Orthotopic T-Cell Receptor Replacement—An “Enabler” for TCR-Based Therapies


{kind=link}
{kind=link}
{kind=link}
Abstract
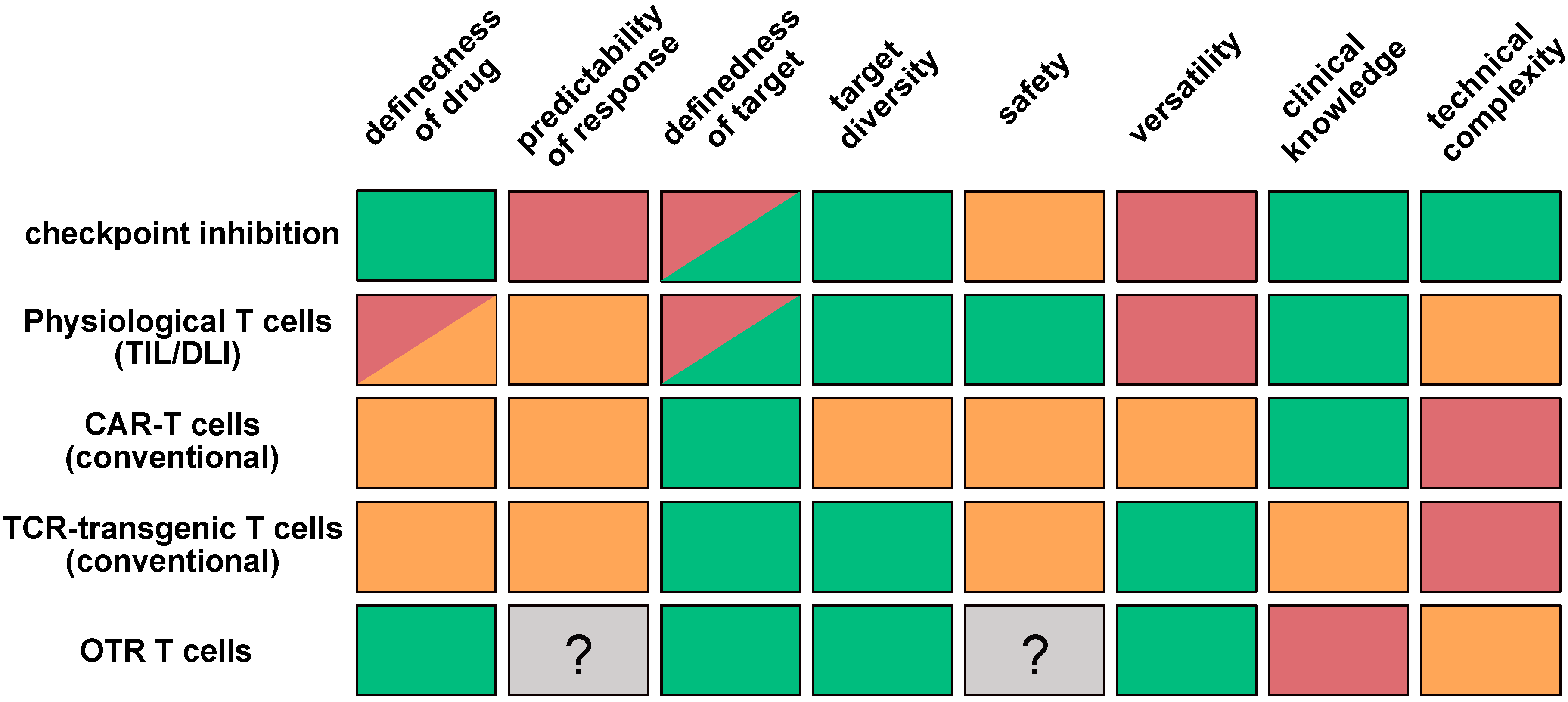
1. Introduction
2. Role of the Remaining Endogenous TCR
2.1. TCR Mispairing
2.2. Risks and Opportunities of Multiplexed Gene Editing
2.3. Deliberate Conservation of Endogenous TCRs
3. True TCR Replacement through Orthotopic Editing
3.1. OTR Enables Engineering of Near-Physiological T Cells
3.2. TCR Regulation
3.3. Defined Antigen Receptor Expression through OTR
3.4. Functional Consequences of OTR
3.5. OTR Mouse Models
3.6. Defined TCR Insertion outside the Endogenous TCR Locus
4. Technology Development
4.1. gRNA Cas9 Ribonucleoproteins
4.2. Off-Target Effects
4.3. Non-Viral DNA Template Delivery
4.4. DNA Template Design
4.4.1. Gene Locus
4.4.2. Homology Arms
4.4.3. Cutting Site
4.4.4. Splice Donor/Acceptor
4.4.5. Transgenic TCR Constant Regions
4.5. HDR Enhancement
5. Toward Clinical Application of OTR
5.1. GMP Process
5.2. Cell Phenotype and Activation
5.3. Cell Numbers and Selection
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Price, D.A.; West, S.M.; Betts, M.R.; Ruff, L.E.; Brenchley, J.M.; Ambrozak, D.R.; Edghill-Smith, Y.; Kuroda, M.J.; Bogdan, D.; Kunstman, K.; et al. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity 2004, 21, 793–803. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Riddell, S.R.; Watanabe, K.S.; Goodrich, J.M.; Li, C.R.; Agha, M.E.; Greenberg, P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science 1992, 257, 238–241. [Google Scholar] [CrossRef]
- Cobbold, M.; Khan, N.; Pourgheysari, B.; Tauro, S.; McDonald, D.; Osman, H.; Assenmacher, M.; Billingham, L.; Steward, C.; Crawley, C.; et al. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. J. Exp. Med. 2005, 202, 379–386. [Google Scholar] [CrossRef]
- Neuenhahn, M.; Albrecht, J.; Odendahl, M.; Schlott, F.; Dössinger, G.; Schiemann, M.; Lakshmipathi, S.; Martin, K.; Bunjes, D.; Harsdorf, S.; et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia 2017, 31, 2161–2171. [Google Scholar] [CrossRef]
- Dembić, Z.; Haas, W.; Weiss, S.; Mccubrey, J.; Kiefer, H.; von Boehmer, H.; Steinmetz, M. Transfer of specificity by murine α and β T-cell receptor genes. Nature 1986, 320, 232–238. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef]
- Chapuis, A.G.; Egan, D.N.; Bar, M.; Schmitt, T.M.; McAfee, M.S.; Paulson, K.G.; Voillet, V.; Gottardo, R.; Ragnarsson, G.B.; Bleakley, M.; et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat. Med. 2019, 25, 1064–1072. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 153303381983106. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2018, 173–200. [Google Scholar] [CrossRef]
- Engels, B.; Cam, H.; Schüler, T.; Indraccolo, S.; Gladow, M.; Baum, C.; Blankenstein, T.; Uckert, W. Retroviral Vectors for High-Level Transgene Expression in T Lymphocytes. Hum. Gene Ther. 2003, 14, 1155–1168. [Google Scholar] [CrossRef]
- Thomas, S.; Xue, S.-A.; Cesco-Gaspere, M.; San Jose, E.; Hart, D.P.; Wong, V.; Debets, R.; Alarcon, B.; Morris, E.; Stauss, H.J. Targeting the Wilms Tumor Antigen 1 by TCR Gene Transfer: TCR Variants Improve Tetramer Binding but Not the Function of Gene Modified Human T Cells. J. Immunol. 2007, 179, 5803–5810. [Google Scholar] [CrossRef]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; de Witte, M.A.; Jorritsma, A.; Kaiser, A.D.M.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570. [Google Scholar] [CrossRef]
