Bioactive Lipid Signaling in Cardiovascular Disease, Development, and Regeneration


Abstract
1. Introduction
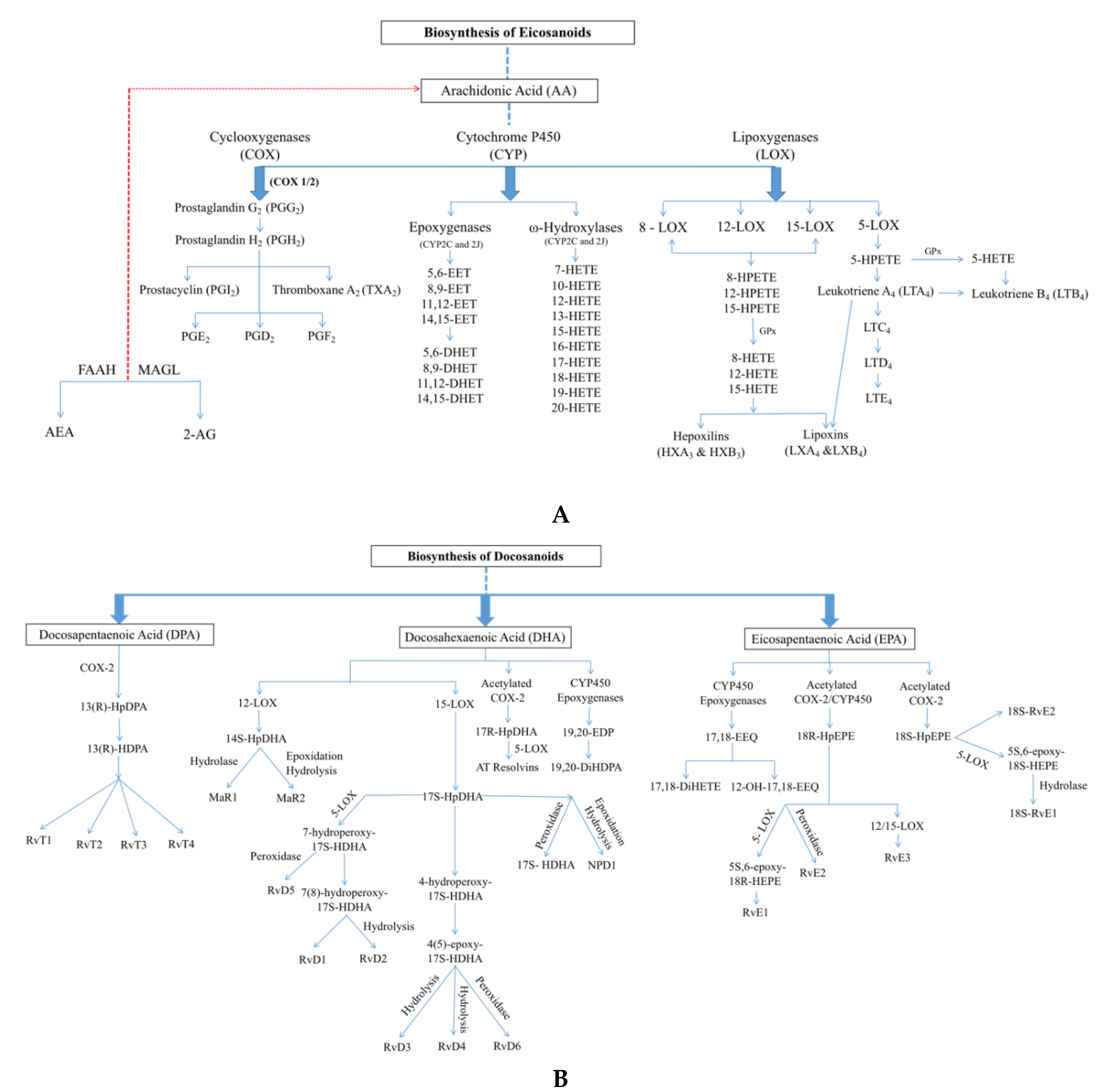
2. Bioactive Lipid Classes
2.1. Oxylipins
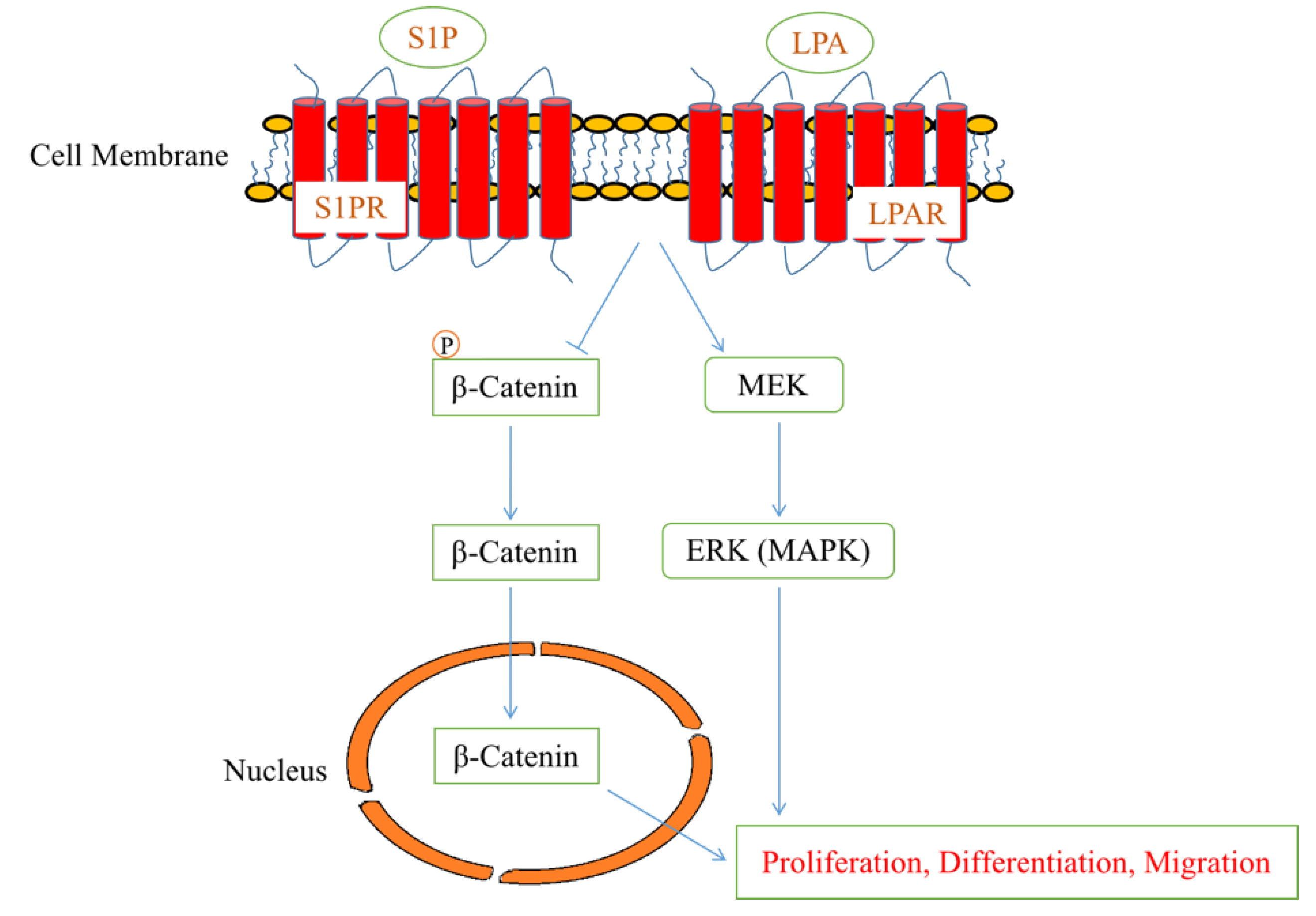
2.2. Lysophospholipids and Sphingolipids
2.3. Endocannabinoids
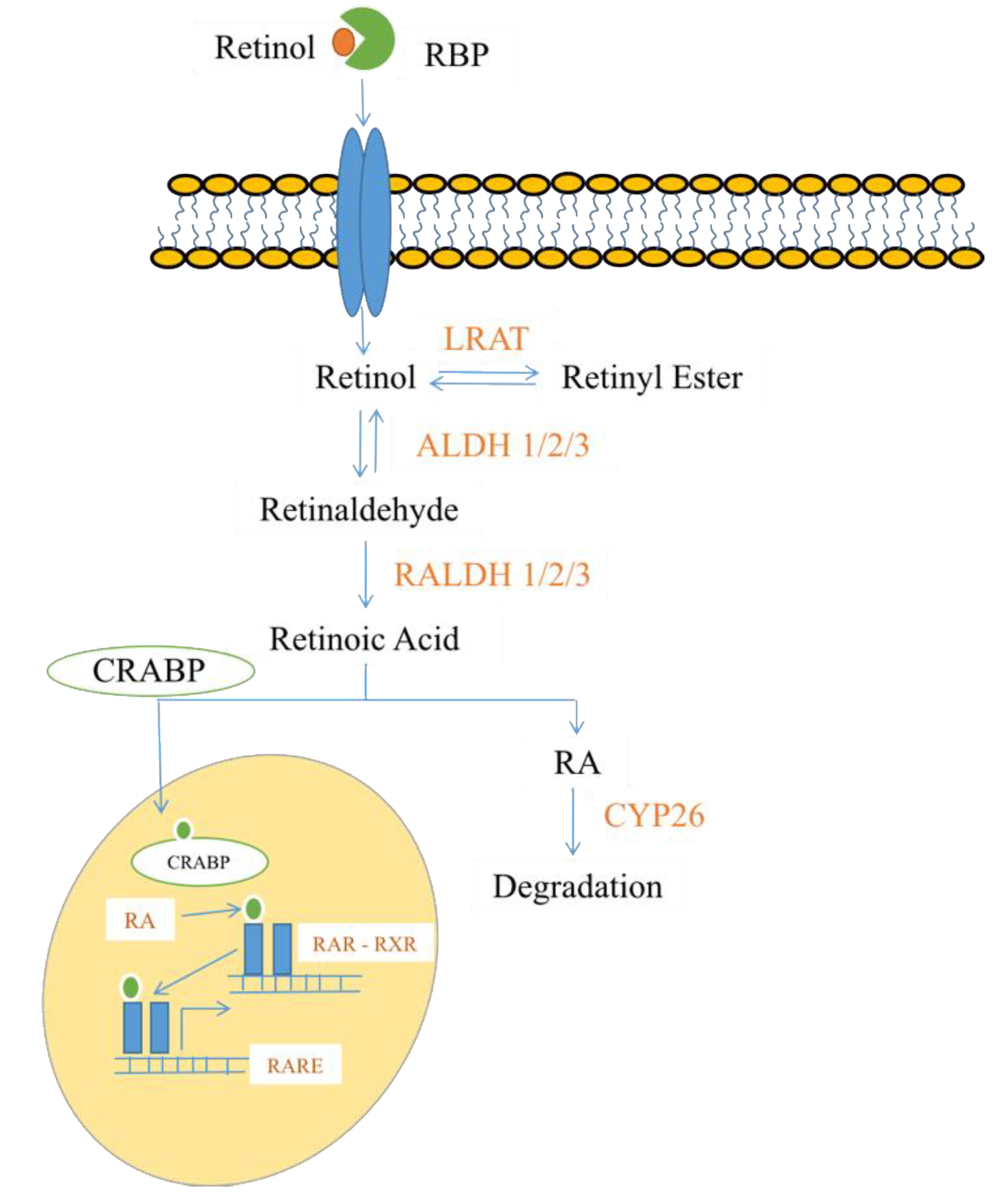
2.4. Steroids and Lipid-Soluble Vitamins
3. Recent Advances in Bioactive Lipids in Cardiac Disease
3.1. Cardioprotective Oxylipins
3.2. Deleterious Effects of Oxylipins
3.3. Other Bioactive Lipids Involved in Cardiac Function
4. Recent Advances in Bioactive Lipids in Cardiac Development
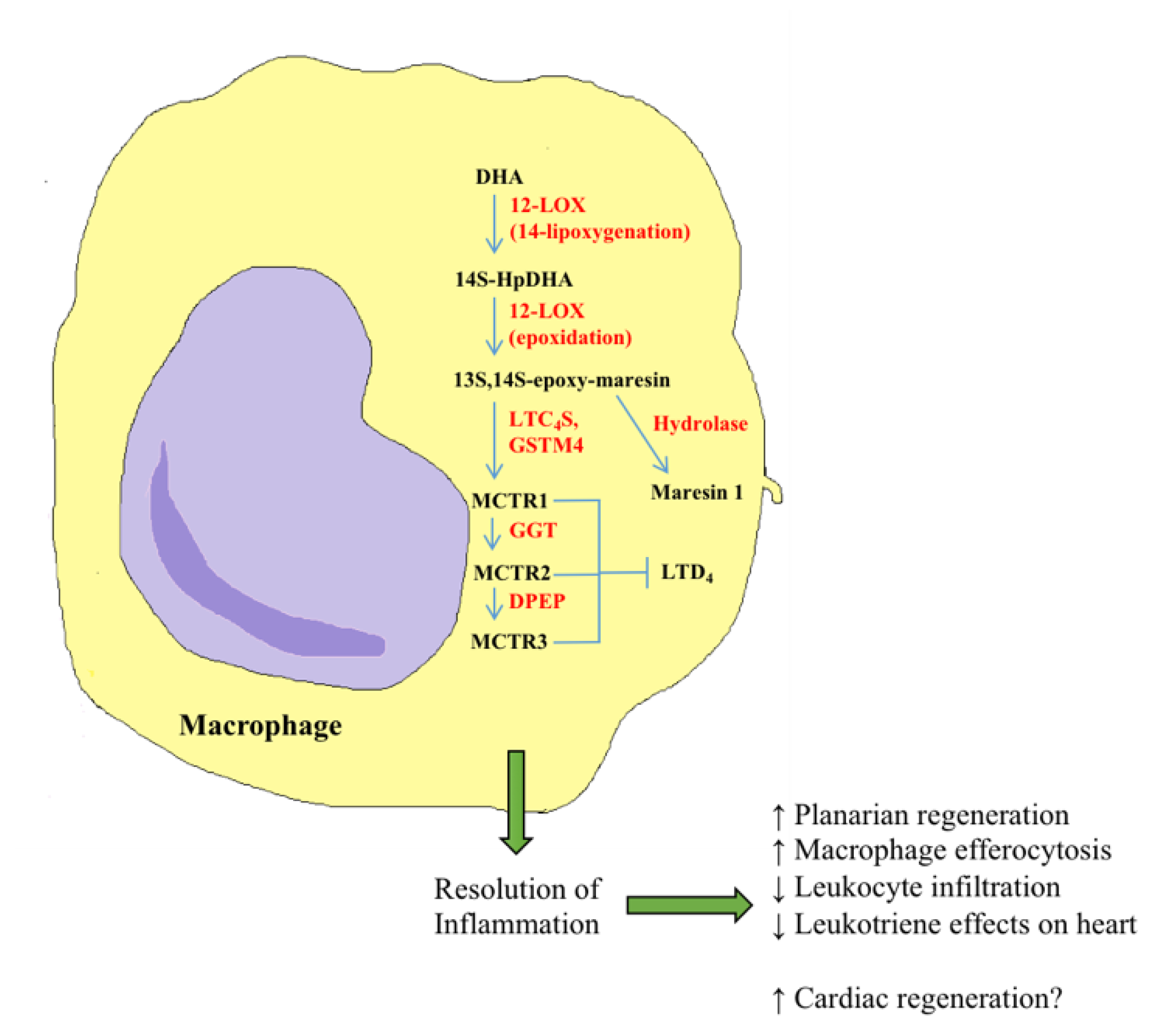
5. Recent Advances in Bioactive Lipids in Cardiac Regeneration
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| diHDHA | dihydroxydocosahexaenoic acid |
| diHOME | dihydroxyoctadecenoic acid |
| EET | epoxyeicosatrienoic acid |
| HEPE | hydroxyeicosapentaenoic acid |
| HETE | hydroxyeicosatetraenoic acid |
| Hx | hepoxilin |
| Lx | lipoxin |
| MaR | maresin |
| MCTR | maresin conjugate in tissue regeneration |
| oxo-ETE | oxo-eicosatetraenoic acid |
| PG | prostaglandin |
| PMN | polymorphonuclear leukocyte |
| Tx | thromboxane |
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2017 Update. Circulation 2017, 135, 146–603. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The various roles of fatty acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef]
- Schmelzer, K.; Fahy, E.; Subramaniam, S.; Dennis, E.A. The Lipid Maps Initiative in Lipidomics. Methods Enzymol. 2007, 432, 171–183. [Google Scholar] [PubMed]
- O’Donnell, V.B.; Dennis, E.A.; Wakelam, M.J.O.; Subramaniam, S. LIPID MAPS: Serving the next generation of lipid researchers with tools, resources, data, and training. Sci. Signal. 2019, 12, 4–7. [Google Scholar] [CrossRef]
- Watrous, J.D.; Niiranen, T.J.; Lagerborg, K.A.; Henglin, M.; Xu, Y.J.; Rong, J.; Sharma, S.; Vasan, R.S.; Larson, M.G.; Armando, A.; et al. Directed Non-targeted Mass Spectrometry and Chemical Networking for Discovery of Eicosanoids and Related Oxylipins. Cell Chem. Biol. 2019, 26, 433–442. [Google Scholar] [CrossRef]
- Fahy, E.; Alvarez-Jarreta, J.; Brasher, C.J.; Nguyen, A.; Hawksworth, J.I.; Rodrigues, P.; Meckelmann, S.; Allen, S.M.; O’Donnell, V.B. LipidFinder on LIPID MAPS: Peak filtering, MS searching and statistical analysis for lipidomics. Bioinformatics 2019, 35, 685–687. [Google Scholar] [CrossRef]
- Chen, B.; McClements, D.J.; Decker, E.A. Design of Foods with Bioactive Lipids for Improved Health. Annu. Rev. Food Sci. Technol. 2013, 4, 35–56. [Google Scholar] [CrossRef]
- Kleger, A.; Liebau, S.; Lin, Q.; Von Wichert, G.; Seufferlein, T. The impact of bioactive lipids on cardiovascular development. Stem Cells Int. 2011, 2011. [Google Scholar] [CrossRef]
- Bieberich, E. It’s a lipid’s world: Bioactive lipid metabolism and signaling in neural stem cell differentiation. Neurochem. Res. 2012, 37, 1208–1229. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Vasquez, V. How Lipids Contribute to Ion Channel Function, a Fat Perspective on Direct and Indirect Interactions. Curr. Opin. Struct. Biol. 2018, 51, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Lee, M.Y.; Ryu, J.M.; Han, H.J. Interaction between PGE2 and EGF receptor through MAPKs in mouse embryonic stem cell proliferation. Cell. Mol. Life Sci. 2009, 66, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Khillan, J.S. A novel signaling by vitamin A/retinol promotes self renewal of mouse embryonic stem cells by activating PI3K/Akt signaling pathway via insulin-like growth factor-1 receptor. Stem Cells 2010, 28, 57–63. [Google Scholar] [PubMed]
- Layden, B.T.; Newman, M.; Chen, F.; Fisher, A.; Lowe, W.L. G protein coupled receptors in embryonic stem cells: A role for Gs-alpha signaling. PLoS ONE 2010, 5, e9105. [Google Scholar] [CrossRef]
- Callihan, P.; Mumaw, J.; MacHacek, D.W.; Stice, S.L.; Hooks, S.B. Regulation of stem cell pluripotency and differentiation by G protein coupled receptors. Pharmacol. Ther. 2011, 129, 290–306. [Google Scholar] [CrossRef]
- Hayek, S.S.; Klyachkin, Y.; Asfour, A.; Ghasemzadeh, N.; Awad, M.; Hesaroieh, I.; Ahmed, H.; Gray, B.; Kim, J.; Waller, E.K.; et al. Bioactive Lipids and Circulating Progenitor Cells in Patients with Cardiovascular Disease. Stem Cells Transl. Med. 2017, 6, 731–735. [Google Scholar] [CrossRef]
- Sharma, A.; Zhang, Y.; Buikema, J.W.; Serpooshan, V.; Chirikian, O.; Kosaric, N.; Churko, J.M.; Dzilic, E.; Shieh, A.; Burridge, P.W.; et al. Stage-specific Effects of Bioactive Lipids on Human iPSC Cardiac Differentiation and Cardiomyocyte Proliferation. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive lipids and chronic inflammation: Managing the fire within. Front. Immunol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- O’Donnell, V.B.; Murphy, R.C.; Watson, S.P. Platelet lipidomics: Modern day perspective on lipid discovery and characterization in platelets. Circ. Res. 2014, 114, 1185–1203. [Google Scholar] [CrossRef]
- Elmasry, K.; Ibrahim, A.S.; Abdulmoneim, S.; Al-Shabrawey, M. Bioactive lipids and pathological retinal angiogenesis. Br. J. Pharmacol. 2019, 176, 93–109. [Google Scholar] [CrossRef]
- Hisano, Y.; Hia, T. Bioactive lysolipids in cancer and angiogenesis. Pharmacol. Ther. 2019, 193, 91–98. [Google Scholar] [CrossRef]
- Chabowski, D.S.; Cohen, K.E.; Abu-Hatoum, O.; Gutterman, D.D.; Freed, J.K. Crossing Signals: Bioactive Lipids in the Microvasculature. Am. J. Physiol. Circ. Physiol. 2020, 318, H1185–H1197. [Google Scholar] [CrossRef]
- Abdelbaset-Ismail, A.; Cymer, M.; Borkowska-Rzeszotek, S.; Brzeźniakiewicz-Janus, K.; Rameshwar, P.; Kakar, S.S.; Ratajczak, J.; Ratajczak, M.Z. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev. Rep. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Lopategi, A.; López-Vicario, C.; Alcaraz-Quiles, J.; García-Alonso, V.; Rius, B.; Titos, E.; Clària, J. Role of bioactive lipid mediators in obese adipose tissue inflammation and endocrine dysfunction. Mol. Cell. Endocrinol. 2016, 419, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Is there a role for bioactive lipids in the pathobiology of diabetes mellitus? Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Sonoshita, M.; Takaku, K.; Sasaki, N.; Sugimoto, Y.; Ushikubi, F.; Narumiya, S.; Oshima, M.; Taketo, M.M. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in ApcΔ716 knockout mice. Nat. Med. 2001, 7, 1048–1051. [Google Scholar] [CrossRef]
- Leishman, E.; Kunkler, P.E.; Hurley, J.H.; Miller, S.; Bradshaw, H.B. Bioactive Lipids in Cancer, Inflammation and Related Diseases: Acute and Chronic Mild Traumatic Brain Injury Differentially Changes Levels of Bioactive Lipids in the CNS Associated with Headache. Adv. Exp. Med. Biol. 2019, 1161, 193–217. [Google Scholar]
- Sokoła-Wysoczańska, E.; Wysoczański, T.; Wagner, J.; Czyż, K.; Bodkowski, R.; Lochyński, S.; Patkowska-Sokoła, B. Polyunsaturated fatty acids and their potential therapeutic role in cardiovascular system disorders—A review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef]
- Bruins, M.J.; Dane, A.D.; Strassburg, K.; Vreeken, R.J.; Newman, J.W.; Salem, N.; Tyburczy, C.; Brenna, J.T. Plasma oxylipin profiling identifies polyunsaturated vicinal diols as responsive to arachidonic acid and docosahexaenoic acid intake in growing piglets. J. Lipid Res. 2013, 54, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Liakh, I.; Pakiet, A.; Sledzinski, T.; Mika, A. Modern methods of sample preparation for the analysis of oxylipins in biological samples. Molecules 2019, 24, 1639. [Google Scholar] [CrossRef]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Aukema, H.M. Advances in Our Understanding of Oxylipins. Am. Soc. Nutr. 2015, 6, 513–540. [Google Scholar]
- Rand, A.A.; Rajamani, A.; Kodani, S.D.; Harris, T.R.; Schlatt, L.; Barnych, B.; Passerini, A.G.; Hammock, B.D. Epoxyeicosatrienoic acid (EET)-stimulated angiogenesis is mediated by epoxy hydroxyeicosatrienoic acids (EHETs) formed from COX-2. J. Lipid Res. 2019, 60, 1996–2005. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W.; Edin, M.L.; Maeyer, R.P.H.D.; Bystrom, J.; Newson, J.; Lih, F.B.; Stables, M.; Zeldin, D.C.; Bishop-Bailey, D. CYP450-derived oxylipins mediate inflammatory resolution. Proc. Natl. Acad. Sci. USA 2016, 113, E3240–E3249. [Google Scholar] [CrossRef]
- Tourdot, B.E.; Ahmed, I.; Holinstat, M. The emerging role of oxylipins in thrombosis and diabetes. Front. Pharmacol. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kurzrok, R.; Lieb, C.C. Biochemical Studies of Human Semen - The Action of Semen on the Human Uterus. Proc. Soc. Exp. Biol. Med. 1930, 26, 268–272. [Google Scholar] [CrossRef]
- Euler, U. The specific hypotensive substance from the secretions of the human prostate vesicles. Klin. Wochschr. 1935, 14, 1182–1183. [Google Scholar] [CrossRef]
