorhinolaryngologica Italica 2016, 36, 415–420. [Google Scholar] [CrossRef]
- Furlano, M.; Arlandis, R.; Venegas, M.D.P.; Novelli, S.; Crespi, J.; Bullich, G.; Ayasreh, N.; Remacha, A.; Ruiz, P.; Lorente, L.; et al. MYH9 Associated nephropathy. Nefrol. Publ. Off. Soc. Esp. Nefrol. 2019, 39, 133–140. [Google Scholar] [CrossRef]
- Tabibzadeh, N.; Fleury, D.; Labatut, D.; Bridoux, F.; Lionet, A.; Jourde-Chiche, N.; Vrtovsnik, F.; Schlegel, N.; Vanhille, P. MYH9-related disorders display heterogeneous kidney involvement and outcome. Clin. Kidney J. 2019, 12, 494–502. [Google Scholar] [CrossRef]
- Aoki, T.; Kunishima, S.; Yamashita, Y.; Minamitani, K.; Ota, S. Macrothrombocytopenia With Congenital Bilateral Cataracts: A Phenotype of MYH9 Disorder With Exon 24 Indel Mutations. J. Pediatric Hematol. /Oncol. 2018, 40, 76–78. [Google Scholar] [CrossRef]
- Pecci, A.; Biino, G.; Fierro, T.; Bozzi, V.; Mezzasoma, A.; Noris, P.; Ramenghi, U.; Loffredo, G.; Fabris, F.; Momi, S.; et al. Alteration of liver enzymes is a feature of the MYH9-related disease syndrome. PLoS ONE 2012, 7, e35986. [Google Scholar] [CrossRef]
- De Rocco, D.; Zieger, B.; Platokouki, H.; Heller, P.G.; Pastore, A.; Bottega, R.; Noris, P.; Barozzi, S.; Glembotsky, A.C.; Pergantou, H.; et al. MYH9-related disease: Five novel mutations expanding the spectrum of causative mutations and confirming genotype/phenotype correlations. Eur. J. Med. Genet. 2013, 56, 7–12. [Google Scholar] [CrossRef]
- Balduini, C.L.; Pecci, A.; Savoia, A. Recent advances in the understanding and management of MYH9-related inherited thrombocytopenias. Br. J. Haematol. 2011, 154, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Han, K.H.; Lee, H.; Kang, H.G.; Moon, K.C.; Lee, J.H.; Park, Y.S.; Ha, I.S.; Ahn, H.S.; Choi, Y.; Cheong, H.I. Renal manifestations of patients with MYH9-related disorders. Pediatric Nephrol. 2011, 26, 549–555. [Google Scholar] [CrossRef]
- Saposnik, B.; Binard, S.; Fenneteau, O.; Nurden, A.; Nurden, P.; Hurtaud-Roux, M.F.; Schlegel, N.; French MYH9 Network. Mutation spectrum and genotype-phenotype correlations in a large French cohort of MYH9-Related Disorders. Mol. Genet. Genom. Med. 2014, 2, 297–312. [Google Scholar] [CrossRef]
- Suzuki, H.; Stafford, W.F., 3rd; Slayter, H.S.; Seidel, J.C. A conformational transition in gizzard heavy meromyosin involving the head-tail junction, resulting in changes in sedimentation coefficient, ATPase activity, and orientation of heads. J. Biol. Chem. 1985, 260, 14810–14817. [Google Scholar] [PubMed]
- Adelstein, R.S.; Conti, M.A. Phosphorylation of platelet myosin increases actin-activated myosin ATPase activity. Nature 1975, 256, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Onishi, H.; Takahashi, K.; Watanabe, S. Structure and function of chicken gizzard myosin. J. Biochem. 1978, 84, 1529–1542. [Google Scholar] [CrossRef]
- Wendt, T.; Taylor, D.; Trybus, K.M.; Taylor, K. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. USA 2001, 98, 4361–4366. [Google Scholar] [CrossRef]
- Burgess, S.A.; Yu, S.; Walker, M.L.; Hawkins, R.J.; Chalovich, J.M.; Knight, P.J. Structures of smooth muscle myosin and heavy meromyosin in the folded, shutdown state. J. Mol. Biol. 2007, 372, 1165–1178. [Google Scholar] [CrossRef]
- Jung, H.S.; Komatsu, S.; Ikebe, M.; Craig, R. Head-head and head-tail interaction: A general mechanism for switching off myosin II activity in cells. Mol. Biol. Cell 2008, 19, 3234–3242. [Google Scholar] [CrossRef]