- Van Loenen, M.M.; de Boer, R.; Amir, A.L.; Hagedoorn, R.S.; Volbeda, G.L.; Willemze, R.; van Rood, J.J.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 10972–10977. [Google Scholar] [CrossRef]
- Schrum, A.G.; Turka, L.A.; Palmer, E. Surface T-cell antigen receptor expression and availability for long-term antigenic signaling. Immunol. Rev. 2003, 196, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, A.M.; Xiong, H.; Leiner, I.M.; Sušac, B.; Glickman, M.S.; Pamer, E.G.; van Heijst, J.W.J. Control of T cell antigen reactivity via programmed TCR downregulation. Nat. Immunol. 2016, 17, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Van Loenen, M.M.; Hagedoorn, R.S.; De Boer, R.; Van Egmond, E.H.M.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Rapid re-expression of retrovirally introduced versus endogenous TCRs in engineered T cells afterantigen-specific stimulation. J. Immunother. 2011, 34, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Schober, K.; Müller, T.R.; Gökmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Ahmadi, M.; King, J.W.; Xue, S.-A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Mineno, J.; Ikeda, H.; Fujiwara, H.; Yasukawa, M.; Shiku, H.; Kato, I. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009, 69, 9003–9011. [Google Scholar] [CrossRef]
- Bunse, M.; Bendle, G.M.; Linnemann, C.; Bies, L.; Schulz, S.; Schumacher, T.N.; Uckert, W. RNAi-mediated TCR Knockdown Prevents Autoimmunity in Mice Caused by Mixed TCR Dimers Following TCR Gene Transfer. Mol. Ther. 2014, 22, 1983–1991. [Google Scholar] [CrossRef]
- Stanislawski, T.; Voss, R.H.; Lotz, C.; Sadovnikova, E.; Willemsen, R.A.; Kuball, J.; Ruppert, T.; Bolhuis, R.L.H.; Melief, C.J.; Huber, C.; et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat. Immunol. 2001, 2, 962–970. [Google Scholar] [CrossRef]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.-H.; Fowler, C.; Greenberg, P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef]
- Voss, R.-H.; Thomas, S.; Pfirschke, C.; Hauptrock, B.; Klobuch, S.; Kuball, J.; Grabowski, M.; Engel, R.; Guillaume, P.; Romero, P.; et al. Coexpression of the T-cell receptor constant alpha domain triggers tumor reactivity of single-chain TCR-transduced human T cells. Blood 2010, 115, 5154–5163. [Google Scholar] [CrossRef]
- Bethune, M.T.; Gee, M.H.; Bunse, M.; Lee, M.S.; Gschweng, E.H.; Pagadala, M.S.; Zhou, J.; Cheng, D.; Heath, J.R.; Kohn, D.B.; et al. Domain-swapped t cell receptors improve the safety of TCR gene therapy. Elife 2016, 5, 1–24. [Google Scholar] [CrossRef]