- Bergström, S.; Danielsson, H.; Samuelsson, B. The enzymatic formation of prostaglandin E2 from arachidonic acid prostaglandins and related factors 32. Biochim. Biophys. Acta 1964, 90, 207–210. [Google Scholar] [CrossRef]
- Piper, P.; Vane, J. The Release of Prostaglandins From Lung And Other Tissues. Ann. N. Y. Acad. Sci. 1971, 180, 363–385. [Google Scholar] [CrossRef]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits—A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef]
- Caligiuri, S.P.B.; Parikh, M.; Stamenkovic, A.; Pierce, G.N.; Aukema, H.M. Dietary modulation of oxylipins in cardiovascular disease and aging. Am. J. Physiol. Hear. Circ. Physiol. 2017, 313, H903–H918. [Google Scholar] [CrossRef]
- Nayeem, M.A. Role of oxylipins in cardiovascular diseases review-article. Acta Pharmacol. Sin. 2018, 39, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Hawley, M.; Holinstat, M. The expansive role of oxylipins on platelet biology. J. Mol. Med. 2017, 95, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Nasjletti, A. The role of eicosanoids in angiotensin-dependent hypertension. Hypertension 1997, 31, 194–200. [Google Scholar] [CrossRef]
- Wolfe, L.S. Eicosanoids: Prostaglandins, Thromboxanes, Leukotrienes, and Other Derivatives of Carbon-20 Unsaturated Fatty Acids. J. Neurochem. 1982, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef]
- Ho, A.T.V.; Palla, A.R.; Blake, M.R.; Yucel, N.D.; Wang, Y.X.; Magnusson, K.E.G.; Holbrook, C.A.; Kraft, P.E.; Delp, S.L.; Blau, H.M. Prostaglandin E2 is essential for efficacious skeletal muscle stem-cell function, augmenting regeneration & strength. Proc. Natl. Acad. Sci. USA 2017, 114, 6675–6684. [Google Scholar]
- Smyth, E.M. Thromboxane and the thromboxane receptor in cardiovascular disease. Clin. Lipidol. 2010, 5, 209–219. [Google Scholar] [CrossRef]
- Bäck, M.; Weber, C.; Lutgens, E. Regulation of atherosclerotic plaque inflammation. J. Intern. Med. 2015, 278, 462–482. [Google Scholar] [CrossRef]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals Through G-Protein-Coupled Receptor GPR75 (Gq) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776–1788. [Google Scholar] [CrossRef]
- Das, U.N. Arachidonic acid in health and disease with focus on hypertension and diabetes mellitus: A review. J. Adv. Res. 2018, 11, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.A.; Blobe, G.C.; Hannun, Y.A. Arachidonic acid and free fatty acids as second messengers and the role of protein kinase C. Cell. Signal. 1995, 7, 171–184. [Google Scholar] [CrossRef]
- Bhagat, K.; Collier, J.; Vallance, P. Vasodilatation to Arachidonic Acid in Humans. Circulation 1995, 92, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Shuldiner, A.R.; Klein, T.E.; Altman, R.B. Platelet aggregation pathway. Pharm. Genom. 2011, 21, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef]
- Hong, S.; Gronert, K.; Devchand, P.R.; Moussignac, R.L.; Serhan, C.N. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells: Autacoids in anti-inflammation. J. Biol. Chem. 2003, 278, 14677–14687. [Google Scholar] [CrossRef]
- Van Ginneken, V.J.T.; Helsper, J.P.F.G.; De Visser, W.; Van Keulen, H.; Brandenburg, W.A. Polyunsaturated fatty acids in various macroalgal species from north Atlantic and tropical seas. Lipids Health Dis. 2011, 10, 4–11. [Google Scholar] [CrossRef]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; de la Rosa, X.; Jouvene, C.C. Cutting Edge: Human Vagus Produces Specialized Proresolving Mediators of Inflammation with Electrical Stimulation Reducing Proinflammatory Eicosanoids. J. Immunol. 2018, 201, 3161–3165. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators Protection versus uncontrolled inflammation: First responders and resolution. J. Clin. Invest. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Tribulova, N.; Bacova, B.S.; Benova, T.E.; Knezl, V.; Barancik, M.; Slezak, J. Omega-3 index and anti-arrhythmic potential of omega-3 PUFAs. Nutrients 2017, 9, 1191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Lyu, B.; Kishi, H.; Kobayashi, S. Omega-3 and omega-6 DPA equally inhibit the sphingosylphosphorylcholine-induced Ca2+-sensitization of vascular smooth muscle contraction via inhibiting Rho-kinase activation and translocation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cottin, S.C.; Alsaleh, A.; Sanders, T.A.B.; Hall, W.L. Lack of effect of supplementation with EPA or DHA on platelet-monocyte aggregates and vascular function in healthy men. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 743–751. [Google Scholar] [CrossRef]
- Nelson, G.J.; Schmidt, P.S.; Bartolini, G.L.; Kelley, D.S.; Kyle, D. The effect of dietary docosahexaenoic acid on platelet function, platelet fatty acid composition, and blood coagulation in humans. Lipids 1997, 32, 1129–1136. [Google Scholar] [CrossRef]
- Von Schacky, C.; Harris, W.S. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc. Res. 2007, 73, 310–315. [Google Scholar] [CrossRef]
- Vergès, B. Pathophysiology of diabetic dyslipidaemia: Where are we? Diabetologia 2015, 58, 886–899. [Google Scholar] [CrossRef]
- Kohli, P.; Levy, B.D. Resolvins and protectins: Mediating solutions to inflammation. Br. J. Pharmacol. 2009, 158, 960–971. [Google Scholar] [CrossRef]
- Serhan Charles, N.; Petasis, N.A. Resolvins and Protectins in Inflammation-Resolution Charles. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef]
- Schwanke, R.C.; Marcon, R.; Bento, A.F.; Calixto, J.B. EPA- and DHA-derived resolvins’ actions in inflammatory bowel disease. Eur. J. Pharmacol. 2016, 785, 156–164. [Google Scholar] [CrossRef]
- Hellman, J.; Tang, Y.; Spite, M. Pro-resolving lipid mediators and diabetic wound healing. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Philippe, R.; Urbach, V. Specialized pro-resolving lipid mediators in cystic fibrosis. Int. J. Mol. Sci. 2018, 19, 2865. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Spite, M. Specialized pro-resolving mediators in cardiovascular diseases. Mol. Aspects Med. 2017, 58, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Depner, C.M.; Tripathy, S. Omega-3 fatty acid supplementation and cardiovascular disease. J. Lipid Res. 2012, 53, 2525–2545. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K. Docosahexaenoic acid regulates vascular endothelial cell function and prevents cardiovascular disease. Lipids Health Dis. 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Richter, C.K.; Skulas-Ray, A.; Kris-Etherton, P.M. Recommended Intake of Fish and Fish Oils Worldwide; Elsevier Inc.: Amsterdam, The Netherlands, 2016; ISBN 9780128028445. [Google Scholar]
- Greenberg, J.A.; Bell, S.J.; Ausdal, W. Van Omega-3 Fatty Acid supplementation during pregnancy. Rev. Obstet. Gynecol. 2008, 1, 162–169. [Google Scholar]
- Mozaffarian, D.; Rimm, E.B. Fish intake, contaminants, and human health evaluating the risks and the benefits. J. Am. Med. Assoc. 2006, 296, 1885–1899. [Google Scholar] [CrossRef]
- Smith, S.C.; Allen, J.; Blair, S.N.; Bonow, R.O.; Brass, L.M.; Fonarow, G.C.; Grundy, S.M.; Hiratzka, L.; Jones, D.; Krumholz, H.M.; et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update. Circulation 2006, 113, 2363–2372. [Google Scholar] [CrossRef]
- Priori, S.G.; Aliot, E.; Blomstrom-Lundqvist, C.; Bossaert, L.; Breithardt, G.; Brugada, P.; Camm, J.A.; Cappato, R.; Cobbe, S.M.; Di Mario, C.; et al. Update of the guidelines on sudden cardiac death of the European Society of Cardiology. Eur. Heart J. 2003, 24, 13–15. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Chaddha, A.; Eagle, K.A. Omega-3 Fatty Acids and Heart Health. Circulation 2015, 132, e350–e352. [Google Scholar] [CrossRef] [PubMed]
- Makide, K.; Uwamizu, A.; Shinjo, Y.; Ishiguro, J.; Okutani, M.; Inoue, A.; Aoki, J. Novel lysophosphoplipid receptors: Their structure and function. J. Lipid Res. 2014, 55, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Pencreac’h, G.; Ergan, F.; Poisson, L. Production of lysophospholipids rich in DHA. Lipid Technol. 2011, 23, 250–252. [Google Scholar] [CrossRef]
- Heringdorf, D.M.Z. Lysophospholipids. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: New York City, NY, USA, 2008; pp. 710–716. [Google Scholar]
- Wang, Y.; Li, Y.; Shi, G. The regulating function of heterotrimeric G proteins in the immune system. Arch. Immunol. Ther. Exp. 2013, 61, 309–319. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef]
- Gardell, S.E.; Dubin, A.E.; Chun, J. Emerging medicinal roles for lysophospholipid signaling. Trends Mol. Med. 2006, 12, 65–75. [Google Scholar] [CrossRef]
- Murakami, M.; Shiraishi, A.; Tabata, K.; Fujita, N. Identification of the orphan GPCR, P2Y10 receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2008, 371, 707–712. [Google Scholar] [CrossRef]
- Parrill, A. LPA receptor agonists and antagonists (WO2010051053). Expert Opin. Ther. Pat. 2011, 21, 281–286. [Google Scholar] [CrossRef]
