The Role of Single-Cell Technology in the Study and Control of Infectious Diseases



Abstract
1. Introduction
2. Uncovering Infection Pathophysiology
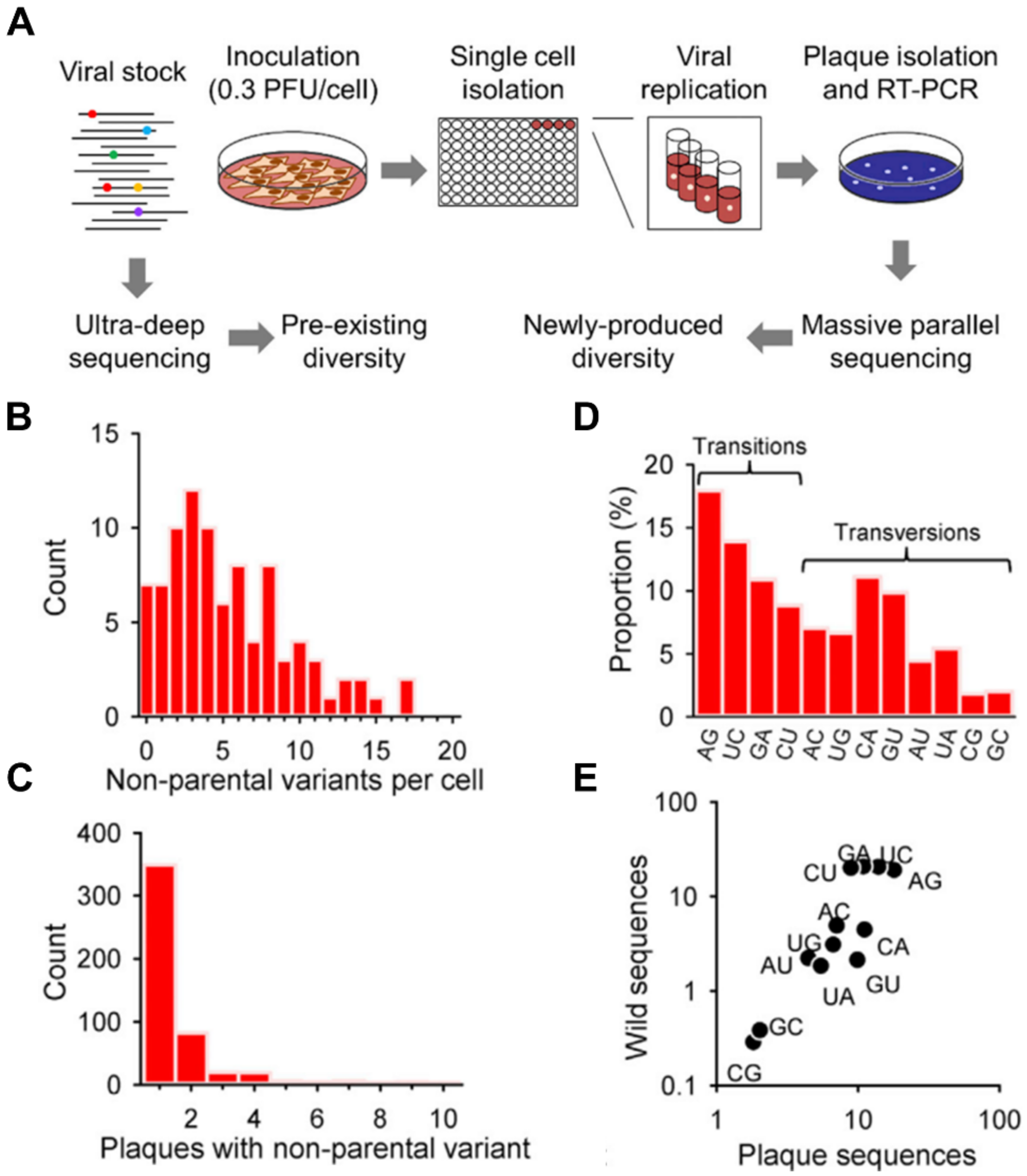
2.1. Pathogen Heterogeneity
2.2. Host Cell Heterogeneity
2.3. Host Immune Responses in Infection
3. Therapeutics Discovery
3.1. Single-Cell Technology in Therapeutics Discovery and Clinical Application
3.2. Vaccine Development
3.3. Antibody Discovery
3.4. Antibiotic Discovery and Antimicrobial Resistance
4. Diagnostics
4.1. Disease Monitoring and Clinical Diagnostics
4.2. Toward Point-of-Care Applications
4.3. Digital Assays
4.4. Label-Free Analysis
5. Considerations in Single-Cell Studies
5.1. Number of Cells and Sequencing Depth
5.2. Reproducibility and Reliability of Data
6. Future Outlook
6.1. General Limitations of Current Single-Cell Platforms
6.2. Techniques on the Horizon
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Worldometer. COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 6 May 2020).
