Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis


 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
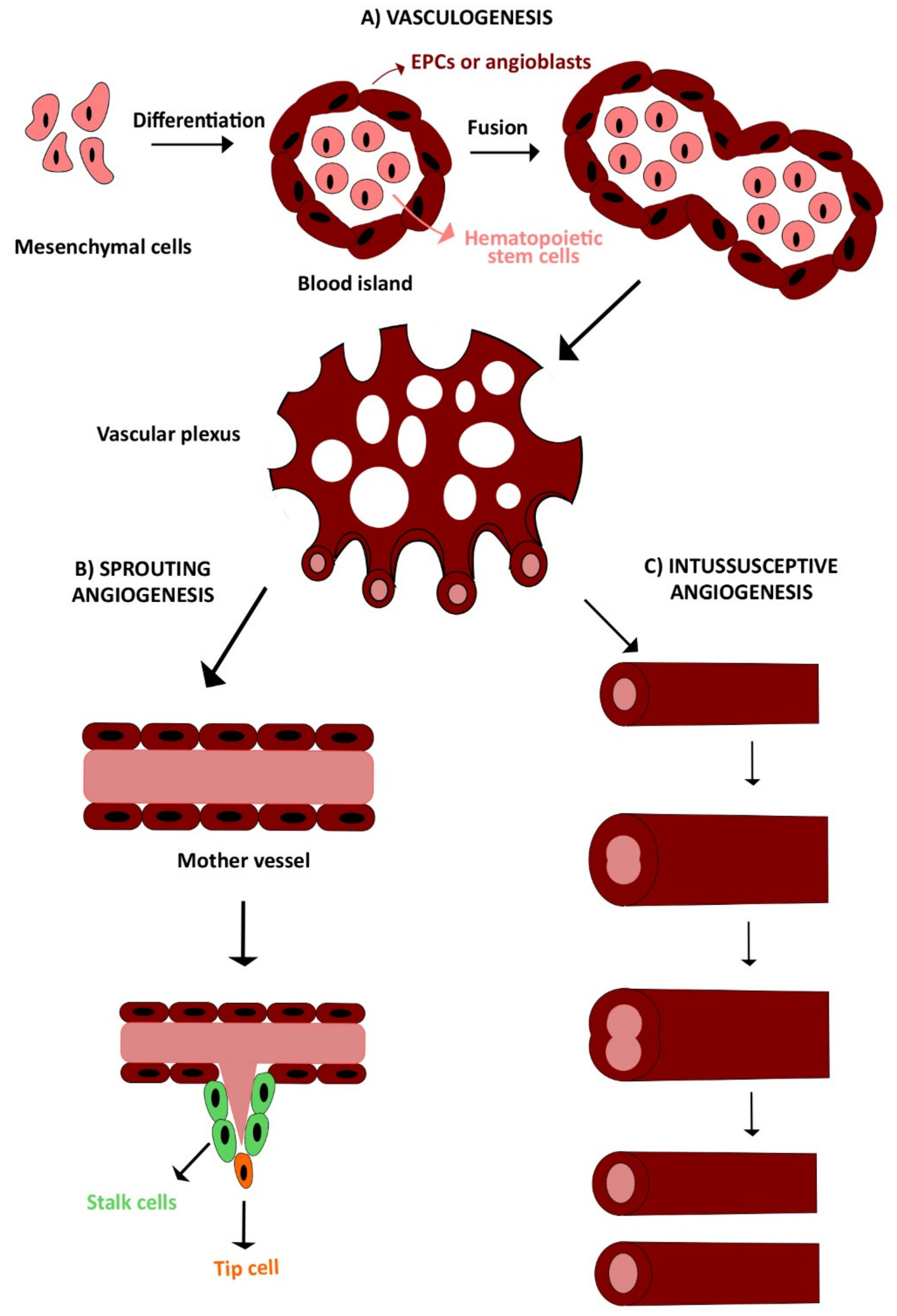
2. Vasculogenesis and Angiogenesis
2.1. Vasculogenesis
2.2. Angiogenesis
2.3. Definition of Endothelial Progenitor Cells (EPCs) in the Adult: Myeloid Angiogenic Cells (MACs) and Endothelial Colony Forming Cells (ECFCs)
2.4. The Signaling Pathways of Angiogenesis and Vasculogenesis: The Role of Endothelial Ca2+ Signaling
3. TRPV1: Molecular Structure and Gating Mechanisms
3.1. TRPV1 Structure
3.2. TRPV1: Biophysical Properties and Gating Mechanisms
4. The Physiological Role of TRPV1 in Vascular Endothelium
4.1. TRPV1 Mediates Endothelium-Dependent Vasodilation
4.2. TRPV1 Ameliorates Endothelial Dysfunction in Diabetes, Atherosclerosis, and Metabolic Syndrome
4.3. TRPV1 Regulates the Blood–Brain Barrier (BBB) Integrity
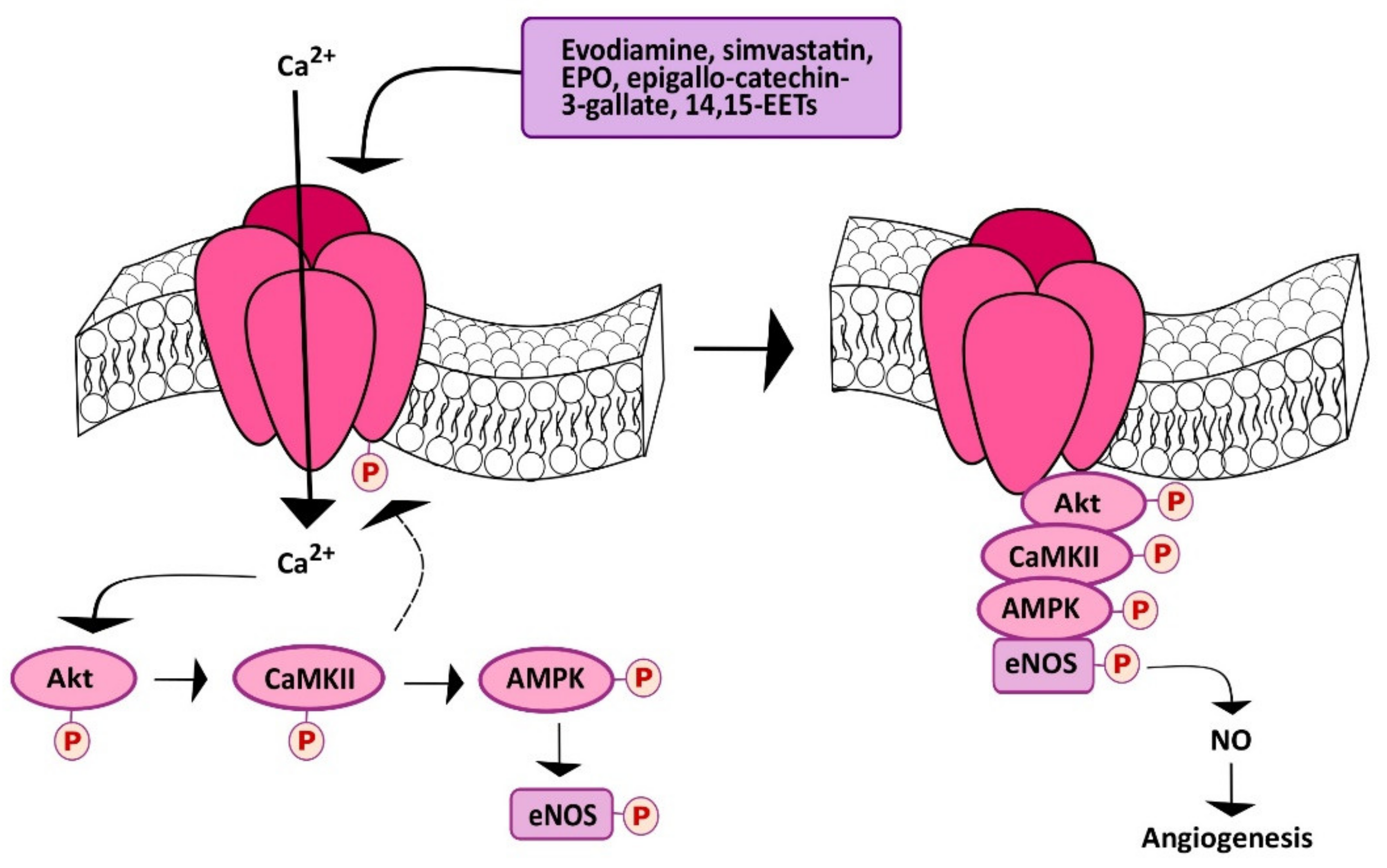
5. Stimulating TRPV1 to Promote Angiogenesis
5.1. TRPV1 Sustains Angiogenesis Independently on VEGF
5.2. TRPV1 Stimulates Re-Endothelialization Following Vascular Injury
5.3. Chemical Stimulation of TRPV1 Promotes Angiogenesis in a Ca2+-Dependent Manner
5.4. Is There a Role for TRPV1 in Heat-Induced Angiogenesis?
6. TRPV1 Controls the Angiogenic Activity in ECFCs
6.1. TRPV1 may Stimulate ECFC Proliferation in a Ca2+-Independent Manner
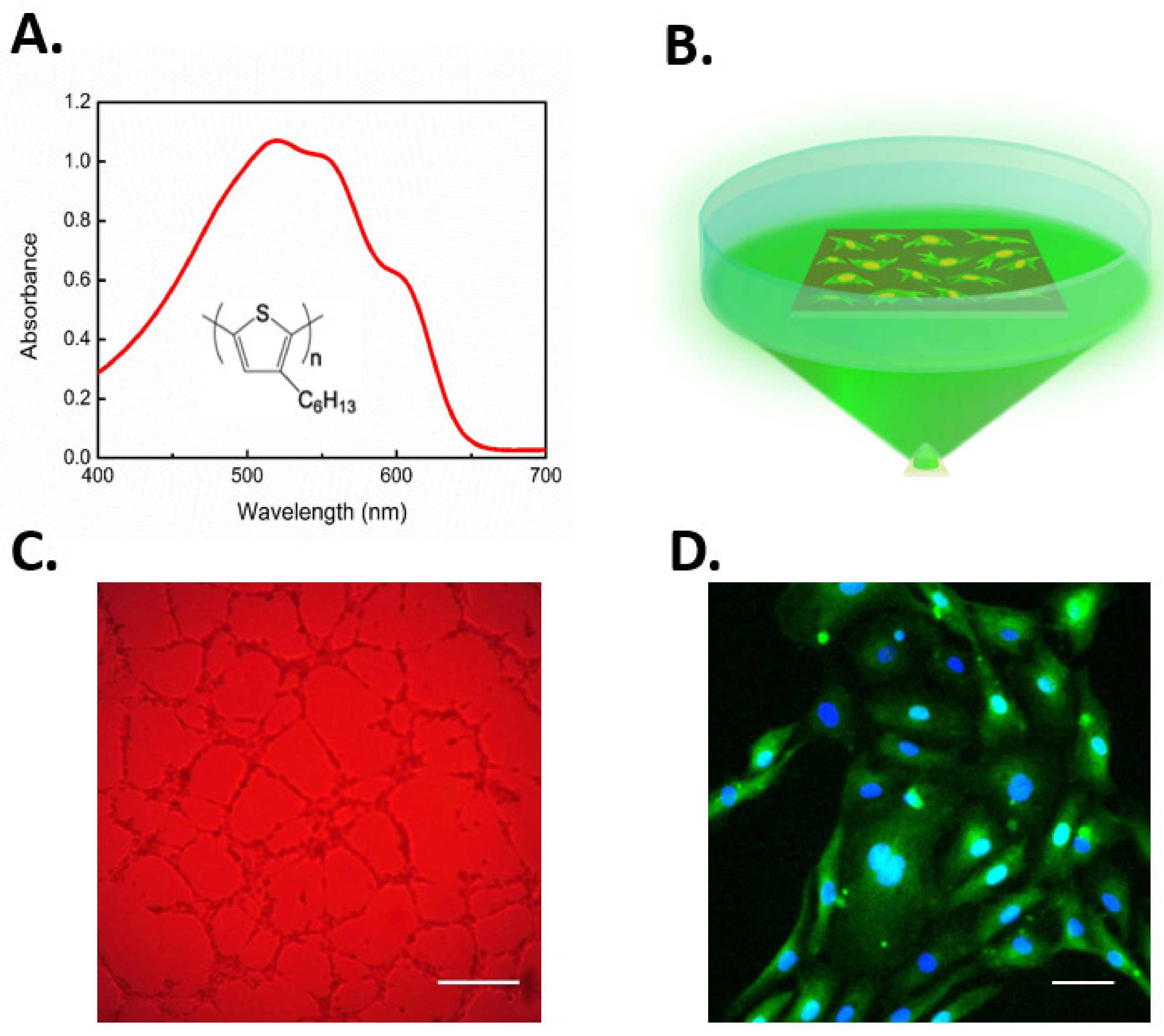
6.2. Gene-Less Opto-Stimulation of TRPV1 Leads to In Vitro Modulation of ECFC Fate
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCarron, J.G.; Lee, M.D.; Wilson, C. The Endothelium Solves problems that endothelial cells do not know exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef] [PubMed]