- Yang, S.; Lee, K.H.; Woodhead, J.L.; Sato, O.; Ikebe, M.; Craig, R. The central role of the tail in switching off 10S myosin II activity. J. Gen. Physiol. 2019, 151, 1081–1093. [Google Scholar] [CrossRef]
- Trybus, K.M.; Lowey, S. Conformational states of smooth muscle myosin. Effects of light chain phosphorylation and ionic strength. J. Biol. Chem. 1984, 259, 8564–8571. [Google Scholar] [PubMed]
- Salzameda, B.; Facemyer, K.C.; Beck, B.W.; Cremo, C.R. The N-terminal lobes of both regulatory light chains interact with the tail domain in the 10 S-inhibited conformation of smooth muscle myosin. J. Biol. Chem. 2006, 281, 38801–38811. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, M.; Hartshorne, D.J. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J. Biol. Chem. 1985, 260, 10027–10031. [Google Scholar] [PubMed]
- Amano, M.; Ito, M.; Kimura, K.; Fukata, Y.; Chihara, K.; Nakano, T.; Matsuura, Y.; Kaibuchi, K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 1996, 271, 20246–20249. [Google Scholar] [CrossRef] [PubMed]
- Sandquist, J.C.; Swenson, K.I.; Demali, K.A.; Burridge, K.; Means, A.R. Rho kinase differentially regulates phosphorylation of nonmuscle myosin II isoforms A and B during cell rounding and migration. J. Biol. Chem. 2006, 281, 35873–35883. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Ito, M.; Amano, M.; Chihara, K.; Fukata, Y.; Nakafuku, M.; Yamamori, B.; Feng, J.; Nakano, T.; Okawa, K.; et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 1996, 273, 245–248. [Google Scholar] [CrossRef]
- Ikebe, M.; Koretz, J.; Hartshorne, D.J. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J. Biol. Chem. 1988, 263, 6432–6437. [Google Scholar]
- Ikebe, M.; Hartshorne, D.J.; Elzinga, M. Identification, phosphorylation, and dephosphorylation of a second site for myosin light chain kinase on the 20,000-dalton light chain of smooth muscle myosin. J. Biol. Chem. 1986, 261, 36–39. [Google Scholar]
- Vicente-Manzanares, M.; Horwitz, A.R. Myosin light chain mono- and di-phosphorylation differentially regulate adhesion and polarity in migrating cells. Biochem. Biophys. Res. Commun. 2010, 402, 537–542. [Google Scholar] [CrossRef]
- Breckenridge, M.T.; Dulyaninova, N.G.; Egelhoff, T.T. Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol. Biol. Cell 2009, 20, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Kiboku, T.; Katoh, T.; Nakamura, A.; Kitamura, A.; Kinjo, M.; Murakami, Y.; Takahashi, M. Nonmuscle myosin II folds into a 10S form via two portions of tail for dynamic subcellular localization. Genes Cells 2013, 18, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Billington, N.; Shu, S.; Yu, S.H.; Piszczek, G.; Sellers, J.R.; Korn, E.D. Effect of ATP and regulatory light-chain phosphorylation on the polymerization of mammalian nonmuscle myosin II. Proc. Natl. Acad. Sci. USA 2017, 114, E6516–E6525. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Sellers, J.R.; Adelstein, R.S.; Hidaka, H. Protein kinase C modulates in vitro phosphorylation of the smooth muscle heavy meromyosin by myosin light chain kinase. J. Biol. Chem. 1984, 259, 8808–8814. [Google Scholar] [PubMed]
- Komatsu, S.; Ikebe, M. The phosphorylation of myosin II at the Ser1 and Ser2 is critical for normal platelet-derived growth factor induced reorganization of myosin filaments. Mol. Biol. Cell 2007, 18, 5081–5090. [Google Scholar] [CrossRef]