- Smyth, S.S.; Cheng, H.; Miriyala, S.; Panchatcharam, M.; Morris, A.J. Roles of Lysophosphatidic Acid in Cardiovascular Physiology and Disease Susan. Biochim. Biophys. Acta 2008, 1781, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.J.; Smyth, S.S. Lysophosphatidic acid and cardiovascular disease: Seeing is believing. J. Lipid Res. 2013, 54, 1153–1155. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, S.; Hashimoto, T.; Kano, K.; Aoki, J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J. Biochem. 2015, 157, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.; Li, R.S.; Samuel, S.B.; Cueto, R.; Li, X.Y.; Wang, H.; Yang, X.F. Lysophospholipids and their G protein-coupled receptors in atherosclerosis. Front. Biosci. Landmark 2016, 21, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA 2/LPC and ATX/LPA axes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Bas, J.M.D.; Caimari, A.; Rodriguez-Naranjo, M.I.; Childs, C.E.; Chavez, C.P.; West, A.L.; Miles, E.A.; Arola, L.; Calder, P.C. Impairment of lysophospholipid metabolism in obesity: Altered plasma profile and desensitization to the modulatory properties of n-3 polyunsaturated fatty acids in a randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 266–279. [Google Scholar]
- Bourgoin, S.G.; Zhao, C. Autotaxin and Lysophospholipids in Rheumatoid Arthritis. Curr. Opin. Investig. Drugs 2010, 11, 515–526. [Google Scholar]
- Linkous, A.G.; Yazlovitskaya, E.M.; Hallahan, D.E. Cytosolic phospholipase a2 and lysophospholipids in tumor angiogenesis. J. Natl. Cancer Inst. 2010, 102, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Kuwajima, K.; Sumitani, M.; Kurano, M.; Kano, K.; Nishikawa, M.; Uranbileg, B.; Tsuchida, R.; Ogata, T.; Aoki, J.; Yatomi, Y.; et al. Lysophosphatidic acid is associated with neuropathic pain intensity in humans: An exploratory study. PLoS ONE 2018, 13, e0207310. [Google Scholar] [CrossRef]
- Ye, X.; Chun, J. Lysophosphatidic Acid (LPA) Signaling in Vertebrate Reproduction. Trends Endocrinol. Metab. 2010, 21, 1–19. [Google Scholar] [CrossRef]
- Schober, A.; Siess, W. Lysophosphatidic acid in atherosclerotic diseases. Br. J. Pharmacol. 2012, 167, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Lopez, C.M.; Tucker, A.L.; Lynch, K.R. Lysophosphatidic acid (LPA) and angiogenesis. Angiogenesis 2008, 11, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular Effects and Benefits of Exercise. Front. Cardiovasc. Med. 2018, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ signaling in angiogenesis. Front. Cell Dev. Biol. 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.Z. Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin. Lipidol. 2011, 6, 413–426. [Google Scholar] [CrossRef]
- Teo, S.T.; Yung, Y.C.; Herr, D.R.; Chun, J. Lysophosphatidic acid in vascular development and disease. IUBMB Life 2009, 61, 791–799. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, X.-L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef]
- Smyth, S.S.; Mueller, P.; Yang, F.; Brandon, J.A.; Morris, A.J. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 479–486. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528. [Google Scholar] [CrossRef]
- Merrill, A.H. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef]
- De Faria Poloni, J.; Chapola, H.; Feltes, B.C.; Bonatto, D. The importance of sphingolipids and reactive oxygen species in cardiovascular development. Biol. Cell 2014, 106, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Borodzicz, S.; Czarzasta, K.; Kuch, M.; Cudnoch-Jedrzejewska, A. Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis. 2015, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.F. Sphingolipids in inflammation: Pathological implications and potential therapeutic targets. Br. J. Pharmacol. 2009, 158, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Jernigan, P.L.; Makley, A.T.; Hoehn, R.S.; Edwards, M.J.; Pritts, T.A. The role of sphingolipids in endothelial barrier function. Biol. Chem. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Michel, T. Sphingosine-1-phosphate and modulation of vascular tone. Cardiovasc. Res. 2009, 82, 212–220. [Google Scholar] [CrossRef]
- Tobia, C.; Chiodelli, P.; Nicoli, S.; Dell’Era, P.; Buraschi, S.; Mitola, S.; Foglia, E.; Van Loenen, P.B.; Alewijnse, A.E.; Presta, M. Sphingosine-1-phosphate receptor-1 controls venous endothelial barrier integrity in zebrafish. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 104–116. [Google Scholar] [CrossRef]
- Bieberich, E. Ceramide in Stem Cell Differentiation and Embryo Development: Novel Functions of a Topological Cell-Signaling Lipid and the Concept of Ceramide Compartments. J. Lipids 2011, 2011. [Google Scholar] [CrossRef]
- Wang, G.; Spassieva, S.S.; Bieberich, E. Ceramide and S1P signaling in embryonic stem cell differentiation. Methods Mol. Biol. 2018, 153–171. [Google Scholar] [CrossRef]
- Simón, M.V.; Prado Spalm, F.H.; Vera, M.S.; Rotstein, N.P. Sphingolipids as emerging mediators in retina degeneration. Front. Cell. Neurosci. 2019, 13, 1–25. [Google Scholar] [CrossRef]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar]
- Karine, M.; Kyuno, J.I.; Bhamra, S.; Jones, E.A. The lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P) receptor gene families: Cloning and comparative expression analysis in Xenopus laevis. Int. J. Dev. Biol. 2010, 54, 1361–1374. [Google Scholar]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine kinases and sphingosine 1-phosphate receptors: Signaling and actions in the cardiovascular system. Front. Pharmacol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Mehmood, A.; Linardi, D.; Sadiq, S.; Tessari, M.; Meo, S.A.; Rehman, R.; Hajjar, W.M.; Muhammad, N.; Iqbal, M.P.; et al. Cardioprotective Effects of Sphingosine-1-Phosphate Receptor Immunomodulator FTY720 in a Clinically Relevant Model of Cardioplegic Arrest and Cardiopulmonary Bypass. Front. Pharmacol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Kleger, A.; Busch, T.; Liebau, S.; Prelle, K.; Paschke, S.; Beil, M.; Rolletschek, A.; Wobus, A.; Wolf, E.; Adler, G.; et al. The bioactive lipid sphingosylphosphorylcholine induces differentiation of mouse embryonic stem cells and human promyelocytic leukaemia cells. Cell. Signal. 2007, 19, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Z.; Zhao, X.; Pan, F.; Cai, M.; Wang, T.; Zhang, H.; Lu, J.R.; Lei, M. Sphingosine-1-phosphate promotes the differentiation of human umbilical cord mesenchymal stem cells into cardiomyocytes under the designated culturing conditions. J. Biomed. Sci. 2011, 18, 37. [Google Scholar] [CrossRef]
- Silvestri, C.; Di Marzo, V. The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab. 2013, 17, 475–490. [Google Scholar] [CrossRef]
- Oláh, A.; Szekanecz, Z.; Bíró, T. Targeting cannabinoid signaling in the immune system: “High”-ly exciting questions, possibilities, and challenges. Front. Immunol. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Cascio, M.G.; Marini, P. Biosynthesis and Fate of Endocannabinoids. Handb. Exp. Pharmacol. 2015, 231, 39–58. [Google Scholar]
- Chye, Y.; Christensen, E.; Solowij, N.; Yücel, M. The Endocannabinoid System and Cannabidiol’s Promise for the Treatment of Substance Use Disorder. Front. Psychiatry 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Chen, C. Endocannabinoids in Synaptic Plasticity and Neuroprotection. Neuroscientist 2015, 21, 152–168. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dragunow, M.; Faull, R.L.M. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; LE Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.R.; Laviolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef]
- Bátkai, S.; Pacher, P. Endocannabinoids and cardiac contractile function: Pathophysiological implications. Pharmacol. Res. 2009, 60, 99–106. [Google Scholar] [CrossRef]
- Tata, J.R. One hundred years of hormones. EMBO Rep. 2005, 6, 490–496. [Google Scholar] [CrossRef]
- Holst, J.P.; Soldin, O.P.; Guo, T.; Soldin, S.J. Steroid hormones: Relevance and measurement in the clinical laboratory. Clin. Lab. Med. 2004, 24, 105–118. [Google Scholar] [CrossRef]
- Basit, S. Vitamin D in health and disease: A literature review. Br. J. Biomed. Sci. 2013, 70, 161–172. [Google Scholar] [CrossRef]
- Cheng, J.B.; Motola, D.L.; Mangelsdorf, D.J.; Russell, D.W. De-orphanization of cytochrome P450 2R1: A microsomal vitamin D 25-hydroxylase. J. Biol. Chem. 2003, 278, 38084–38093. [Google Scholar] [CrossRef]
- Ohyama, Y.; Yamasaki, T. Eight Cytochrome P450s Catalyze Vitamin D Metabolism. Front. Biosci. 2004, 9, 3007–3018. [Google Scholar] [CrossRef]
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Van Cromphaut, S.; Carmeliet, G. Intestinal calcium absorption: Molecular vitamin D mediated mechanisms. J. Cell. Biochem. 2003, 88, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Makin, G.; Lohnes, D.; Byford, V.; Ray, R.; Jones, G. Target cell metabolism of 1, 25-dihydroxyvitamin D3 to calcitroic acid. Biochem. J. 1989, 262, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Zierold, C.; Darwish, H.M.; Deluca, H.F. Identification of a vitamin D-response element in the rat calcidiol (25-hydroxyvitamin Da) 24-hydroxylase gene. Proc. Natl. Acad. Sci. USA 1994, 91, 900–902. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Angeline, M.E.; Gee, A.O.; Shindle, M.; Warren, R.F.; Rodeo, S.A. The effects of vitamin d deficiency in athletes. Am. J. Sports Med. 2013, 41, 461–464. [Google Scholar] [CrossRef]