- World Trade Organization. Trade Set to Plunge as COVID-19 Pandemic Upends Global Economy. Available online: https://www.wto.org/english/news_e/pres20_e/pr855_e.htm (accessed on 8 April 2020).
- Microbiology by numbers. Nat. Rev. Microbiol. 2011, 9, 628. [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Cox, F.E.G. History of the discovery of the malaria parasites and their vectors. Parasites Vectors 2010, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Cristinelli, S.; Ciuffi, A. The use of single-cell RNA-Seq to understand virus-host interactions. Curr. Opin. Virol. 2018, 29, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Avraham, R. Breaking the population barrier by single cell analysis: One host against one pathogen. Curr. Opin. Microbiol. 2017, 36, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Heldt, F.S.; Kupke, S.Y.; Dorl, S.; Reichl, U.; Frensing, T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection. Nat. Commun. 2015, 6, 8938. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.B.; Trapnell, C.; Bloom, J.D. Extreme heterogeneity of influenza virus infection in single cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Akpinar, F.; Timm, A.; Yin, J. High-Throughput Single-Cell Kinetics of Virus Infections in the Presence of Defective Interfering Particles. J. Virol. 2016, 90, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.B.; Elshina, E.; Kowalsky, J.R.; Te Velthuis, A.J.W.; Bloom, J.D. Single-Cell Virus Sequencing of Influenza Infections That Trigger Innate Immunity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Combe, M.; Garijo, R.; Geller, R.; Cuevas, J.M.; Sanjuan, R. Single-Cell Analysis of RNA Virus Infection Identifies Multiple Genetically Diverse Viral Genomes within Single Infectious Units. Cell Host. Microbe 2015, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.E.; Li, L.; Westermann, A.J.; Appenzeller, S.; Stapels, D.A.; Schulte, L.N.; Helaine, S.; Vogel, J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2016, 2, 16206. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Haseley, N.; Brown, D.; Penaranda, C.; Jijon, H.B.; Trombetta, J.J.; Satija, R.; Shalek, A.K.; Xavier, R.J.; Regev, A.; et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell 2015, 162, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Claudi, B.; Sprote, P.; Chirkova, A.; Personnic, N.; Zankl, J.; Schurmann, N.; Schmidt, A.; Bumann, D. Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 2014, 158, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Wang, H.; Han, L.; Wang, M.; Fang, H.; Hao, Y.; Li, J.; Zhang, H.; Zheng, C.; Shen, C. Single-Cell Analysis of the Impact of Host Cell Heterogeneity on Infection with Foot-and-Mouth Disease Virus. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, J.T.; Upadhyay, A.A.; Wolabaugh, A.; Patel, N.B.; Bosinger, S.E.; Suthar, M.S. West Nile Virus-Inclusive Single-Cell RNA Sequencing Reveals Heterogeneity in the Type I Interferon Response within Single Cells. J. Virol. 2019, 93, e01778-18. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, S.; Caglar, M.U.; Mao, Z.; Liu, W.; Woodman, A.; Arnold, J.J.; Wilke, C.O.; Huang, T.J.; Cameron, C.E. Single-Cell Virology: On-Chip Investigation of Viral Infection Dynamics. Cell Rep. 2017, 21, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Franke, V.; Menegatti, J.; Kocks, C.; Boltengagen, A.; Praktiknjo, S.; Walch-Ruckheim, B.; Bosse, J.; Rajewsky, N.; Grasser, F.; et al. Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 2019, 10, 4878. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, Y.; Cohen, M.; Peshes-Yaloz, N.; Valadarsky, L.; Cohn, O.; David, E.; Frishberg, A.; Mayo, L.; Bacharach, E.; Amit, I.; et al. Dissection of Influenza Infection In Vivo by Single-Cell RNA Sequencing. Cell Syst. 2018, 6, 679–691.e674. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Pu, S.Y.; Bekerman, E.; Einav, S.; Quake, S.R. Single-cell transcriptional dynamics of flavivirus infection. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.A.-O.; Scheffler, K.A.-O.; Halpern, A.L.; Bekritsky, M.A.-O.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.A.-O.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Y.; Zhang, L.; Li, Z.; Fang, Q.; Gao, R.; Zhang, Z.A.-O. Systematic comparative analysis of single-nucleotide variant detection methods from single-cell RNA sequencing data. Genome Biol. 2019, 20, 242. [Google Scholar] [CrossRef] [PubMed]