- Asokan, S.B.; Johnson, H.E.; Rahman, A.; King, S.J.; Rotty, J.D.; Lebedeva, I.P.; Haugh, J.M.; Bear, J.E. Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCgamma/PKCalpha pathway. Dev. Cell 2014, 31, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Cuenca, R.; Llorente-Gonzalez, C.; Chapman, J.R.; Talayero, V.C.; Garrido-Casado, M.; Delgado-Arevalo, C.; Millan-Salanova, M.; Shabanowitz, J.; Hunt, D.F.; Sellers, J.R.; et al. Tyrosine Phosphorylation of the Myosin Regulatory Light Chain Controls Non-muscle Myosin II Assembly and Function in Migrating Cells. Curr. Biol. 2020. [Google Scholar] [CrossRef]
- Cohen, C.; Parry, D.A. A conserved C-terminal assembly region in paramyosin and myosin rods. J. Struct. Biol. 1998, 122, 180–187. [Google Scholar] [CrossRef]
- Dulyaninova, N.G.; Bresnick, A.R. The heavy chain has its day: Regulation of myosin-II assembly. Bioarchitecture 2013, 3, 77–85. [Google Scholar] [CrossRef]
- Ricketson, D.; Johnston, C.A.; Prehoda, K.E. Multiple tail domain interactions stabilize nonmuscle myosin II bipolar filaments. Proc. Natl. Acad. Sci. USA 2010, 107, 20964–20969. [Google Scholar] [CrossRef]
- Kaufmann, T.; Schwarz, U.S. Electrostatic and bending energies predict staggering and splaying in nonmuscle myosin II minifilaments. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nakasawa, T.; Takahashi, M.; Matsuzawa, F.; Aikawa, S.; Togashi, Y.; Saitoh, T.; Yamagishi, A.; Yazawa, M. Critical regions for assembly of vertebrate nonmuscle myosin II. Biochemistry 2005, 44, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Zareno, J.; Whitmore, L.; Choi, C.K.; Horwitz, A.F. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J. Cell Biol. 2007, 176, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, P.T.; Goyal, R.K. Calponin binds to the 20-kilodalton regulatory light chain of myosin. Biochemistry 1999, 38, 3778–3784. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, G.; Zhang, Y.; Schimerlik, M.I.; Hau, A.M.; Yang, J.; Filtz, T.M.; Kioussi, C.; Ishmael, J.E. N-methyl-D-aspartate receptor subunits are non-myosin targets of myosin regulatory light chain. J. Biol. Chem. 2009, 284, 1252–1266. [Google Scholar] [CrossRef]
- Guzik-Lendrum, S.; Heissler, S.M.; Billington, N.; Takagi, Y.; Yang, Y.; Knight, P.J.; Homsher, E.; Sellers, J.R. Mammalian myosin-18A, a highly divergent myosin. J. Biol. Chem. 2013, 288, 9532–9548. [Google Scholar] [CrossRef]
- Liu, X.; Shu, S.; Billington, N.; Williamson, C.D.; Yu, S.; Brzeska, H.; Donaldson, J.G.; Sellers, J.R.; Korn, E.D. Mammalian Nonmuscle Myosin II Binds to Anionic Phospholipids with Concomitant Dissociation of the Regulatory Light Chain. J. Biol. Chem. 2016, 291, 24828–24837. [Google Scholar] [CrossRef]
- Clark, K.; Middelbeek, J.; Dorovkov, M.V.; Figdor, C.G.; Ryazanov, A.G.; Lasonder, E.; van Leeuwen, F.N. The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. Febs. Lett. 2008, 582, 2993–2997. [Google Scholar] [CrossRef]
- Even-Faitelson, L.; Ravid, S. PAK1 and aPKCzeta regulate myosin II-B phosphorylation: A novel signaling pathway regulating filament assembly. Mol. Biol. Cell 2006, 17, 2869–2881. [Google Scholar] [CrossRef]
- Ludowyke, R.I.; Elgundi, Z.; Kranenburg, T.; Stehn, J.R.; Schmitz-Peiffer, C.; Hughes, W.E.; Biden, T.J. Phosphorylation of nonmuscle myosin heavy chain IIA on Ser1917 is mediated by protein kinase C beta II and coincides with the onset of stimulated degranulation of RBL-2H3 mast cells. J. Immunol. 2006, 177, 1492–1499. [Google Scholar] [CrossRef]