- Giovannucci, E.; Liu, Y.; Hollis, B.W.; Rimm, E.B. 25-Hydroxyvitamin D and risk of myocardial infarction in men: A prospective study. Arch. Intern. Med. 2008, 168, 1174–1180. [Google Scholar] [CrossRef]
- Dobnig, H.; Pilz, S.; Scharnagl, H.; Renner, W.; Seelhorst, U. Independent Association of Low Serum. Arch. Intern. Med. 2008, 168, 1340–1349. [Google Scholar] [CrossRef]
- Ross, A.C.; Manson, J.A.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Danik, J.S.; Manson, J.E. Vitamin D and Cardiovascular Disease. Curr. Treat. Options Cardiovasc. Med. 2012, 14, 414–424. [Google Scholar] [CrossRef]
- Kwon, S.; Hermayer, K.L. Glucocorticoid-induced hyperglycemia. Am. J. Med. Sci. 2013, 345, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Wallerath, T.; Witte, K.; Schafer, S.C.; Schwarz, P.M.; Prellwitz, W.; Wohlfart, P.; Kleinert, H.; Lehr, H.A.; Lemmer, B.; Forstermann, U. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc. Natl. Acad. Sci. USA 1999, 96, 13357–13362. [Google Scholar] [CrossRef]
- Krausz, Y.; Bar-On, H.; Shafrir, E. Dose-dependent bimodal changes in serum lipids and lipoproteins in relation to hepatic lipogenesis and tissue lipoprotein lipase activity. Biochim. Biophys. Acta 1981, 663, 69–82. [Google Scholar] [CrossRef]
- Santana, P.; Akana, S.F.; Hanson, E.S.; Strack, A.M.; Sebastian, R.J.; Dallman, M.F. Aldosterone and Dexamethasone Both Stimulate Energy Acquisition Whereas Only the Glucocorticoid Alters Energy Storage. Endocrinology 1995, 136, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.J.; Tilbrook, A. The glucocorticoid contribution to obesity. Stress 2011, 14, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Berard, A.; Van Staa, T.P.; Cooper, C.; Egberts, A.C.G.; Leufkens, H.G.M.; Walker, B.R. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 2004, 90, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a Selective Aldosterone Blocker, in Patients with Left Ventricular Dysfunction after Myocardial Infarction Bertram. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Hagihara, H.; Nomoto, A.; Mutoh, S.; Yamaguchi, I.; Ono, T. Role of inflammatory responses in initiation of atherosclerosis: Effects of anti-inflammatory drugs on cuff-induced leukocyte accumulation and intimal thickening of rabbit carotid artery. Atherosclerosis 1991, 91, 107–116. [Google Scholar] [CrossRef]
- Shen, J.Z.; Young, M.J. Corticosteroids, heart failure, and hypertension: A role for immune cells? Endocrinology 2012, 153, 5692–5700. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. Glucocorticoid signaling in the heart: A cardiomyocyte perspective. J. Steroid Biochem. Mol. Biol. 2015, 153, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Rog-Zielinska, E.A.; Craig, M.A.; Manning, J.R.; Richardson, R.V.; Gowans, G.J.; Dunbar, D.R.; Gharbi, K.; Kenyon, C.J.; Holmes, M.C.; Hardie, D.G.; et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: A role for PGC-1α. Cell Death Differ. 2015, 22, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Kam, R.K.T.; Deng, Y.; Chen, Y.; Zhao, H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kedishvilli, N.Y. Retinoic Acid Synthesis and Degradation. Subcell Biochem. 2016, 81, 127–161. [Google Scholar]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef]
- Cañete, A.; Cano, E.; Muñoz-Chápuli, R.; Carmona, R. Role of vitamin a/retinoic acid in regulation of embryonic and adult hematopoiesis. Nutrients 2017, 9, 159. [Google Scholar] [CrossRef]
- Karrer, P.; Morf, R.; Schopp, K. Zur Kenntnis des Vitamins-A aus Fischtranen II. Helv. Chim. Acta 1931, 14, 1431–1436. [Google Scholar] [CrossRef]
- Semba, R.D. On the “discovery” of vitamin a. Ann. Nutr. Metab. 2012, 61, 192–198. [Google Scholar] [CrossRef]
- Wolf, G. Multiple functions of vitamin A. Physiol. Rev. 1984, 64, 873–937. [Google Scholar] [CrossRef]
- Ratnayake, K.; Payton, J.L.; Lakmal, O.H.; Karunarathne, A. Blue light excited retinal intercepts cellular signaling. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Vilhais-Neto, C.; Pourquie, O. Retinoic acid Essay The origins of behavioral genetics. Curr. Biol. 2008, 18, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Xu, F.; Song, W.; Wang, X.; Hu, P.; Yang, Y.; Gao, X.; Zhao, Q. A novel cytochrome P450, zebrafish Cyp26D1, is involved in metabolism of all-trans retinoic acid. Mol. Endocrinol. 2006, 20, 1661–1672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Janesick, A.; Wu, S.C.; Blumberg, B. Retinoic acid signaling and neuronal differentiation. Cell. Mol. Life Sci. 2015, 72, 1559–1576. [Google Scholar] [CrossRef]
- Xavier-Neto, J.; Sousa Costa, Â.M.; Figueira, A.C.M.; Caiaffa, C.D.; Amaral, F.N.D.; Peres, L.M.C.; da Silva, B.S.P.; Santos, L.N.; Moise, A.R.; Castillo, H.A. Signaling through retinoic acid receptors in cardiac development: Doing the right things at the right times. Biochim. Biophys. Acta Gene Regul. Mech. 2015, 1849, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef]
- Lin, S.C.; Dollé, P.; Ryckebüsch, L.; Noseda, M.; Zaffran, S.; Schneider, M.D.; Niederreithera, K. Endogenous retinoic acid regulates cardiac progenitor differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 9234–9239. [Google Scholar] [CrossRef]
- Vermot, J.; Niederreither, K.; Garnier, J.M.; Chambon, P.; Dollé, P. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1763–1768. [Google Scholar] [CrossRef]
- Yutzey, K.E. Digeorge syndrome, Tbx1, and retinoic acid signaling come full circle. Circ. Res. 2010, 106, 630–632. [Google Scholar] [CrossRef]
- Zhou, W.; Lin, J.; Chen, H.; Wang, J.; Liu, Y.; Xia, M. Retinoic acid induces macrophage cholesterol efflux and inhibits atherosclerotic plaque formation in apoE-deficient mice. Br. J. Nutr. 2015, 114, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, H.; Mu, D.; Li, D.; Zhong, Y.; Jiang, N.; Zhang, Y.; Xia, M. Association of Serum Retinoic Acid with Risk of Mortality in Patients with Coronary Artery Disease. Circ. Res. 2016, 119, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Grimsgaard, S.; Bonna, K.H.; Hansen, J.-B.; Nordoy, A. Highly purified eicosapentaenoic acid and docosahexaenoic acid in humans have similar effects but divergent effects serum fatty acids. Am. J. Clin. Nutr. 1997, 66, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Heydari, B.; Abdullah, S.; Pottala, J.V.; Shah, R.; Abbasi, S.; Mandry, D.; Francis, S.A.; Lumish, H.; Ghoshhajra, B.B.; Hoffmann, U.; et al. Effect of omega-3 acid ethyl esters on left ventricular remodeling after acute myocardial infarction. Circulation 2016, 134, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Colas, R.A.; Souza, P.R.; Walker, M.E.; Burton, M.; Zasłona, Z.; Curtis, A.M.; Marques, R.M.; Dalli, J. Impaired production and diurnal regulation of vascular RvD n-3 DPA increase systemic inflammation and cardiovascular disease. Circ. Res. 2018, 122, 855–863. [Google Scholar] [CrossRef]
- Shabani, P.; Ghazizadeh, Z.; Gorgani-Firuzjaee, S.; Molazem, M.; Rajabi, S.; Vahdat, S.; Azizi, Y.; Doosti, M.; Aghdami, N.; Baharvand, H. Cardioprotective effects of omega-3 fatty acids and ascorbic acid improve regenerative capacity of embryonic stem cell-derived cardiac lineage cells. BioFactors 2019, 45, 427–438. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Sumanth, D.; Serhan, C.N.; Joshi, M.; Halade, G. V Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell. Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Kain, V.; Liu, F.; Kozlovskaya, V.; Ingle, K.A.; Bolisetty, S.; Agarwal, A.; Khedkar, S.; Prabhu, S.D.; Kharlampieva, E.; Halade, G.V. Resolution Agonist 15-epi-Lipoxin A4 Programs Early Activation of Resolving Phase in Post-Myocardial Infarction Healing. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Kain, V.; Ingle, K.A.; Kabarowski, J.; Barnes, S.; Limdi, N.A.; Prabhu, S.D.; Halade, G.V. Genetic deletion of 12/15 lipoxygenase promotes effective resolution of inflammation following myocardial infarction. J. Mol. Cell. Cardiol. 2018, 118, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Cherpokova, D.; Jouvene, C.C.; Libreros, S.; DeRoo, E.P.; Chu, L.; De La Rosa, X.; Norris, P.C.; Wagner, D.D.; Serhan, C.N. Resolvin D4 attenuates the severity of pathological thrombosis in mice. Blood 2019, 134, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Elkhatali, S.; El-Sherbeni, A.A.; Elshenawy, O.H.; Abdelhamid, G.; El-Kadi, A.O.S. 19-Hydroxyeicosatetraenoic acid and isoniazid protect against angiotensin II-induced cardiac hypertrophy. Toxicol. Appl. Pharmacol. 2015, 289, 550–559. [Google Scholar] [CrossRef] [PubMed]