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Yoder, M.C. Tissue regeneration using endothelial colony-forming cells: Promising cells for vascular repair. Pediatr. Res. 2018, 83, 283–290. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Randi, A.M. Recent Advances in endothelial colony forming cells toward their use in clinical translation. Front. Med. 2018, 5, 295. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.L.; McLoughlin, K.J.; Chambers, S.E.J.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. The Vasoreparative potential of endothelial colony forming cells: A journey through pre-clinical studies. Front. Med. 2018, 5, 273. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Poletto, V.; Rosti, V.; Biggiogera, M.; Guerra, G.; Moccia, F.; Porta, C. The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit. Rev. Oncol. Hematol. 2018, 132, 89–99. [Google Scholar] [CrossRef]
- Fischer, C.; Schneider, M.; Carmeliet, P. Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb. Exp. Pharmacol. 2006, 157–212. [Google Scholar]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Madeddu, P. Therapeutic angiogenesis and vasculogenesis for tissue regeneration. Exp. Physiol. 2005, 90, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Tasev, D.; Koolwijk, P.; van Hinsbergh, V.W. Therapeutic potential of human-derived endothelial colony-forming cells in animal models. Tissue Eng. Part B Rev. 2016, 22, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Mitsos, S.; Katsanos, K.; Koletsis, E.; Kagadis, G.C.; Anastasiou, N.; Diamantopoulos, A.; Karnabatidis, D.; Dougenis, D. Therapeutic angiogenesis for myocardial ischemia revisited: Basic biological concepts and focus on latest clinical trials. Angiogenesis 2012, 15, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Qadura, M.; Terenzi, D.C.; Verma, S.; Al-Omran, M.; Hess, D.A. Concise review: Cell therapy for critical limb ischemia: An integrated review of preclinical and clinical studies. Stem Cells 2018, 36, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Rosti, V. Manipulating intracellular Ca2+ signals to stimulate therapeutic angiogenesis in cardiovascular disorders. Curr. Pharm. Biotechnol. 2018, 19, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef]