- Prashant, N.M.; Liu, H.; Bousounis, P.; Spurr, L.; Alomran, N.; Ibeawuchi, H.; Sein, J.; Reece-Stremtan, D.; Horvath, A. Estimating the Allele-Specific Expression of SNVs From 10× Genomics Single-Cell RNA-Sequencing Data. Genes 2020, 11, 240. [Google Scholar]
- Van den Berge, K.; Roux de Bézieux, H.; Street, K.; Saelens, W.; Cannoodt, R.; Saeys, Y.; Dudoit, S.; Clement, L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat. Commun. 2020, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Spurr, L.F.; Alomran, N.; Bousounis, P.; Reece-Stremtan, D.; Prashant, N.M.; Liu, H.; Słowiński, P.; Li, M.; Zhang, Q.; Sein, J.; et al. ReQTL: Identifying correlations between expressed SNVs and gene expression using RNA-sequencing data. Bioinformatics 2019, 36, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Dai, Y.; Wang, Y.; Yang, Q.; Guo, J.; Wei, C.; Chen, W.; Huang, H.; Zhu, J.; Zhang, C.; et al. Single-cell transcriptomics of blood reveals a natural killer cell subset depletion in tuberculosis. EBioMedicine 2020, 53, 102686. [Google Scholar] [CrossRef] [PubMed]
- Kazer, S.W.; Aicher, T.P.; Muema, D.M.; Carroll, S.L.; Ordovas-Montanes, J.; Miao, V.N.; Tu, A.A.; Ziegler, C.G.K.; Nyquist, S.K.; Wong, E.B.; et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat. Med. 2020, 26, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Robinson, M.L.; Croote, D.; Sahoo, M.K.; Sanz, A.M.; Ortiz-Lasso, E.; Albornoz, L.L.; Rosso, F.; Montoya, J.G.; Goo, L.; et al. Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Natl. Acad. Sci. USA 2018, 115, E12363–E12369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Amodio, M.; Vander Wyk, B.; Gerritsen, B.; Kumar, M.M.; van Dijk, D.; Moon, K.; Wang, X.; Malawista, A.; Richards, M.M.; et al. Single cell immune profiling of dengue virus patients reveals intact immune responses to Zika virus with enrichment of innate immune signatures. PLoS Negl. Trop. Dis. 2020, 14, e0008112. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, R.E.; Rahman, A.; Pak, T.R.; Maringer, K.; Mena, I.; Bernal-Rubio, D.; Potla, U.; Maestre, A.M.; Fredericks, A.C.; Amir, E.D.; et al. High-dimensional CyTOF analysis of dengue virus-infected human DCs reveals distinct viral signatures. JCI Insight 2017, 2, e92424. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, Q.; Eisele, M.R.; Sulistijo, E.S.; Brower, K.; Han, L.; Amir, E.-A.D.; Pe’er, D.; Miller-Jensen, K.; Fan, R. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc. Natl. Acad. Sci. USA 2015, 112, E607. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lu, Y.; Zhang, K.; Xiao, Y.; Lu, J.; Fan, R. Multiplexed, Sequential Secretion Analysis of the Same Single Cells Reveals Distinct Effector Response Dynamics Dependent on the Initial Basal State. Adv. Sci. 2019, 6, 1801361. [Google Scholar] [CrossRef] [PubMed]
- Buggert, M.A.-O.; Nguyen, S.A.-O.; Salgado-Montes de Oca, G.A.-O.; Bengsch, B.A.-O.X.; Darko, S.; Ransier, A.; Roberts, E.R.; Del Alcazar, D.A.-O.; Brody, I.B.; Vella, L.A.-O.; et al. Identification and characterization of HIV-specific resident memory CD8+ T cells in human lymphoid tissue. Sci. Immunol. 2018, 3, eaar4526. [Google Scholar] [CrossRef]
- Yao, C.; Sun, H.-W.; Lacey, N.E.; Ji, Y.; Moseman, E.A.; Shih, H.-Y.; Heuston, E.F.; Kirby, M.; Anderson, S.; Cheng, J.; et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat. Immunol. 2019, 20, 890–901. [Google Scholar] [CrossRef]
- Michlmayr, D.; Pak, T.R.; Rahman, A.H.; Amir, E.D.; Kim, E.Y.; Kim-Schulze, S.; Suprun, M.; Stewart, M.G.; Thomas, G.P.; Balmaseda, A.; et al. Comprehensive innate immune profiling of chikungunya virus infection in pediatric cases. Mol. Syst. Biol. 2018, 14, e7862. [Google Scholar] [CrossRef] [PubMed]
- Kotliarov, Y.; Sparks, R.; Martins, A.J.; Mule, M.P.; Lu, Y.; Goswami, M.; Kardava, L.; Banchereau, R.; Pascual, V.; Biancotto, A.; et al. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med. 2020, 26, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, Z.E.; Tsalik, E.L.; Woods, C.W.; McClain, M.T. Host-Based Peripheral Blood Gene Expression Analysis for Diagnosis of Infectious Diseases. J. Clin. Microbiol. 2017, 55, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Del Tordello, E.; Rappuoli, R.; Delany, I. Reverse Vaccinology: Exploiting Genomes for Vaccine Design. In Human Vaccines; Chapter 3; Modjarrad, K., Koff, W.C., Eds.; Academic Press: London, UK, 2017; pp. 65–86. [Google Scholar]