- Govers, C.; Sebestyén, Z.; Roszik, J.; van Brakel, M.; Berrevoets, C.; Szöőr, Á.; Panoutsopoulou, K.; Broertjes, M.; Van, T.; Vereb, G.; et al. TCRs Genetically Linked to CD28 and CD3ε Do Not Mispair with Endogenous TCR Chains and Mediate Enhanced T Cell Persistence and Anti-Melanoma Activity. J. Immunol. 2014, 193, 5315–5326. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Mohammed, F.; Reijmers, R.M.; Woolston, A.; Stauss, T.; Kennedy, A.; Stirling, D.; Holler, A.; Green, L.; Jones, D.; et al. Framework engineering to produce dominant T cell receptors with enhanced antigen-specific function. Nat. Commun. 2019, 10, 4451. [Google Scholar] [CrossRef] [PubMed]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Genovese, P.; Magnani, Z.; Ruggiero, E.; Landoni, E.; Camisa, B.; Schiroli, G.; Provasi, E.; Lombardo, A.; Reik, A.; et al. NY-ESO-1 TCR single edited stem and central memory T cells to treat multiple myeloma without graft-versus-host disease. Blood 2017, 130, 606–618. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 522, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Stenger, D.; Stief, T.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Binje, V.; Lotfi, R.; Wagner, B.; Grünewald, T.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020. [Google Scholar]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef]
- Schumann, K.; Lin, S.; Boyer, E.; Simeonov, D.R.; Subramaniam, M.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Bluestone, J.A.; Doudna, J.A.; et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Alcover, A.; Alarcón, B.; Di Bartolo, V. Cell Biology of T Cell Receptor Expression and Regulation. Annu. Rev. Immunol. 2018, 36, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Lanzavecchia, A. T cell activation determined by T cell receptor number and tunable thresholds. Science 1996, 273, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, M.H.M.; Hagedoorn, R.S.; van der Hoorn, M.A.W.G.; van der Veken, L.T.; Hoogeboom, M.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood 2007, 109, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Uckert, W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef]
- Gutierrez-Arcelus, M.; Baglaenko, Y.; Arora, J.; Hannes, S.; Luo, Y.; Amariuta, T.; Teslovich, N.; Rao, D.A.; Ermann, J.; Jonsson, A.H.; et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat. Genet. 2020, 52, 247–253. [Google Scholar] [CrossRef]
- D’Ippolito, E.; Schober, K.; Nauerth, M.; Busch, D.H. T cell engineering for adoptive T cell therapy: Safety and receptor avidity. Cancer Immunol. Immunother. 2019, 68, 1701–1712. [Google Scholar] [CrossRef]
- Hebeisen, M.; Allard, M.; Gannon, P.O.; Schmidt, J.; Speiser, D.E.; Rufer, N. Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Front. Immunol. 2015, 6, 582. [Google Scholar] [CrossRef]
- Kisielow, P.; Blüthmann, H.; Staerz, U.D.; Steinmetz, M.; von Boehmer, H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 1988, 333, 730–733. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Barnden, M.J.; Allison, J.; Heath, W.R.; Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998, 76, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Pircher, H.; Bürki, K.; Lang, R.; Hengartner, H.; Zinkernagel, R.M. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 1989, 342, 559–561. [Google Scholar] [CrossRef]
- Oxenius, A.; Bachmann, M.F.; Zinkernagel, R.M.; Hengartner, H. Virus-specific MHC-class II-restricted TCR-transgenic mice: Effects on humoral and cellular immune responses after viral infection. Eur. J. Immunol. 1998, 28, 390–400. [Google Scholar] [CrossRef]