- Shoieb, S.M.; El-Kadi, A.O.S. S-enantiomer of 19-hydroxyeicosatetraenoic acid preferentially protects against angiotensin II-induced cardiac hypertrophy. Drug Metab. Dispos. 2018, 46, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Tunaru, S.; Chennupati, R.; Nusing, R.M.; Offermanns, S. Arachidonic acid metabolite 19(S)-HETE induces vasorelaxation and platelet inhibition by activating prostacyclin (IP) receptor. PLoS ONE 2016, 11, e0163633. [Google Scholar] [CrossRef] [PubMed]
- Aliwarga, T.; Evangelista, E.A.; Sotoodehnia, N.; Lemaitre, R.N.; Totah, R.A. Regulation of CYP2J2 and EET levels in cardiac disease and diabetes. Int. J. Mol. Sci. 2018, 19, 1916. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zeng, H.; Wen, Z.; Chen, C.; Wang, D.W. CYP2J2 and its metabolites (epoxyeicosatrienoic acids) attenuate cardiac hypertrophy by activating AMPKα2 and enhancing nuclear translocation of Akt1. Aging Cell 2016, 15, 940–952. [Google Scholar] [PubMed]
- Liu, W.; Wang, T.; He, X.; Liu, X.; Wang, B.; Liu, Y.; Li, Z.; Tan, R.; Ding, C.; Wang, H.; et al. CYP2J2 Overexpression Increases EETs and Protects Against HFD-Induced Atherosclerosis in ApoE-/- Mice. J. Cardiovasc. Pharmacol. 2016, 67, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat. Rev. Drug Discov. 2009, 8, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Islam, O.; Patil, P.; Goswami, S.K.; Razdan, R.; Inamdar, M.N.; Rizwan, M.; Mathew, J.; Inceoglu, B.; Stephen Lee, K.S.; Hwang, S.H.; et al. Inhibitors of soluble epoxide hydrolase minimize ischemia-reperfusion-induced cardiac damage in normal, hypertensive, and diabetic rats. Cardiovasc. Ther. 2017, 35, e12259. [Google Scholar] [CrossRef] [PubMed]
- Revermann, M.; Barbosa-Sicard, E.; Dony, E.; Schermuly, R.T.; Morisseau, C.; Geisslinger, G.; Fleming, I.; Hammock, B.D.; Brandes, R.P. Inhibition of the soluble epoxide hydrolase attenuates monocrotaline- induced pulmonary hypertension in rats. J. Hypertens. 2009, 27, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Laguna-Fernandez, A.; Arnardottir, H.; Wheelock, C.E.; Perretti, M.; Hansson, G.K.; Bäck, M. Aspirin-triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E−/− mice. Br. J. Pharmacol. 2017, 174, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.H.; Laguna-Fernandez, A.; Tseng, C.N.; Hedin, U.; Perretti, M.; Bäck, M. Aspirin-triggered 15-epi-lipoxin A4 signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int. J. Cardiol. 2015, 179, 370–372. [Google Scholar] [CrossRef]
- Petri, M.H.; Thul, S.; Andonova, T.; Lindquist-Liljeqvist, M.; Jin, H.; Skenteris, N.T.; Arnardottir, H.; Maegdefessel, L.; Caidahl, K.; Perretti, M.; et al. Resolution of Inflammation Through the Lipoxin and ALX/FPR2 Receptor Pathway Protects Against Abdominal Aortic Aneurysms. JACC Basic Transl. Sci. 2018, 3, 719–727. [Google Scholar] [CrossRef]
- Makino, Y.; Miyahara, T.; Nitta, J.; Miyahara, K.; Seo, A.; Kimura, M.; Suhara, M.; Akai, A.; Akagi, D.; Yamamoto, K.; et al. Proresolving lipid mediators resolvin D1 and protectin D1 isomer attenuate neointimal hyperplasia in the rat carotid artery balloon injury model. J. Surg. Res. 2019, 233, 104–110. [Google Scholar] [CrossRef]
- Viola, J.R.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M.; et al. Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ. Res. 2016, 119, 1030–1038. [Google Scholar] [CrossRef]
- Caligiuri, S.P.B.; Rodriguez-Leyva, D.; Aukema, H.M.; Ravandi, A.; Weighell, W.; Guzman, R.; Pierce, G.N. Dietary Flaxseed Reduces Central Aortic Blood Pressure Without Cardiac Involvement but Through Changes in Plasma Oxylipins. Hypertension 2016, 68, 1031–1038. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health implications of high dietary omega-6 polyunsaturated fatty acids. J. Nutr. Metab. 2012. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; O’Keefe, J.H. Effects of dietary fats on blood lipids: A review of direct comparison trials. Open Hear. 2018, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- De Hoog, V.C.; Bovens, S.M.; De Jager, S.C.A.; Van Middelaar, B.J.; Van Duijvenvoorde, A.; Doevendans, P.A.; Pasterkamp, G.; De Kleijn, D.P.V.; Timmers, L. BLT1 antagonist LSN2792613 reduces infarct size in a mouse model of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2015, 108, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Becher, U.M.; Ghanem, A.; Tiyerili, V.; Fürst, D.O.; Nickenig, G.; Mueller, C.F.H. Inhibition of leukotriene C4 action reduces oxidative stress and apoptosis in cardiomyocytes and impedes remodeling after myocardial injury. J. Mol. Cell. Cardiol. 2011, 50, 570–577. [Google Scholar] [CrossRef]
- Wiedermann, C.J.; Kiechl, S.; Dunzendorfer, S.; Schratzberger, P.; Egger, G.; Oberhollenzer, F.; Willeit, J. Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: Prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 1999, 34, 1975–1981. [Google Scholar] [CrossRef]
- Mawhin, M.A.; Tilly, P.; Zirka, G.; Charles, A.L.; Slimani, F.; Vonesch, J.L.; Michel, J.B.; Bäck, M.; Norel, X.; Fabre, J.E. Neutrophils recruited by leukotriene B4 induce features of plaque destabilization during endotoxaemia. Cardiovasc. Res. 2018, 114, 1656–1666. [Google Scholar] [CrossRef]
- Pacher, P.; Steffens, S. The emerging role of the endocannabinoid system in cardiovascular disease. Semin. Immunopathol. 2009, 31, 63–77. [Google Scholar] [CrossRef]
- Pacher, P.; Haskó, G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 2008, 153, 252–262. [Google Scholar] [CrossRef]
- Pacher, P.; Mukhopadhyay, P.; Mohanraj, R.; Godlewski, G.; Bátkai, S.; Kunos, G. Modulation of the endocannabinoid system in cardiovascular disease: Therapeutic potential and limitations. Hypertension 2008, 52, 601–607. [Google Scholar] [CrossRef]
- Upadhyay, R.K. Emerging risk biomarkers in cardiovascular diseases and disorders. J. Lipids 2015, 2015, 971453. [Google Scholar] [CrossRef]
- Steffens, S.; Veillard, N.R.; Arnaud, C.; Pelli, G.; Burger, F.; Staub, C.; Zimmer, A.; Frossard, J.-L.; Mach, F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature 2005, 434, 782–786. [Google Scholar] [CrossRef]
- Steffens, S.; Pacher, P. Targeting cannabinoid receptor CB2 in cardiovascular disorders: Promises and controversies. Br. J. Pharmacol. 2012, 167, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Bátkai, S.; Rajesh, M.; Mukhopadhyay, P.; Haskó, G.; Liaudet, L.; Cravatt, B.F.; Csiszár, A.; Ungvári, Z.; Pacher, P. Decreased age-related cardiac dysfunction, myocardial nitrative stress, inflammatory gene expression, and apoptosis in mice lacking fatty acid amide hydrolase. Am. J. Physiol. Hear. Circ. Physiol. 2007, 293. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease-Successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef]
- Fulmer, M.L.; Thewke, D.P. The Endocannabinoid System and Heart Disease: The Role of Cannabinoid Receptor Tye 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Pacher, P.; Kogan, N.M.; Mechoulam, R. Beyond THC and Endocannabinoids. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 637–659. [Google Scholar] [CrossRef]
- Rajesh, M.; Mukhopadhyay, P.; Btkai, S.; Patel, V.; Saito, K.; Matsumoto, S.; Kashiwaya, Y.; Horvth, B.; Mukhopadhyay, B.; Becker, L.; et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 2115–2125. [Google Scholar] [CrossRef]
- Migliazza, L.; Xia, H.M.; Arnaiz, A.; Alvarez, J.I.; Alfonso, L.F.; Diez-Pardo, J.A.; Valls I Soler, A.; Tovar, J.A. Prenatal dexamethasone rescues heart hypoplasia in fetal rats with congenital diaphragmatic hernia. J. Pediatr. Surg. 2000, 35, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Takeba, Y.; Matsumoto, N.; Tsuzuki, Y.; Asoh, K.; Takagi, M.; Kobayashi, S.; Yamamoto, H. Antenatal glucocorticoid therapy accelerates ATP production with creatine kinase increase in the growth-enhanced fetal rat heart. Circ. J. 2010, 74, 171–180. [Google Scholar] [CrossRef]
- Xu, B.; Strom, J.; Chen, Q.M. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur. J. Pharmacol. 2011, 668, 194–200. [Google Scholar] [CrossRef]
- Kuropka, P.; Dobrzyński, M.; Gamian, A.; Gostomska-Pampuch, K.; Kuryszko, J.; Całkosiński, I. Effect of Glucocorticoids on Ultrastructure of Myocardial Muscle in the Course of Experimentally Induced Acute Myocardial Ischemia. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Agnew, E.J.; Ivy, J.R.; Stock, S.J.; Chapman, K.E. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J. Mol. Endocrinol. 2018, 61, R61–R73. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cruz-Topete, D.; He, B.; Foley, J.F.; Myers, P.H.; Xu, X.; Gomez-Sanchez, C.E.; Chambon, P.; Willis, M.S.; Cidlowski, J.A. Cardiomyocyte glucocorticoid and mineralocorticoid receptors directly and antagonistically regulate heart disease in mice. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bedja, D.; Mishra, S.; Amuzie, C.; Avolio, A.; Kass, D.A.; Berkowitz, D.; Renehan, M. Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in apolipoprotein E-/-Mice and rabbits fed a high-fat and -cholesterol diet. Circulation 2014, 129, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Bedja, D.; Amuzie, C.; Foss, C.A.; Pomper, M.G.; Bhattacharya, R.; Yarema, K.J.; Chatterjee, S. Improved intervention of atherosclerosis and cardiac hypertrophy through biodegradable polymer-encapsulated delivery of glycosphingolipid inhibitor. Biomaterials 2015, 64, 125–135. [Google Scholar] [CrossRef]