- Rappuoli, R.; Bottomley, M.J.; D’Oro, U.; Finco, O.; De Gregorio, E. Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med. 2016, 213, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A.; Gilbert, P. 3—Correlates of Protection. In Plotkin’s Vaccines (Seventh Edition); Plotkin, S.A., Orenstein, W.A., Offit, P.A., Edwards, K.M., Eds.; Elsevier: Philadelphia, PA, USA, 2018; pp. 35–40.e34. [Google Scholar]
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. Cvi. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, A.; Ewer, K.J. Methods for Measuring T-Cell Memory to Vaccination: From Mouse to Man. Vaccines 2018, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Updates on immunologic correlates of vaccine-induced protection. Vaccine 2020, 38, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B. Systems vaccinology: Probing humanity’s diverse immune systems with vaccines. Proc. Natl. Acad. Sci. USA 2014, 111, 12300–12306. [Google Scholar] [CrossRef]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Chang, H.Y.; Satpathy, A.T. Tracking the immune response with single-cell genomics. Vaccine 2019, 38, 4487–4490. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Victor, K.; Li, T.; Hatch, K.; Rutvisuttinunt, W.; Medin, C.; Gabriel, B.; Jarman, R.G.; Friberg, H.; Currier, J.R. Dissecting the heterogeneity of DENV vaccine-elicited cellular immunity using single-cell RNA sequencing and metabolic profiling. Nat. Commun. 2019, 10, 3666. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.A.; Gierahn, T.M.; Monian, B.; Morgan, D.M.; Mehta, N.K.; Ruiter, B.; Shreffler, W.G.; Shalek, A.K.; Love, J.C. TCR sequencing paired with massively parallel 3’ RNA-seq reveals clonotypic T cell signatures. Nat. Immunol. 2019, 20, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, E.A. Antibodies: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2013. [Google Scholar]
- Yu, X.; McGraw, P.A.; House, F.S.; Crowe, J.E., Jr. An optimized electrofusion-based protocol for generating virus-specific human monoclonal antibodies. J. Immunol. Methods 2008, 336, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Traggiai, E.; Becker, S.; Subbarao, K.; Kolesnikova, L.; Uematsu, Y.; Gismondo, M.R.; Murphy, B.R.; Rappuoli, R.; Lanzavecchia, A. An efficient method to make human monoclonal antibodies from memory B cells: Potent neutralization of SARS coronavirus. Nat. Med. 2004, 10, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Lanzavecchia, A. Efficient Methods To Isolate Human Monoclonal Antibodies from Memory B Cells and Plasma Cells. Microbiol. Spectr 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Bonsignori, M.; Hwang, K.K.; Chen, X.; Tsao, C.Y.; Morris, L.; Gray, E.; Marshall, D.J.; Crump, J.A.; Kapiga, S.H.; Sam, N.E.; et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J. Virol. 2011, 85, 9998–10009. [Google Scholar] [CrossRef] [PubMed]
- Bonsignori, M.; Kreider, E.F.; Fera, D.; Meyerhoff, R.R.; Bradley, T.; Wiehe, K.; Alam, S.M.; Aussedat, B.; Walkowicz, W.E.; Hwang, K.K.; et al. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci. Transl. Med. 2017, 9, eaai7514. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Morbach, H.; Eichhorn, E.M.; Liese, J.G.; Girschick, H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010, 162, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.; Mouquet, H.; Feldhahn, N.; Walker, B.; Pereyra, F.; Cutrell, E.; Seaman, M.; Mascola, J.; Wyatt, R.; Wardemann, H.; et al. A method for identification of HIV gp140 binding memory B cells in human blood. J. Immunol. Methods 2009, 343, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Pinder, C.L.; Kratochvil, S.; Cizmeci, D.; Muir, L.; Guo, Y.; Shattock, R.J.; McKay, P.F. Isolation and Characterization of Antigen-Specific Plasmablasts Using a Novel Flow Cytometry-Based Ig Capture Assay. J. Immunol. 