- Seder, R.A.; Paul, W.E.; Davis, M.M.; Fazekas de St Groth, B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 1992, 176, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, Y.; Uno, Y.; Yoshimi, K.; Kunihiro, Y.; Yoshimura, T.; Tanaka, T.; Ishikubo, H.; Hiraoka, Y.; Takemoto, N.; Tanaka, T.; et al. CLICK: One-step generation of conditional knockout mice. BMC Genom. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Quadros, R.M.; Miura, H.; Harms, D.W.; Akatsuka, H.; Sato, T.; Aida, T.; Redder, R.; Richardson, G.P.; Inagaki, Y.; Sakai, D.; et al. Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017, 18, 92. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Graf, R.; Sommermann, T.; Petsch, K.; Sack, U.; Volchkov, P.; Rajewsky, K.; Kühn, R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016, 16, 4. [Google Scholar] [CrossRef]
- Tröder, S.E.; Ebert, L.K.; Butt, L.; Assenmacher, S.; Schermer, B.; Zevnik, B. An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS ONE 2018, 13, e0196891. [Google Scholar] [CrossRef]
- Wefers, B.; Bashir, S.; Rossius, J.; Wurst, W.; Kühn, R. Gene editing in mouse zygotes using the CRISPR/Cas9 system. Methods 2017, 121–122, 55–67. [Google Scholar] [CrossRef]
- Teixeira, M.; Py, B.F.; Bosc, C.; Laubreton, D.; Moutin, M.-J.; Marvel, J.; Flamant, F.; Markossian, S. Electroporation of mice zygotes with dual guide RNA/Cas9 complexes for simple and efficient cloning-free genome editing. Sci. Rep. 2018, 8, 474. [Google Scholar] [CrossRef] [PubMed]
- Simeonov, D.R.; Marson, A. CRISPR-Based Tools in Immunity. Annu. Rev. Immunol. 2019, 37, 571–597. [Google Scholar] [CrossRef] [PubMed]
- Beil-Wagner, J.; Dössinger, G.; Schober, K.; vom Berg, J.; Tresch, A.; Grandl, M.; Palle, P.; Mair, F.; Gerhard, M.; Becher, B.; et al. T cell-specific inactivation of mouse CD2 by CRISPR/Cas9. Sci. Rep. 2016, 6, 21377. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.; Vignali, K.M.; Burton, A.R.; Vignali, D.A.A. Rapid analysis of T-cell selection in vivo using T cell-receptor retrogenic mice. Nat. Methods 2006, 3, 191–197. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef]
- Sachdeva, M.; Busser, B.W.; Temburni, S.; Jahangiri, B.; Gautron, A.S.; Maréchal, A.; Juillerat, A.; Williams, A.; Depil, S.; Duchateau, P.; et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Domínguez-Andrés, J.; Netea, M.G. Impact of Historic Migrations and Evolutionary Processes on Human Immunity. Trends Immunol. 2019, 40, 1105–1119. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2015, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Heisenberg, W. Über den anschaulichen Inhalt der quantentheoretischen Kinematik und Mechanik. Zeitschrift für Phys. 1927, 43, 172–198. [Google Scholar] [CrossRef]
- De Vree, P.J.P.; de Wit, E.; Yilmaz, M.; van de Heijning, M.; Klous, P.; Verstegen, M.J.A.M.; Wan, Y.; Teunissen, H.; Krijger, P.H.L.; Geeven, G.; et al. Targeted sequencing by proximity ligation for comprehensive variant detection and local haplotyping. Nat. Biotechnol. 2014, 32, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Poirot, L.; Philip, B.; Schiffer-Mannioui, C.; Le Clerre, D.; Chion-Sotinel, I.; Derniame, S.; Bas, C.; Potrel, P.; Lemaire, L.; Duclert, A.; et al. Multiplex genome edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015, 75, 3853–3864. [Google Scholar] [CrossRef]