- Potenza, D.M.; Guerra, G.; Avanzato, D.; Poletto, V.; Pareek, S.; Guido, D.; Gallanti, A.; Rosti, V.; Munaron, L.; Tanzi, F.; et al. Hydrogen sulphide triggers VEGF-induced intracellular Ca(2+) signals in human endothelial cells but not in their immature progenitors. Cell Calcium 2014, 56, 225–234. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca(2+) entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca(2+) signaling, angiogenesis and vasculogenesis: Just what it takes to make a blood vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, ra20. [Google Scholar] [CrossRef]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Savage, A.M.; Kurusamy, S.; Chen, Y.; Jiang, Z.; Chhabria, K.; MacDonald, R.B.; Kim, H.R.; Wilson, H.L.; van Eeden, F.J.M.; Armesilla, A.L.; et al. tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 2019, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca(2+)-dependent activation of the extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef]
- Balbi, C.; Lodder, K.; Costa, A.; Moimas, S.; Moccia, F.; van Herwaarden, T.; Rosti, V.; Campagnoli, F.; Palmeri, A.; De Biasio, P.; et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int. J. Cardiol. 2019, 287, 87–95. [Google Scholar] [CrossRef]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic acid evokes an increase in Intracellular Ca(2+) concentration and nitric oxide production in endothelial cells from human brain microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Earley, S. Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The role of endothelial Ca(2+) signaling in neurovascular coupling: A view from the lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Yu, P.C.; Gu, S.Y.; Bu, J.W.; Du, J.L. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 2010, 106, 1221–1232. [Google Scholar] [CrossRef]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient receptor potential channel expression signatures in tumor-derived endothelial cells: Functional roles in prostate cancer angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Kanugula, A.K.; Adapala, R.K.; Midha, P.; Cappelli, H.C.; Meszaros, J.G.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019, 33, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef]