- Mishra, S.; Bedja, D.; Amuzie, C.; Avolio, A.; Chatterjee, S. Prevention of cardiac hypertrophy by the use of a glycosphingolipid synthesis inhibitor in ApoE-/- mice. Biochem. Biophys. Res. Commun. 2015, 465, 159–164. [Google Scholar] [CrossRef]
- Ahuja, G.; Bartsch, D.; Yao, W.; Geissen, S.; Frank, S.; Aguirre, A.; Russ, N.; Messling, J.; Dodzian, J.; Lagerborg, K.A.; et al. Loss of genomic integrity induced by lysosphingolipid imbalance drives ageing in the heart. EMBO Rep. 2019, 20, e47407. [Google Scholar] [CrossRef]
- Mariotti, L.G.; Pirovano, G.; Savage, K.I.; Ghita, M.; Ottolenghi, A.; Prise, K.M.; Schettino, G. Use of the γ-H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PLoS ONE 2013, 8, e0079541. [Google Scholar] [CrossRef]
- Liu, G.H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature 2011, 472, 221–227. [Google Scholar] [CrossRef]
- Krishnan, V.; Chow, M.Z.Y.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef]
- Hisano, Y.; Ota, S.; Takada, S.; Kawahara, A.K. Functional cooperation of spns2 and fibronectin in cardiac and lower jaw development. Biol. Open 2013, 2, 789–794. [Google Scholar] [CrossRef]
- Kupperman, E.; An, S.; Osborne, N.; Waldron, S.; Stainier, D.Y.R. A sphingosine-1-phosphate receptor regulates cell migration during vertebrate heart development. Nature 2000, 406, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Shoham, A.B.; Malkinson, G.; Krief, S.; Shwartz, Y.; Ely, Y.; Ferrara, N.; Yaniv, K.; Zelzer, E. S1P1 inhibits sprouting angiogenesis during vascular development. Development 2012, 139, 3859–3869. [Google Scholar] [CrossRef] [PubMed]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Siemsen, B.L.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nyström, S.; Rymo, S.; Chen, L.L.; et al. The Sphingosine-1-Phosphate Receptor S1PR1 Restricts Sprouting Angiogenesis by Regulating the Interplay between VE-Cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Hisano, Y.; Inoue, A.; Taimatsu, K.; Ota, S.; Ohga, R.; Kotani, H.; Muraki, M.; Aoki, J.; Kawahara, A. Comprehensive analysis of sphingosine-1-phosphate receptor mutants during zebrafish embryogenesis. Genes Cells 2015, 20, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Holper, S.; Kruger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef]
- Mendelson, K.; Zygmunt, T.; Torres-Vázquez, J.; Evans, T.; Hla, T. Sphingosine 1-phosphate receptor signaling regulates proper embryonic vascular patterning. J. Biol. Chem. 2013, 288, 2143–2156. [Google Scholar] [CrossRef]
- Guzzolino, E.; Chiavacci, E.; Ahuja, N.; Mariani, L.; Evangelista, M.; Ippolito, C.; Rizzo, M.; Garrity, D.; Cremisi, F.; Pitto, L. Post-transcriptional modulation of sphingosine-1-phosphate receptor 1 by miR-19a affects cardiovascular development in zebrafish. Front. Cell Dev. Biol. 2018, 6, 58. [Google Scholar] [CrossRef]
- Clay, H.; Wilsbacher, L.D.; Wilson, S.J.; Duong, D.N.; McDonald, M.; Lam, I.; Park, K.E.; Chun, J.; Coughlin, S.R. Sphingosine 1-phosphate receptor-1 in cardiomyocytes is required for normal cardiac development. Dev. Biol. 2016, 418, 157–165. [Google Scholar] [CrossRef]
- Lai, S.L.; Yao, W.L.; Tsao, K.C.; Houben, A.J.S.; Albers, H.M.H.G.; Ovaa, H.; Moolenaar, W.H.; Lee, S.J. Autotaxin/Lpar3 signaling regulates Kupffer’s vesicle formation and left-right asymmetry in zebrafish. Development 2012, 139, 4439–4448. [Google Scholar] [CrossRef]
- Frisca, F.; Colquhoun, D.; Goldshmit, Y.; Änkö, M.L.; Pébay, A.; Kaslin, J. Role of ectonucleotide pyrophosphatase/phosphodiesterase 2 in the midline axis formation of zebrafish. Sci. Rep. 2016, 6, 37678. [Google Scholar] [CrossRef]
- Olley, P.M.; Coceani, F.; Bodach, E.V.A. E-Type Prostaglandins: A New Emergency Therapy for Certain Cyanotic Congenital Heart Malformations. Circulation 1976, 53, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Srivastava, D. The paradoxical patent ductus arteriosus. J. Clin. Invest. 2006, 116, 2863–2866. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Locker, J.; Lu, R.; Schuster, V.L. Failure of postnatal ductus arteriosus closure in prostaglandin transporter-deficient mice. Circulation 2010, 121, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Nagasawa, K.; Akaike, T.; Oshima, T.; Kato, T.; Minamisawa, S. CCN3 secreted by prostaglandin E2 inhibits intimal cushion formation in the rat ductus arteriosus. Biochem. Biophys. Res. Commun. 2018, 503, 3242–3247. [Google Scholar] [CrossRef]
- Ugwuagbo, K.C.; Maiti, S.; Omar, A.; Hunter, S.; Nault, B.; Northam, C.; Majumder, M. Prostaglandin E2 promotes embryonic vascular development and maturation in zebrafish. Biol. Open 2019, 8, bio039768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; He, L.; Liu, Y.; Zhang, J.; Zeng, Q.; Wang, S.; Fan, Z.; Fang, F.; Chen, L.; Lv, Y.; et al. Prostaglandin E2 Is Required for BMP4-Induced Mesoderm Differentiation of Human Embryonic Stem Cells. Stem Cell Rep. 2018, 10, 905–919. [Google Scholar] [CrossRef]
- Li, P.; Pashmforoush, M.; Sucov, H.M. Retinoic Acid Regulates Differentiation of the Secondary Heart Field and TGFβ-Mediated Outflow Tract Septation. Dev. Cell 2010, 18, 480–485. [Google Scholar] [CrossRef]
- Sakabe, M.; Kokubo, H.; Nakajima, Y.; Saga, Y. Ectopic retinoic acid signaling affects outflow tract cushion development through suppression of the myocardial Tbx2-Tgfβ2 pathway. Development 2012, 139, 385–395. [Google Scholar] [CrossRef]
- Rydeen, A.B.; Waxman, J.S. Cyp26 Enzymes Facilitate Second Heart Field Progenitor Addition and Maintenance of Ventricular Integrity. PLoS Biol. 2016, 14, e2000504. [Google Scholar] [CrossRef]
- Song, Y.C.; Dohn, T.E.; Rydeen, A.B.; Nechiporuk, A.V.; Waxman, J.S. HDAC1-mediated repression of the retinoic acid-responsive gene ripply3 promotes second heart field development. PLoS Genet. 2019, 15, e1008165. [Google Scholar] [CrossRef]
- El Robrini, N.; Etchevers, H.C.; Ryckebüsch, L.; Faure, E.; Eudes, N.; Niederreither, K.; Zaffran, S.; Bertrand, N. Cardiac outflow morphogenesis depends on effects of retinoic acid signaling on multiple cell lineages. Dev. Dyn. 2016, 245, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, A.; Manova, K.; Anderson, K.V. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc. Natl. Acad. Sci. USA 1998, 95, 7485–7490. [Google Scholar] [CrossRef]
- Sugrue, K.F.; Sarkar, A.A.; Leatherbury, L.; Zohn, I.E. The ubiquitin ligase HECTD1 promotes retinoic acid signaling required for development of the aortic arch. DMM Dis. Model. Mech. 2019, 12, dmm036491. [Google Scholar] [CrossRef] [PubMed]
- Rydeen, A.B.; Waxman, J.S. Cyp26 enzymes are required to balance the cardiac and vascular lineages within the anterior lateral plate mesoderm. Development 2014, 141, 1638–1648. [Google Scholar] [CrossRef] [PubMed]
- Brade, T.; Kumar, S.; Cunningham, T.J.; Chatzi, C.; Zhao, X.; Cavallero, S.; Li, P.; Sucov, H.M.; Ruiz-Lozano, P.; Duester, G. Retinoic acid stimulates myocardial expansion by induction of hepatic erythropoietin which activates epicardial Igf2. Development 2011, 138, 139–148. [Google Scholar] [CrossRef]
- Azambuja, A.P.; Portillo-Sánchez, V.; Rodrigues, M.V.; Omae, S.V.; Schechtman, D.; Strauss, B.E.; Costanzi-Strauss, E.; Krieger, J.E.; Perez-Pomares, J.M.; Xavier-Neto, J. Retinoic acid and VEGF delay smooth muscle relative to endothelial differentiation to coordinate inner and outer coronary vessel wall morphogenesis. Circ. Res. 2010, 107, 204–216. [Google Scholar] [CrossRef]
- Lu, L.; Liu, M.; Sun, R.R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biochem. Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/reperfusion injury following acute myocardial infarction: A critical issue for clinicians and forensic pathologists. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef]
- Aguirre, A.; Sancho-Martinez, I.; Izpisua Belmonte, J.C. Reprogramming toward heart regeneration: Stem cells and beyond. Cell Stem Cell 2013, 12, 275–284. [Google Scholar] [CrossRef]
- Bar, A.; Cohen, S. Inducing Endogenous Cardiac Regeneration: Can Biomaterials Connect the Dots? Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.L.; Lee, R.T. Regeneration of the heart. EMBO Mol. Med. 2011, 3, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Braga, L.; Giacca, M. Cardiac regeneration and remodelling of the cardiomyocyte cytoarchitecture. FEBS J. 2020, 287, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Hansson, E.M.; Lindsay, M.E.; Chien, K.R. Regeneration Next: Toward Heart Stem Cell Therapeutics. Cell Stem Cell 2009, 5, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Montserrat, N.; Zacchigna, S.; Nivet, E.; Hishida, T.; Krause, M.N.; Kurian, L.; Ocampo, A.; Vazquez-Ferrer, E.; Rodriguez-Esteban, C.; et al. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 2014, 15, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Moradi, S.; Mahdizadeh, H.; Šarić, T.; Kim, J.; Harati, J.; Shahsavarani, H.; Greber, B.; Moore, J.B. Research and therapy with induced pluripotent stem cells (iPSCs): Social, legal, and ethical considerations. Stem Cell Res. Ther. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Ratajczak, J. Extracellular Microvesicles as Game Changers in Better Understanding the Complexity of Cellular Interactions—From Bench to Clinical Applications. Am. J. Med. Sci. 2017, 354, 449–452. [Google Scholar] [CrossRef]