2017, 199, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Gerard, A.; Woolfe, A.; Mottet, G.; Reichen, M.; Castrillon, C.; Menrath, V.; Ellouze, S.; Poitou, A.; Doineau, R.; Briseno-Roa, L.; et al. High-throughput single-cell activity-based screening and sequencing of antibodies using droplet microfluidics. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Ozawa, T.; Tajiri, K.; Obata, T.; Kishi, H.; Muraguchi, A. Rapid isolation of antigen-specific antibody-secreting cells using a chip-based immunospot array. Nat. Protoc. 2011, 6, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Love, J.C.; Ronan, J.L.; Grotenbreg, G.M.; van der Veen, A.G.; Ploegh, H.L. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat. Biotechnol. 2006, 24, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Ogunniyi, A.O.; Story, C.M.; Papa, E.; Guillen, E.; Love, J.C. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat. Protoc. 2009, 4, 767–782. [Google Scholar] [CrossRef] [PubMed]
- Story, C.M.; Papa, E.; Hu, C.C.; Ronan, J.L.; Herlihy, K.; Ploegh, H.L.; Love, J.C. Profiling antibody responses by multiparametric analysis of primary B cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17902–17907. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, V.; Manning, B.; O’Donnell, B.; O’Reilly, B.; O’Sullivan, D.; O’Kennedy, R.; Leonard, P. Exploiting highly ordered subnanoliter volume microcapillaries as microtools for the analysis of antibody producing cells. Anal. Chem. 2015, 87, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Setliff, I.; Shiakolas, A.R.; Pilewski, K.A.; Murji, A.A.; Mapengo, R.E.; Janowska, K.; Richardson, S.; Oosthuysen, C.; Raju, N.; Ronsard, L.; et al. High-Throughput Mapping of B Cell Receptor Sequences to Antigen Specificity. Cell 2019, 179, 1636–1646.e1615. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; DeKosky, B.J.; Timm, M.R.; Lee, J.; Normandin, E.; Misasi, J.; Kong, R.; McDaniel, J.R.; Delidakis, G.; Leigh, K.E. Functional interrogation and mining of natively paired human V H: V L antibody repertoires. Nat. Biotechnol. 2018, 36, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, C.J.; Koglin, M.; Marshall, F.H. Therapeutic antibodies directed at G protein-coupled receptors. mAbs 2010, 2, 594–606. [Google Scholar] [CrossRef] [PubMed]
- De Alwis, R.; Smith, S.A.; Olivarez, N.P.; Messer, W.B.; Huynh, J.P.; Wahala, W.M.; White, L.J.; Diamond, M.S.; Baric, R.S.; Crowe, J.E., Jr.; et al. Identification of human neutralizing antibodies that bind to complex epitopes on dengue virions. Proc. Natl. Acad. Sci. USA 2012, 109, 7439–7444. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.L.; Crooks, E.T.; Porter, L.; Zhu, P.; Cayanan, C.S.; Grise, H.; Corcoran, P.; Zwick, M.B.; Franti, M.; Morris, L.; et al. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol. 2006, 80, 2515–2528. [Google Scholar] [CrossRef]
- Sanders, R.W.; Vesanen, M.; Schuelke, N.; Master, A.; Schiffner, L.; Kalyanaraman, R.; Paluch, M.; Berkhout, B.; Maddon, P.J.; Olson, W.C.; et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 2002, 76, 8875–8889. [Google Scholar] [CrossRef] [PubMed]
- Binley, J.M.; Ditzel, H.J.; Barbas, C.F., 3rd; Sullivan, N.; Sodroski, J.; Parren, P.W.; Burton, D.R. Human antibody responses to HIV type 1 glycoprotein 41 cloned in phage display libraries suggest three major epitopes are recognized and give evidence for conserved antibody motifs in antigen binding. Aids Res. Hum. Retrovir. 1996, 12, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, G.D.; Yates, N.L.; Liu, P.; Qin, L.; Fouda, G.G.; Chavez, L.L.; Decamp, A.C.; Parks, R.J.; Ashley, V.C.; Lucas, J.T.; et al. Initial B-cell responses to transmitted human immunodeficiency virus type 1: Virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J. Virol. 2008, 82, 12449–12463. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Alter, G. Systems serology: Profiling vaccine induced humoral immunity against HIV. Retrovirology 2017, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Debs, B.E.; Utharala, R.; Balyasnikova, I.V.; Griffiths, A.D.; Merten, C.A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. USA 2012, 109, 11570–11575. [Google Scholar] [CrossRef]
- Roberts, J.P. Single-Cell Analysis Deepens Antibody Discovery. Available online: https://www.genengnews.com/insights/single-cell-analysis-deepens-antibody-discovery/ (accessed on 22 April 2020).