- Ferreira-Martins, J.; Rondon-Clavo, C.; Tugal, D.; Korn, J.A.; Rizzi, R.; Padin-Iruegas, M.E.; Ottolenghi, S.; De Angelis, A.; Urbanek, K.; Ide-Iwata, N.; et al. Spontaneous calcium oscillations regulate human cardiac progenitor cell growth. Circ. Res. 2009, 105, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.T.; Wagner, M.B.; Davis, M.E. Electrically induced calcium handling in cardiac progenitor cells. Stem Cells Int. 2016, 2016, 8917380. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca(2+) signals to reconstruct a broken heart: Still a theoretical approach? Curr. Drug Targets 2015, 16, 793–815. [Google Scholar] [CrossRef]
- Udan, R.S.; Culver, J.C.; Dickinson, M.E. Understanding vascular development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 327–346. [Google Scholar] [CrossRef]
- Goldie, L.C.; Nix, M.K.; Hirschi, K.K. Embryonic vasculogenesis and hematopoietic specification. Organogenesis 2008, 4, 257–263. [Google Scholar] [CrossRef]
- Eichmann, A.; Yuan, L.; Moyon, D.; Lenoble, F.; Pardanaud, L.; Breant, C. Vascular development: From precursor cells to branched arterial and venous networks. Int. J. Dev. Biol. 2005, 49, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Iba, T.; Takakura, N. Mechanisms of new blood vessel formation and proliferative heterogeneity of endothelial cells. Int. Immunol. 2000. [Google Scholar] [CrossRef] [PubMed]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C. Endothelial stem and progenitor cells (stem cells): (2017 Grover Conference Series). Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef]