- Dulyaninova, N.G.; House, R.P.; Betapudi, V.; Bresnick, A.R. Myosin-IIA heavy-chain phosphorylation regulates the motility of MDA-MB-231 carcinoma cells. Mol. Biol. Cell 2007, 18, 3144–3155. [Google Scholar] [CrossRef] [PubMed]
- Dulyaninova, N.G.; Malashkevich, V.N.; Almo, S.C.; Bresnick, A.R. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry 2005, 44, 6867–6876. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Swift, J.; Dingal, P.C.; Shah, P.; Shin, J.W.; Discher, D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 2012, 199, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, M.; Ambartsumian, N.; Lukanidin, E. Metastasis-inducing S100A4 protein: Implication in non-malignant human pathologies. Curr. Mol. Med. 2008, 8, 492–496. [Google Scholar] [CrossRef]
- Li, Z.H.; Dulyaninova, N.G.; House, R.P.; Almo, S.C.; Bresnick, A.R. S100A4 regulates macrophage chemotaxis. Mol. Biol. Cell 2010, 21, 2598–2610. [Google Scholar] [CrossRef]
- Dulyaninova, N.G.; Ruiz, P.D.; Gamble, M.J.; Backer, J.M.; Bresnick, A.R. S100A4 regulates macrophage invasion by distinct myosin-dependent and myosin-independent mechanisms. Mol. Biol. Cell 2018, 29, 632–642. [Google Scholar] [CrossRef]
- Davies, B.R.; O’Donnell, M.; Durkan, G.C.; Rudland, P.S.; Barraclough, R.; Neal, D.E.; Mellon, J.K. Expression of S100A4 protein is associated with metastasis and reduced survival in human bladder cancer. J. Pathol. 2002, 196, 292–299. [Google Scholar] [CrossRef]
- Conti, M.A.; Even-Ram, S.; Liu, C.; Yamada, K.M.; Adelstein, R.S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J. Biol. Chem. 2004, 279, 41263–41266. [Google Scholar] [CrossRef]
- Smutny, M.; Cox, H.L.; Leerberg, J.M.; Kovacs, E.M.; Conti, M.A.; Ferguson, C.; Hamilton, N.A.; Parton, R.G.; Adelstein, R.S.; Yap, A.S. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 2010, 12, 696–702. [Google Scholar] [CrossRef]
- Spudich, J.A. The myosin swinging cross-bridge model. Nat. Rev. Mol. Cell Biol. 2001, 2, 387–392. [Google Scholar] [CrossRef]
- Hu, A.; Wang, F.; Sellers, J.R. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J. Biol. Chem. 2002, 277, 46512–46517. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Burridge, K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 1996, 133, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Dumbauld, D.W.; Shin, H.; Gallant, N.D.; Michael, K.E.; Radhakrishna, H.; Garcia, A.J. Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. J. Cell. Physiol. 2010, 223, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Spektor, A.; Varlamova, O.; Bresnick, A.R. Mts1 regulates the assembly of nonmuscle myosin-IIA. Biochemistry 2003, 42, 14258–14266. [Google Scholar] [CrossRef]
- Bao, J.; Jana, S.S.; Adelstein, R.S. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J. Biol. Chem. 2005, 280, 19594–19599. [Google Scholar] [CrossRef]
- Tuzovic, L.; Yu, L.; Zeng, W.; Li, X.; Lu, H.; Lu, H.M.; Gonzalez, K.D.; Chung, W.K. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis. 2013, 1, e26144. [Google Scholar] [CrossRef]
- Olney, J.J.; Sellers, J.R.; Cremo, C.R. Structure and function of the 10 S conformation of smooth muscle myosin. J. Biol. Chem. 1996, 271, 20375–20384. [Google Scholar] [CrossRef]