- Liu, X.; Painter, R.E.; Enesa, K.; Holmes, D.; Whyte, G.; Garlisi, C.G.; Monsma, F.J.; Rehak, M.; Craig, F.F.; Smith, C.A. High-throughput screening of antibiotic-resistant bacteria in picodroplets. Lab. Chip 2016, 16, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Janossy, G.; Shapiro, H. Simplified cytometry for routine monitoring of infectious diseases. Cytom. Part. B Clin. Cytom. 2008, 74B, S6–S10. [Google Scholar] [CrossRef] [PubMed]
- Grimberg, B.T. Methodology and application of flow cytometry for investigation of human malaria parasites. J. Immunol. Methods 2011, 367, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Barnett, D.; Walker, B.; Landay, A.; Denny, T.N. CD4 immunophenotyping in HIV infection. Nat. Rev. Microbiol. 2008, 6, S7–S15. [Google Scholar] [CrossRef] [PubMed]
- Riou, C.; Berkowitz, N.; Goliath, R.; Burgers, W.A.; Wilkinson, R.J. Analysis of the Phenotype of Mycobacterium tuberculosis-Specific CD4+ T Cells to Discriminate Latent from Active Tuberculosis in HIV-Uninfected and HIV-Infected Individuals. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Frickmann, H.; Zautner, A.E.; Moter, A.; Kikhney, J.; Hagen, R.M.; Stender, H.; Poppert, S. Fluorescence in situ hybridization (FISH) in the microbiological diagnostic routine laboratory: A review. Crit. Rev. Microbiol. 2017, 43, 263–293. [Google Scholar] [CrossRef] [PubMed]
- Makristathis, A.; Riss, S.; Hirschl, A.M. A novel fluorescence in situ hybridization test for rapid pathogen identification in positive blood cultures. Clin. Microbiol. Infect. 2014, 20, O760–O763. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Mark, O.; Weltman, H.; Barcelo, N.; Lo, W.; Wronska, D.; Kakkilaya, S.; Rao, A.; Bhat, S.T.; Sinha, R.; et al. Fluorescence In Situ Hybridization (FISH) Assays for Diagnosing Malaria in Endemic Areas. PLoS ONE 2015, 10, e0136726. [Google Scholar] [CrossRef] [PubMed]
- Prudent, E.; Raoult, D. Fluorescence in situ hybridization, a complementary molecular tool for the clinical diagnosis of infectious diseases by intracellular and fastidious bacteria. Fems Microbiol. Rev. 2018, 43, 88–107. [Google Scholar] [CrossRef] [PubMed]
- Arrigucci, R.; Bushkin, Y.; Radford, F.; Lakehal, K.; Vir, P.; Pine, R.; Martin, D.; Sugarman, J.; Zhao, Y.; Yap, G.S.; et al. FISH-Flow, a protocol for the concurrent detection of mRNA and protein in single cells using fluorescence in situ hybridization and flow cytometry. Nat. Protoc. 2017, 12, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Grau-Expósito, J.; Serra-Peinado, C.; Miguel, L.; Navarro, J.; Curran, A.; Burgos, J.; Ocaña, I.; Ribera, E.; Torrella, A.; Planas, B.; et al. A Novel Single-Cell FISH-Flow Assay Identifies Effector Memory CD4+ T cells as a Major Niche for HIV-1 Transcription in HIV-Infected Patients. mBio 2017, 8, e00876-17. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.X.; Urosevic, N.; Inglis, T.J.J. Accelerated bacterial detection in blood culture by enhanced acoustic flow cytometry (AFC) following peptide nucleic acid fluorescence in situ hybridization (PNA-FISH). PLoS ONE 2019, 14, e0201332. [Google Scholar] [CrossRef] [PubMed]
- Vembadi, A.; Menachery, A.; Qasaimeh, M.A. Cell Cytometry: Review and Perspective on Biotechnological Advances. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Dekker, S.; Isgor, P.K.; Feijten, T.; Segerink, L.I.; Odijk, M. From chip-in-a-lab to lab-on-a-chip: A portable Coulter counter using a modular platform. Microsyst. Nanoeng. 2018, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Subramanian, G.; Duan, J.; Gao, S.; Bai, L.; Chandramohanadas, R.; Ai, Y. A portable image-based cytometer for rapid malaria detection and quantification. PLoS ONE 2017, 12, e0179161. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Yang, D.; Huang, Z.; Sha, H.; Chang, H. Cellular immunity monitoring in long-duration spaceflights based on an automatic miniature flow cytometer. Sens. Actuators B Chem. 2018, 267, 419–429. [Google Scholar] [CrossRef]
- Kuupiel, D.; Bawontuo, V.; Mashamba-Thompson, T.P. Improving the Accessibility and Efficiency of Point-of-Care Diagnostics Services in Low- and Middle-Income Countries: Lean and Agile Supply Chain Management. Diagnostics 2017, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.M.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef]
- Jagannadh, V.K.; Murthy, R.S.; Srinivasan, R.; Gorthi, S.S. Field-Portable Microfluidics-Based Imaging Flow Cytometer. J. Lightwave Technol. 2015, 33, 3469–3474. [Google Scholar] [CrossRef]
- Choi, H.; Jeon, C.S.; Hwang, I.; Ko, J.; Lee, S.; Choo, J.; Boo, J.-H.; Kim, H.C.; Chung, T.D. A flow cytometry-based submicron-sized bacterial detection system using a movable virtual wall. Lab. A Chip. 2014, 14, 2327–2333. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Xue, C.; Wang, X.; He, S.; Wu, L.; Yan, X. Rapid quantification of pathogenic Salmonella Typhimurium and total bacteria in eggs by nano-flow cytometry. Talanta 2020, 217, 121020. [Google Scholar] [CrossRef] [PubMed]