- Uygur, A.; Lee, R.T. Mechanisms of Cardiac Regeneration. Dev. Cell 2016, 36, 362–374. [Google Scholar] [CrossRef]
- Beffagna, G. Zebrafish as a Smart Model to Understand Regeneration After Heart Injury: How Fish Could Help Humans. Front. Cardiovasc. Med. 2019, 6, 107. [Google Scholar] [CrossRef]
- Han, Y.; Chen, A.; Umansky, K.B.; Oonk, K.A.; Choi, W.Y.; Dickson, A.L.; Ou, J.; Cigliola, V.; Yifa, O.; Cao, J.; et al. Vitamin D Stimulates Cardiomyocyte Proliferation and Controls Organ Size and Regeneration in Zebrafish. Dev. Cell 2019, 48, 853.e5–863.e5. [Google Scholar] [CrossRef]
- Choi, W.Y.; Gemberling, M.; Wang, J.; Holdway, J.E.; Shen, M.C.; Karlstrom, R.O.; Poss, K.D. In vivo monitoring of cardiomyocyte proliferation to identify chemical modifiers of heart regeneration. Development 2013, 140, 660–666. [Google Scholar] [CrossRef] [PubMed]
- D’Uva, G.; Aharonov, A.; Lauriola, M.; Kain, D.; Yahalom-Ronen, Y.; Carvalho, S.; Weisinger, K.; Bassat, E.; Rajchman, D.; Yifa, O.; et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat. Cell Biol. 2015, 17, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.K.; Leisegang, M.S.; Kruse, C.; Vogel, J.; Schürmann, C.; Dehne, N.; Weigert, A.; Herrmann, E.; Brüne, B.; Shah, A.M.; et al. Vitamin D promotes vascular regeneration. Circulation 2014, 130, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Heeschen, C.; Aicher, A.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Relevance of Monocytic Features for Neovascularization Capacity of Circulating Endothelial Progenitor Cells. Circulation 2003, 108, 2511–2516. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.J.; O’Neill, C.L.; O’Doherty, T.M.; Knott, H.; Guduric-Fuchs, J.; Gardiner, T.A.; Stitt, A.W. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol. Med. 2011, 17, 1045–1055. [Google Scholar] [CrossRef]
- Duelen, R.; Sampaolesi, M. Stem Cell Technology in Cardiac Regeneration: A Pluripotent Stem Cell Promise. EBioMedicine 2017, 16, 30–40. [Google Scholar] [CrossRef]
- Santini, M.P.; Forte, E.; Harvey, R.P.; Kovacic, J.C. Developmental origin and lineage plasticity of endogenous cardiac stem cells. Development 2016, 143, 1242–1258. [Google Scholar] [CrossRef]
- Monsanto, M.M.; White, K.S.; Kim, T.; Wang, B.J.; Fisher, K.; Ilves, K.; Khalafalla, F.G.; Casillas, A.; Broughton, K.; Mohsin, S.; et al. Concurrent Isolation of 3 Distinct Cardiac Stem Cell Populations from a Single Human Heart Biopsy. Circ. Res. 2017, 121, 113–124. [Google Scholar] [CrossRef]
- Müller, P.; Lemcke, H.; David, R. Stem Cell Therapy in Heart Diseases-Cell Types, Mechanisms and Improvement Strategies. Cell. Physiol. Biochem. 2018, 48, 2607–2655. [Google Scholar] [CrossRef]
- Hsueh, Y.; Wu, J.M.F.; Yu, C.; Wu, K.K.; Hsieh, P.C.H. Prostaglandin E 2 promotes post-infarction cardiomyocyte replenishment by endogenous stem cells. EMBO Mol. Med. 2014, 6, 496–503. [Google Scholar] [CrossRef]
- Hsieh, P.C.H.; Segers, V.F.M.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Lechene, C.P.; Lee, R.T. Mammalian Heart Renewal by Preexisting Cardiomyocytes Samuel. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef]
- Muraoka, N.; Nara, K.; Tamura, F.; Kojima, H.; Yamakawa, H.; Sadahiro, T.; Miyamoto, K.; Isomi, M.; Haginiwa, S.; Tani, H.; et al. Role of cyclooxygenase-2-mediated prostaglandin E2-prostaglandin E receptor 4 signaling in cardiac reprogramming. Nat. Commun. 2019, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic Acid Production by Endocardium and Epicardium Is an Injury Response Essential for Zebrafish Heart Regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Drowley, L.; McPheat, J.; Nordqvist, A.; Peel, S.; Karlsson, U.; Martinsson, S.; Müllers, E.; Dellsén, A.; Knight, S.; Barrett, I.; et al. Discovery of retinoic acid receptor agonists as proliferators of cardiac progenitor cells through a phenotypic screening approach. Stem Cells Transl. Med. 2020, 9, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Drowley, L.; Koonce, C.; Peel, S.; Jonebring, A.; Plowright, A.T.; Kattman, S.J.; Andersson, H.; Anson, B.; Swanson, B.J.; Wang, Q.-D.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiac Progenitor Cells in Phenotypic Screening: A Transforming Growth Factor-β Type 1 Receptor Kinase Inhibitor Induces Efficient Cardiac Differentiation. Stem Cells Transl. Med. 2016, 5, 164–174. [Google Scholar] [CrossRef]
- Wang, S.; Yu, J.; Jones, J.W.; Pierzchalski, K.; Kane, M.A.; Trainor, P.A.; Xavier-Neto, J.; Moise, A.R. Retinoic acid signaling promotes the cytoskeletal rearrangement of embryonic epicardial cells. FASEB J. 2018, 32, 3765–3781. [Google Scholar] [CrossRef]
- Riley, P.R. An Epicardial Floor Plan for Building and Rebuilding the Mammalian Heart. Curr. Top. Dev. Biol. 2012, 100, 233–251. [Google Scholar]
- Krithika, S.R.; Spees, J.L. Harnessing Epicardial Progenitor Cells and Their Derivatives for Rescue and Repair of Cardiac Tissue After Myocardial Infarction. Curr. Mol. Biol. Rep. 2017, 3, 149–158. [Google Scholar]
- Smits, A.M.; Dronkers, E.; Goumans, M.J. The epicardium as a source of multipotent adult cardiac progenitor cells: Their origin, role and fate. Pharmacol. Res. 2018, 127, 129–140. [Google Scholar] [CrossRef]
- Matrone, G.; Tucker, C.S.; Denvir, M.A. Cardiomyocyte proliferation in zebrafish and mammals: Lessons for human disease. Cell. Mol. Life Sci. 2017, 74, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E.; Wang, G. Bioactive Lipids in Stem Cell Differentiation. In Embryonic Stem Cells—Differentiation and Pluripotent Alternatives; Intech Open: London, UK, 2011; pp. 33–54. [Google Scholar]
- Tucker, S.C.; Honn, K.V. Emerging targets in lipid-based therapy. Biochim. Biophys. Acta 2013, 85, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Aspects Med. 2018, 64, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.; Xu, Z.; Ji, R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Chiang, N.; Serhan, C.N. Identification of 14-series sulfido-conjugated mediators that promote resolution of infection and organ protection. Proc. Natl. Acad. Sci. USA 2014, 111, E4753–E4761. [Google Scholar] [CrossRef]
- Dalli, J.; Sanger, J.M.; Rodriguez, A.R.; Chiang, N.; Spur, B.W.; Serhan, C.N. Identification and actions of a novel third maresin conjugate in tissue regeneration: MCTR3. PLoS ONE 2016, 11, e0149319. [Google Scholar] [CrossRef]
- Dalli, J.; Vlasakov, I.; Riley, I.R.; Rodriguez, A.R.; Spur, B.W.; Petasis, N.A.; Chiang, N.; Serhan, C.N. Maresin conjugates in tissue regeneration Biosynthesis enzymes in human macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 12232–12237. [Google Scholar] [CrossRef]
- Chiang, N.; Riley, I.R.; Dalli, J.; Rodriguez, A.R.; Spur, B.W.; Serhan, C.N. New maresin conjugates in tissue regeneration pathway counters leukotriene D4–stimulated vascular responses. FASEB J. 2018, 32, 4043–4052. [Google Scholar] [CrossRef]
- Pozzi, A.; Macias-Perez, I.; Abair, T.; Wei, S.; Su, Y.; Zent, R.; Falck, J.R.; Capdevila, J.H. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J. Biol. Chem. 2005, 280, 27138–27146. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, A.; Gruenloh, S.K.; Buonaccorsi, J.N.; Zhang, R.; Gross, G.J.; Falck, J.R.; Patel, P.K.; Jacobs, E.R.; Medhora, M. Multiple anti-apoptotic targets of the PI3K-Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am. J. Physiol. Hear. Circ. Physiol. 2008, 294, H724–H735. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, H.J.M.; Van Buul-Wortelboer, M.F.; Van Mourik, J.A. Involvement of cyclooxygenase- and lipoxygenase-mediated conversion of arachidonic acid in controlling human vascular smooth muscle cell proliferation. Thromb. Haemost. 1990, 63, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Takata, S.; Papayianni, A.; Matsubara, M.; Jimenez, W.; Pronovost, P.H.; Brady, H.R. 15-Hydroxyeicosatetraenoic acid inhibits neutrophil migration across cytokine-activated endothelium. Am. J. Pathol. 1994, 145, 541–549. [Google Scholar]
- Uddin, M.R.; Muthalif, M.M.; Karzoun, N.A.; Benter, I.F.; Malik, K.U. Cytochrome P-450 Metabolites Norepinephrine-Induced Mitogenic Mediate Signaling. Hypertension 1998, 31, 242–247. [Google Scholar] [CrossRef]
- Ishizuka, T.; Cheng, J.; Singh, H.; Vitto, M.D.; Manthati, V.L.; Falck, J.R.; Laniado-Schwartzman, M. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-κB activation and the production of inflammatory cytokines in human endothelial cells. J. Pharmacol. Exp. Ther. 2008, 324, 103–110. [Google Scholar] [CrossRef]