- Heinke, J.; Patterson, C.; Moser, M. Life is a pattern: Vascular assembly within the embryo. Front. Biosci. 2012, 4, 2269–2288. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Djonov, V.; Baum, O.; Burri, P.H. Vascular remodeling by intussusceptive angiogenesis. Cell Tissue Res. 2003, 314, 107–117. [Google Scholar] [CrossRef]
- Fitzgerald, G.; Soro-Arnaiz, I.; De Bock, K. The warburg effect in endothelial cells and its potential as an anti-angiogenic target in cancer. Front. Cell Dev. Biol. 2018, 6, 100. [Google Scholar] [CrossRef]
- Caduff, J.H.; Fischer, L.C.; Burri, P.H. Scanning electron microscope study of the developing microvasculature in the postnatal rat lung. Anat. Rec. 1986, 216, 154–164. [Google Scholar] [CrossRef]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Liao, S.; Luo, C.; Cao, B.; Hu, H.; Wang, S.; Yue, H.; Chen, L.; Zhou, Z. Endothelial progenitor cells for ischemic stroke: Update on basic research and application. Stem Cells Int. 2017, 2017, 2193432. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Cinelli, M.; Bonetti, E.; Guerra, G.; Rosti, V. Endothelial progenitor cells support tumour growth and metastatisation: Implications for the resistance to anti-angiogenic therapy. Tumour. Biol. 2015, 36, 6603–6614. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.; Perrotta, F.; Testa, G. Circulating endothelial progenitor cells biology and regenerative medicine in pulmonary vascular diseases. Curr. Pharm. Biotechnol. 2018, 19, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Edwards, N.; Langford-Smith, A.W.W.; Wilkinson, F.L.; Alexander, M.Y. Endothelial progenitor cells: New targets for therapeutics for inflammatory conditions with high cardiovascular risk. Front. Med. 2018, 5, 200. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef]
- Yoder, M.C. Human endothelial progenitor cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006692. [Google Scholar] [CrossRef]
- Yoder, M.C. Is endothelium the origin of endothelial progenitor cells? Arter. Thromb. Vasc. Biol. 2010, 30, 1094–1103. [Google Scholar] [CrossRef]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef]