- Pai, N.P.; Vadnais, C.; Denkinger, C.; Engel, N.; Pai, M. Point-of-care testing for infectious diseases: Diversity, complexity, and barriers in low- and middle-income countries. PLoS Med. 2012, 9, e1001306. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.W.; Chan, S.N.; Wu, A.R. Microfluidic single-cell analysis—Toward integration and total on-chip analysis. Biomicrofluidics 2020, 14, 021502. [Google Scholar] [CrossRef]
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by shrinking: Empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet. 2017, 18, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. Slas Technol. Transl. Life Sci. Innov. 2017, 22, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Maurer, F.P.; Christner, M.; Hentschke, M.; Rohde, H. Advances in Rapid Identification and Susceptibility Testing of Bacteria in the Clinical Microbiology Laboratory: Implications for Patient Care and Antimicrobial Stewardship Programs. Infect. Dis. Rep. 2017, 9, 6839. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, A.; Bachmann, T.T.; Lüdke, G.; Lisby, J.G.; Kahlmeter, G.; Mohess, A.; Becker, K.; Hays, J.P.; Woodford, N.; Mitsakakis, K.; et al. Developmental roadmap for antimicrobial susceptibility testing systems. Nat. Rev. Microbiol. 2019, 17, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, N.G.; Schlappi, T.S.; Curtis, M.S.; Butkovich, S.S.; Miller, S.; Humphries, R.M.; Ismagilov, R.F. Rapid pathogen-specific phenotypic antibiotic susceptibility testing using digital LAMP quantification in clinical samples. Sci. Transl. Med. 2017, 9, eaal3693. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.-T.; Kaminski, T.S.; Postek, W.; Guzowski, J.; Makuch, K.; Ruszczak, A.; von Stetten, F.; Zengerle, R.; Garstecki, P. Gravity-driven microfluidic assay for digital enumeration of bacteria and for antibiotic susceptibility testing. Lab. A Chip. 2020, 20, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Boitard, L.; Broyer, P.; Chareire, A.C.; Bourne-Branchu, P.; Mahé, P.; Tournoud, M.; Franceschi, C.; Zambardi, G.; Baudry, J.; et al. Digital antimicrobial susceptibility testing using the MilliDrop technology. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, R.R.; Li, J.Z. The Alphabet Soup of HIV Reservoir Markers. Curr. Hiv Aids Rep. 2017, 14, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Yucha, R.W.; Hobbs, K.S.; Hanhauser, E.; Hogan, L.E.; Nieves, W.; Ozen, M.O.; Inci, F.; York, V.; Gibson, E.A.; Thanh, C.; et al. High-throughput Characterization of HIV-1 Reservoir Reactivation Using a Single-Cell-in-Droplet PCR Assay. EBioMedicine 2017, 20, 217–229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Charlebois, R.; Massanella, M.; Brassard, N.; Alsahafi, N.; Delgado, G.-G.; et al. Single-Cell Characterization of Viral Translation-Competent Reservoirs in HIV-Infected Individuals. Cell Host Microbe 2016, 20, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Honrado, C.; Ciuffreda, L.; Spencer, D.; Ranford-Cartwright, L.; Morgan, H. Dielectric characterization of Plasmodium falciparum-infected red blood cells using microfluidic impedance cytometry. J. R. Soc. Interface 2018, 15, 20180416. [Google Scholar] [CrossRef] [PubMed]
- Carey, T.R.; Cotner, K.L.; Li, B.; Sohn, L.L. Developments in label-free microfluidic methods for single-cell analysis and sorting. Wires Nanomed. Nanobiotechnol. 2019, 11, e1529. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.S.; Honrado, C.; Spencer, D.; Horton, B.; Bridle, H.L.; Morgan, H. Analysis of Parasitic Protozoa at the Single-cell Level using Microfluidic Impedance Cytometry. Sci. Rep. 2017, 7, 2601. [Google Scholar] [CrossRef] [PubMed]
- Sinjab, F.; Elsheikha, H.M.; Dooley, M.; Notingher, I. Induction and measurement of the early stage of a host-parasite interaction using a combined optical trapping and Raman microspectroscopy system. J. Biophoton. 2020, 13, e201960065. [Google Scholar] [CrossRef] [PubMed]
- Hebert, C.G.; DiNardo, N.; Evans, Z.L.; Hart, S.J.; Hachmann, A.-B. Rapid quantification of vesicular stomatitis virus in Vero cells using Laser Force Cytology. Vaccine 2018, 36, 6061–6069. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Chin, L.K.; Ser, W.; Ayi, T.C.; Yap, P.H.; Bourouina, T.; Leprince-Wang, Y. An optofluidic imaging system to measure the biophysical signature of single waterborne bacteria. Lab. A Chip. 2014, 14, 4237–4243. [Google Scholar] [CrossRef] [PubMed]