- Qasim, W.; Amrolia, P.J.; Samarasinghe, S.; Ghorashian, S.; Zhan, H.; Stafford, S.; Butler, K.; Ahsan, G.; Gilmour, K.; Adams, S.; et al. First Clinical Application of Talen Engineered Universal CAR19 T Cells in B-ALL. Blood 2015, 126, 2046. [Google Scholar] [CrossRef]
- Webber, B.R.; Lonetree, C.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.S.; et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019, 10, 5222. [Google Scholar] [CrossRef]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.-T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra53. [Google Scholar] [CrossRef]
- Schober, K.; Buchholz, V.R.; Busch, D.H. TCR repertoire evolution during maintenance of CMV-specific T-cell populations. Immunol. Rev. 2018, 283, 113–128. [Google Scholar] [CrossRef]
- Schober, K.; Voit, F.; Grassmann, S.; Müller, T.R.; Eggert, J.; Jarosch, S.; Weißbrich, B.; Hoffmann, P.; Borkner, L.; Nio, E.; et al. Reverse TCR repertoire evolution toward dominant low-affinity clones during chronic CMV infection. Nat. Immunol. 2020. [Google Scholar] [CrossRef]
- Richardson, C.D.; Ray, G.J.; DeWitt, M.A.; Curie, G.L.; Corn, J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016, 34, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, Y.; Tian, Q.; Lan, Y.; Yeh, H.; Zhang, M.; Tasan, I.; Jain, S.; Zhao, H. An efficient gene knock-in strategy using 5′-modified double-stranded DNA donors with short homology arms. Nat. Chem. Biol. 2019, 16, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Maricic, T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat. Commun. 2018, 9, 2164. [Google Scholar] [CrossRef] [PubMed]
- Chicaybam, L.; Sodre, A.L.; Curzio, B.A.; Bonamino, M.H. An Efficient Low Cost Method for Gene Transfer to T Lymphocytes. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 1–7. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2015, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Invest. 2005, 115, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Aksoy, B.A.; Czech, E.; Hammerbacher, J. Electroporation characteristics of human primary T cells. bioRxiv 2019. [Google Scholar] [CrossRef]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8+ Central Memory T Cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef]
- Stemberger, C.; Huster, K.M.; Koffler, M.; Anderl, F.; Schiemann, M.; Wagner, H.; Busch, D.H. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity 2007, 27, 985–997. [Google Scholar] [CrossRef]
- Stemberger, C.; Graef, P.; Odendahl, M.; Albrecht, J.; Dössinger, G.; Anderl, F.; Buchholz, V.R.; Gasteiger, G.; Schiemann, M.; Grigoleit, G.U.; et al. Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood 2014, 124, 628–637. [Google Scholar] [CrossRef]
- Buchholz, V.R.; Schumacher, T.N.M.; Busch, D.H. T Cell Fate at the Single-Cell Level. Annu. Rev. Immunol. 2016, 34, 65–92. [Google Scholar] [CrossRef]
- Buchholz, V.R.; Flossdorf, M.; Hensel, I.; Kretschmer, L.; Weissbrich, B.; Gräf, P.; Verschoor, A.; Schiemann, M.; Höfer, T.; Busch, D.H. Disparate individual fates compose robust CD8+ T cell immunity. Science 2013, 340, 630–635. [Google Scholar] [CrossRef]