- Sata, M.; Stafford, W.F., 3rd; Mabuchi, K.; Ikebe, M. The motor domain and the regulatory domain of myosin solely dictate enzymatic activity and phosphorylation-dependent regulation, respectively. Proc. Natl. Acad. Sci. USA 1997, 94, 91–96. [Google Scholar] [CrossRef]
- Beach, J.R.; Shao, L.; Remmert, K.; Li, D.; Betzig, E.; Hammer, J.A., 3rd. Nonmuscle Myosin II Isoforms Coassemble in Living Cells. Curr. Biol. 2014, 24, 1160–1166. [Google Scholar] [CrossRef]
- Billington, N.; Beach, J.R.; Heissler, S.M.; Remmert, K.; Guzik-Lendrum, S.; Nagy, A.; Takagi, Y.; Shao, L.; Li, D.; Yang, Y.; et al. Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr. Biol. 2015, 25, 942–948. [Google Scholar] [CrossRef]
- Shutova, M.S.; Spessott, W.A.; Giraudo, C.G.; Svitkina, T. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr. Biol. 2014, 24, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Kovacs, M.; Hu, A.; Limouze, J.; Harvey, E.V.; Sellers, J.R. Kinetic mechanism of non-muscle myosin IIB: Functional adaptations for tension generation and maintenance. J. Biol. Chem. 2003, 278, 27439–27448. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Wang, F.; Hu, A.; Zhang, Y.; Sellers, J.R. Functional divergence of human cytoplasmic myosin II: Kinetic characterization of the non-muscle IIA isoform. J. Biol. Chem. 2003, 278, 38132–38140. [Google Scholar] [CrossRef] [PubMed]
- Jiu, Y.; Kumari, R.; Fenix, A.M.; Schaible, N.; Liu, X.; Varjosalo, M.; Krishnan, R.; Burnette, D.T.; Lappalainen, P. Myosin-18B Promotes the Assembly of Myosin II Stacks for Maturation of Contractile Actomyosin Bundles. Curr. Biol. 2019, 29, 81–92.e5. [Google Scholar] [CrossRef]
- Buschman, M.D.; Field, S.J. MYO18A: An unusual myosin. Adv. Biol. Regul. 2018, 67, 84–92. [Google Scholar] [CrossRef]
- Wang, A.; Ma, X.; Conti, M.A.; Adelstein, R.S. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem. Soc. Trans. 2011, 39, 1131–1135. [Google Scholar] [CrossRef]
- Sandquist, J.C.; Means, A.R. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell 2008, 19, 5156–5167. [Google Scholar] [CrossRef]
- Taneja, N.; Bersi, M.R.; Baillargeon, S.M.; Fenix, A.M.; Cooper, J.A.; Ohi, R.; Gama, V.; Merryman, W.D.; Burnette, D.T. Precise Tuning of Cortical Contractility Regulates Cell Shape during Cytokinesis. Cell Rep. 2020, 31, 107477. [Google Scholar] [CrossRef]
- Chandrasekar, I.; Goeckeler, Z.M.; Turney, S.G.; Wang, P.; Wysolmerski, R.B.; Adelstein, R.S.; Bridgman, P.C. Nonmuscle myosin II is a critical regulator of clathrin-mediated endocytosis. Traffic 2014, 15, 418–432. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asensio-Juárez, G.; Llorente-González, C.; Vicente-Manzanares, M. Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells. Cells 2020, 9, 1458. https://doi.org/10.3390/cells9061458
Asensio-Juárez G, Llorente-González C, Vicente-Manzanares M. Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells. Cells. 2020; 9(6):1458. https://doi.org/10.3390/cells9061458
Chicago/Turabian StyleAsensio-Juárez, Gloria, Clara Llorente-González, and Miguel Vicente-Manzanares. 2020. "Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells" Cells 9, no. 6: 1458. https://doi.org/10.3390/cells9061458
APA StyleAsensio-Juárez, G., Llorente-González, C., & Vicente-Manzanares, M. (2020). Linking the Landscape of MYH9-Related Diseases to the Molecular Mechanisms that Control Non-Muscle Myosin II-A Function in Cells. Cells, 9(6), 1458. https://doi.org/10.3390/cells9061458