- Warkiani, M.E.; Tay, A.K.P.; Khoo, B.L.; Xiaofeng, X.; Han, J.; Lim, C.T. Malaria detection using inertial microfluidics. Lab. A Chip. 2015, 15, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Z.; Shin, D.M.; Chen, Z.G.; Cho, Y.; Kim, Y.-J.; Han, A. A continuous-flow acoustofluidic cytometer for single-cell mechanotyping. Lab. A Chip. 2019, 19, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Sajeesh, P.; Raj, A.; Doble, M.; Sen, A.K. Characterization and sorting of cells based on stiffness contrast in a microfluidic channel. RSC Adv. 2016, 6, 74704–74714. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef] [PubMed]
- See, P.; Lum, J.; Chen, J.; Ginhoux, F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e634. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Shuga, J.; Wang, X.; Leyrat, A.A.; Lui, J.H.; Li, N.; Szpankowski, L.; Fowler, B.; Chen, P.; et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 2014, 32, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J.; et al. Immune cell profiling of COVID-19 patients in the recovery stageby single-cell sequencing. Cell Discov. 2020, 6, 31. [Google Scholar] [CrossRef]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kołodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Pervolarakis, N.; Nee, K.; Kessenbrock, K. Experimental Considerations for Single-Cell RNA Sequencing Approaches. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Büttner, M.; Miao, Z.; Wolf, F.A.; Teichmann, S.A.; Theis, F.J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 2019, 16, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Severson, D.T.; Owen, R.P.; White, M.J.; Lu, X.; Schuster-Böckler, B. BEARscc determines robustness of single-cell clusters using simulated technical replicates. Nat. Commun. 2018, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Dal Molin, A.; Di Camillo, B. How to design a single-cell RNA-sequencing experiment: Pitfalls, challenges and perspectives. Brief. Bioinform. 2018, 20, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2006, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef] [PubMed]
- Nichterwitz, S.; Chen, G.; Aguila Benitez, J.; Yilmaz, M.; Storvall, H.; Cao, M.; Sandberg, R.; Deng, Q.; Hedlund, E. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat. Commun. 2016, 7, 12139. [Google Scholar] [CrossRef] [PubMed]
- Junker, J.P.; Noel, E.S.; Guryev, V.; Peterson, K.A.; Shah, G.; Huisken, J.; McMahon, A.P.; Berezikov, E.; Bakkers, J.; van Oudenaarden, A. Genome-wide RNA Tomography in the zebrafish embryo. Cell 2014, 159, 662–675. [Google Scholar] [CrossRef] [PubMed]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Kelbauskas, L.; Glenn, H.; Anderson, C.; Messner, J.; Lee, K.B.; Song, G.; Houkal, J.; Su, F.; Zhang, L.; Tian, Y.; et al. A platform for high-throughput bioenergy production phenotype characterization in single cells. Sci. Rep. 2017, 7, 45399. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Claas, A.M.; Sarkar, A.; Lauffenburger, D.A.; Han, J. High-throughput protease activity cytometry reveals dose-dependent heterogeneity in PMA-mediated ADAM17 activation. Integr. Biol. 2015, 7, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Robert, L.; Ollion, J.; Elez, M. Real-time visualization of mutations and their fitness effects in single bacteria. Nat. Protoc. 2019, 14, 3126–3143. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Throughput | Downstream Assay Compatibility | Genetic Information | Epigenetic Information | Proteomic Information | Cell Function Information | Spatial Information | Temporal Information | Cost | |
|---|---|---|---|---|---|---|---|---|---|
| Bulk | |||||||||
| Cell/organ function assays | |||||||||
| Next-Generation Sequencing (NGS) | |||||||||
| Scalable to single cell | |||||||||
| Polymerase Chain Reaction (PCR) | |||||||||
| Microfluidic tools | |||||||||
| Microscopy (including FISH) | |||||||||
| Single cell | |||||||||
| Flow cytometry (FACS) | |||||||||
| Mass Cytometry (CyTOF) | |||||||||
| Single Cell Sequencing | |||||||||
| CITE-seq/REAP-seq | |||||||||
| Imaging mass cytometry | |||||||||
| Spatial Transcriptomics | |||||||||
| Capability | Not Able | Low | Medium | High. | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.N.; Tay, M.Z.; Lu, R.; Liu, Y.; Chen, C.-H.; Cheow, L.F. The Role of Single-Cell Technology in the Study and Control of Infectious Diseases. Cells 2020, 9, 1440. https://doi.org/10.3390/cells9061440
Lin WN, Tay MZ, Lu R, Liu Y, Chen C-H, Cheow LF. The Role of Single-Cell Technology in the Study and Control of Infectious Diseases. Cells. 2020; 9(6):1440. https://doi.org/10.3390/cells9061440
Chicago/Turabian StyleLin, Weikang Nicholas, Matthew Zirui Tay, Ri Lu, Yi Liu, Chia-Hung Chen, and Lih Feng Cheow. 2020. "The Role of Single-Cell Technology in the Study and Control of Infectious Diseases" Cells 9, no. 6: 1440. https://doi.org/10.3390/cells9061440
APA StyleLin, W. N., Tay, M. Z., Lu, R., Liu, Y., Chen, C.-H., & Cheow, L. F. (2020). The Role of Single-Cell Technology in the Study and Control of Infectious Diseases. Cells, 9(6), 1440. https://doi.org/10.3390/cells9061440