- Stemberger, C.; Dreher, S.; Tschulik, C.; Piossek, C.; Bet, J.; Yamamoto, T.N.; Schiemann, M.; Neuenhahn, M.; Martin, K.; Schlapschy, M.; et al. Novel Serial Positive Enrichment Technology Enables Clinical Multiparameter Cell Sorting. PLoS ONE 2012, 7, e35798. [Google Scholar] [CrossRef]
- Mohr, F.; Fischer, J.C.; Nikolaus, M.; Stemberger, C.; Dreher, S.; Verschoor, A.; Haas, T.; Poeck, H.; Busch, D.H. Minimally manipulated murine regulatory T cells purified by reversible Fab Multimers are potent suppressors for adoptive T cell therapy. Eur. J. Immunol. 2017, 47, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Knabel, M.; Franz, T.J.; Schiemann, M.; Wulf, A.; Villmow, B.; Schmidt, B.; Bernhard, H.; Wagner, H.; Busch, D.H. Reversible MHC multimer staining for functional isolation of T-cell populations and effective adoptive transfer. Nat. Med. 2002, 8, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.; Skerra, A. Mutagenesis of a flexible loop in streptavidin leads to higher affinity for the Strep-tag II peptide and improved performance in recombinant protein purification. Protein Eng. 1997, 10, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.G.M.; Skerra, A. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2007, 2, 1528–1535. [Google Scholar] [CrossRef]
- Liu, L.; Sommermeyer, D.; Cabanov, A.; Kosasih, P.; Hill, T.; Riddell, S.R. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat. Biotechnol. 2016, 34, 430–434. [Google Scholar] [CrossRef]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 2016, 126, 4262–4272. [Google Scholar] [CrossRef]
- Hartweger, H.; McGuire, A.T.; Horning, M.; Taylor, J.J.; Dosenovic, P.; Yost, D.; Gazumyan, A.; Seaman, M.S.; Stamatatos, L.; Jankovic, M.; et al. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 2019, 216, 1301–1310. [Google Scholar] [CrossRef]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Nahmad, A.A.D.; Horovitz-fried, M.; Sofer, I.; Akriv, T.; Nataf, D. B cells engineered to express an anti-HIV antibody allow memory retention, class switch recombination and somatic hypermutation in mice. bioRxiv 2020, 1–19. [Google Scholar]
- Deli Huang, A.; Abbott, R.K.; Havenar-Daughton, C.; Skog, P.D.; Al-Kolla, R.; Groschel, B.; Blane, T.R.; Menis, S.; Tuyet Tran, J.; Thinnes, T.C.; et al. B cells expressing authentic naive human VRC01-class BCRs can be primed and recruited to germinal centers in multiple independent mouse models. bioRxiv 2020, 1–25. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schober, K.; Müller, T.R.; Busch, D.H. Orthotopic T-Cell Receptor Replacement—An “Enabler” for TCR-Based Therapies. Cells 2020, 9, 1367. https://doi.org/10.3390/cells9061367
Schober K, Müller TR, Busch DH. Orthotopic T-Cell Receptor Replacement—An “Enabler” for TCR-Based Therapies. Cells. 2020; 9(6):1367. https://doi.org/10.3390/cells9061367
Chicago/Turabian StyleSchober, Kilian, Thomas R. Müller, and Dirk H. Busch. 2020. "Orthotopic T-Cell Receptor Replacement—An “Enabler” for TCR-Based Therapies" Cells 9, no. 6: 1367. https://doi.org/10.3390/cells9061367
APA StyleSchober, K., Müller, T. R., & Busch, D. H. (2020). Orthotopic T-Cell Receptor Replacement—An “Enabler” for TCR-Based Therapies. Cells, 9(6), 1367. https://doi.org/10.3390/cells9061367