- Powell, W.S.; Gravel, S.; Halwani, F. 5-oxo-6,8,11,14-eicosatetraenoic acid is a potent stimulator of L-selectin shedding, surface expression of CD11b, actin polymerization, and calcium mobilization in human eosinophils. Am. J. Respir. Cell Mol. Biol. 1999, 20, 163–170. [Google Scholar] [CrossRef]
- Wei, C.; Zhu, P.; Shah, S.J.; Blair, I.A. 15-Oxo-Eicosatetraenoic Acid, a Metabolite of Macrophage 15-Hydroxyprostaglandin Dehydrogenase That Inhibits Endothelial Cell Proliferation. Mol. Pharmacol. 2009, 76, 516–525. [Google Scholar] [CrossRef]
- Endo, J.; Sano, M.; Isobe, Y.; Fukuda, K.; Kang, J.X.; Arai, H.; Arita, M. 18-HEPE, an n-3 fatty acid metabolite released by macrophages, prevents pressure overload-induced maladaptive cardiac remodeling. J. Exp. Med. 2014, 211, 1673–1687. [Google Scholar] [CrossRef]
- Tian, H.; Lu, Y.; Shah, S.P.; Hong, S. Novel 14S, 21-dihydroxy-docosahexaenoic acid Rescues Wound Healing and Associated Angiogenesis Impaired by Acute Ethanol Intoxication/Exposure. J. Cell Biochem. 2010, 111, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Edin, M.L.; Wang, Z.; Bradbury, J.A.; Graves, J.P.; Lih, F.B.; DeGraff, L.M.; Foley, J.F.; Torphy, R.; Ronnekleiv, O.K.; Tomer, K.B.; et al. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011, 25, 3436–3447. [Google Scholar] [CrossRef] [PubMed]
- Mrsny, R.J.; Gewirtz, A.T.; Siccardi, D.; Savidge, T.; Hurley, B.P.; Madara, J.L.; McCormick, B.A. Identification of hepoxilin A3 in inflammatory events: A required role in neutrophil migration across intestinal epithelia. Proc. Natl. Acad. Sci. USA 2004, 101, 7421–7426. [Google Scholar] [CrossRef]
- Nigam, S.; Fiore, S.; Luscinskas, F.W.; Serhan, C.N. Lipoxin A, and Lipoxin B, Stimulate the Release but Not the Oxygenation of Arachidonic Acid in Human Neutrophils: Dissociation Between Lipid Remodeling and Adhesion. J. Cell. Physiol. 1990, 143, 512–523. [Google Scholar] [CrossRef]
- Morinelli, T.A.; Zhang, L.M.; Newman, W.H.; Meier, K.E. Thromboxane A2/prostaglandin H2-stimulated mitogenesis of coronary artery smooth muscle cells involves activation of mitogen-activated protein kinase and S6 kinase. J. Biol. Chem. 1994, 269, 5693–5698. [Google Scholar] [PubMed]
- Stephenson, D.J.; Hoeferlin, L.A.; Chalfant, C.E. Lipidomics in translational research and the clinical significance of lipid-based biomarkers. Transl. Res. 2017, 189, 13–29. [Google Scholar] [CrossRef]
- Lydic, T.A.; Goo, Y.-H. Lipidomics unveils the complexity of the lipidome in metabolic diseases. Clin. Transl. Med. 2018, 7, 1–13. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxylipins in Cardiovascular Disease | ||
|---|---|---|
| Lipid | Beneficial Function | Reference |
| 11,12-EET | Suppresses cardiac hypertrophy and increases ANP levels in mouse hearts Prevents high-fat diet-induced atherosclerosis in mice | [200] [201] |
| 14,15-EET | Decreases ventricular pressure and reduces pulmonary artery wall thickness in rats | [204] |
| CYP2J-Derived EETs | Improves left ventricular function and reduces collagen accumulation after MI in mice | [193] |
| sEH Inhibitors | Prevent ischemia-reperfusion injury in rat heart isolates | [203] |
| 19-HETE | Ameliorates angiotensin II-induced cardiac hypertrophy in rats Reduces platelet aggregation in mouse blood vessels | [196] [198] |
| 15-epi-lipoxin A4 | Improves ejection fraction and facilitates neutrophil clearance after coronary artery ligation in mice Reduces intimal hyperplasia after carotid artery ligation in mice | [192] [207] |
| ATL A4 | Prevents atherosclerotic lesions in rat aorta by resolving inflammation | [206] |
| EPA/DHA | Lower serum triglyceride levels in patients Reduce adverse ventricular remodeling and myocardial fibrosis in patients Reduce the incidence of ischemia, severe cardiac events, and cardiovascular death in patients Decrease fibrosis accumulation after MI in rat hearts | [185] [186] [187] [189] |
| RvDn-3 DPA | Decreases leukocyte and platelet activation in patient peripheral blood samples | [188] |
| RvD1 | Improves left ventricular function, promotes resolution of inflammation, and reduces collagen deposition after MI in mice Decreases oxidative stress, fibrosis, and necrosis and promotes the stability of atherosclerotic plaques in mice | [190] [194] |
| RvD4 | Improves thrombus resolution in mice | [195] |
| PD1 | Reduces inflammatory cell infiltration and neointimal hyperplasia after carotid artery injury in rats | [209] |
| MaR1 | Reduces necrosis and promotes the stability of atherosclerotic plaques in mice | [210] |
| Lipid | Detrimental Function | Reference |
| 16-HETrE | Increases blood pressure in patients | [211] |
| 5,6-diHETrE | Increases blood pressure in patients | [211] |
| TXB2 | Increases blood pressure in patients | [211] |
| LTB4 | Antagonism reduces infarct size and inflammatory cell accumulation after coronary artery ligation in mice Inhibition increases the stability of atherosclerotic plaques in mice | [214] [217] |
| LTC4 | Receptor inhibition increases ejection fraction and myocardial mass after cardiac cryoinjury in mice | [215] |
| Abbreviations: ANP, atrial natriuretic peptide; ATL, aspirin-triggered lipoxin; CYP, cytochrome P450 enzyme; DHA, docosahexaenoic acid; diHETrE, dihydroxyeicosatrienoic acid; EET, epoxyeicosatrienoic acid; EPA, eicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; HETrE, hydroxyeicosatrienoic acid; LT, leukotriene; MaR, maresin; MI, myocardial infarction; PD, protectin; RV, resolvin; sEH, soluble epoxide hydrolase; TX, thromboxane | ||
| Oxylipins in Cardiovascular Regeneration | ||
|---|---|---|
| Lipid | Function | Reference |
| 5,6-EET 8,9-EET | Stimulate proliferation in murine microvascular endothelial cells and angiogenesis in mice | [315] |
| 8,9-EET | Attenuates apoptosis in primary rat cardiac myocytes after hypoxia and reoxygenation | [316] |
| 11-HETE | Inhibits proliferation of human vascular smooth muscle cells | [317] |
| 15-HETE | Inhibits PMN migration across cytokine-activated human endothelium cells in culture | [318] |
| 20-HETE | Stimulates proliferation of rat aorta vascular smooth muscle cells Stimulates inflammatory cytokine production in human vascular endothelial cells | [319] [320] |
| 5-oxo-ETE | Stimulates human eosinophil migration | [321] |
| 15-oxo-ETE | Inhibits human vascular vein endothelial cell proliferation | [322] |
| 18-HEPE | Inhibits macrophage-mediated activation of murine cardiac fibroblasts and prevents pressure overload-induced cardiac fibrosis and inflammation in mice | [323] |
| 14S,21-diHDHA | Enhances wound healing in murine models | [324] |
| 9,10-diHOME | Decreases left ventricular developed pressure and increases coronary resistance after ischemia/reperfusion injury in mice | [325] |
| HxA3 | Recruits human PMN to sites of inflammation | [326] |
| LxA4 LxB4 | Stimulate phospholipid remodeling without causing aggregation in human neutrophils | [327] |
| PGE2 | Enhances cardiomyocyte replenishment after MI in young mice and rescues cardiomyocyte replenishment after MI in aged mice | [292] |
| Inhibition enhances reprogramming of mouse tail-tip fibroblasts to cardiac cells | [295] | |
| TxA2 | Stimulates mitogenesis of guinea-pig coronary artery smooth muscle cells | [328] |
| MaR1 | Accelerates planarian regeneration, increases human macrophage efferocytosis, decreases PMN infiltration in mice | [310] |
| MCTR1,2,3 | Stimulate planarian tissue regeneration, increase macrophage efferocytosis, reduce neutrophil infiltration, counteract leukotriene effects on the heart | [311,312,314] |
| Abbreviations: diHDHA, dihydroxydocosahexaenoic acid; diHOME, dihydroxyoctadecenoic acid; EET, epoxyeicosatrienoic acid; HEPE, hydroxyeicosapentaenoic acid; HETE, hydroxyeicosatetraenoic acid; Hx, hepoxilin; Lx, lipoxin; MaR, maresin; MCTR, maresin conjugate in tissue regeneration; MI, myocardial infarction; oxo-ETE, oxo-eicosatetraenoic acid; PG, prostaglandin; PMN, polymorphonuclear leukocyte; Tx, thromboxane | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasserman, A.H.; Venkatesan, M.; Aguirre, A. Bioactive Lipid Signaling in Cardiovascular Disease, Development, and Regeneration. Cells 2020, 9, 1391. https://doi.org/10.3390/cells9061391
Wasserman AH, Venkatesan M, Aguirre A. Bioactive Lipid Signaling in Cardiovascular Disease, Development, and Regeneration. Cells. 2020; 9(6):1391. https://doi.org/10.3390/cells9061391
Chicago/Turabian StyleWasserman, Aaron H., Manigandan Venkatesan, and Aitor Aguirre. 2020. "Bioactive Lipid Signaling in Cardiovascular Disease, Development, and Regeneration" Cells 9, no. 6: 1391. https://doi.org/10.3390/cells9061391
APA StyleWasserman, A. H., Venkatesan, M., & Aguirre, A. (2020). Bioactive Lipid Signaling in Cardiovascular Disease, Development, and Regeneration. Cells, 9(6), 1391. https://doi.org/10.3390/cells9061391