- Medina, R.J.; O’Neill, C.L.; Humphreys, M.W.; Gardiner, T.A.; Stitt, A.W. Outgrowth endothelial cells: Characterization and their potential for reversing ischemic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5906–5913. [Google Scholar] [CrossRef]
- Rohban, R.; Pieber, T.R. Mesenchymal stem and progenitor cells in regeneration: Tissue specificity and regenerative potential. Stem Cells Int. 2017, 2017, 5173732. [Google Scholar] [CrossRef]
- Burger, D.; Vinas, J.L.; Akbari, S.; Dehak, H.; Knoll, W.; Gutsol, A.; Carter, A.; Touyz, R.M.; Allan, D.S.; Burns, K.D. Human endothelial colony-forming cells protect against acute kidney injury: Role of exosomes. Am. J. Pathol. 2015, 185, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Gatti, S.; Medica, D.; Figliolini, F.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Biancone, L.; Tetta, C.; Camussi, G. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012, 82, 412–427. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E. “Sprouting angiogenesis”, a reappraisal. Dev. Biol. 2012, 372, 157–165. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.J.; Song, S.; Seo, H.R.; Shin, J.H.; Choi, S.C.; Park, J.H.; Yu, C.W.; Hong, S.J.; Lim, D.S. Human endothelial colony forming cells from adult peripheral blood have enhanced sprouting angiogenic potential through up-regulating VEGFR2 signaling. Int. J. Cardiol. 2015, 197, 33–43. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 2016, 10, 347–354. [Google Scholar] [CrossRef]
- Westhoff, M.A.; Serrels, B.; Fincham, V.J.; Frame, M.C.; Carragher, N.O. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell Biol. 2004, 24, 8113–8133. [Google Scholar] [CrossRef]
- Li, X.; Padhan, N.; Sjostrom, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sainz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef]
- Teixido, J.; Martinez-Moreno, M.; Diaz-Martinez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 121–131. [Google Scholar] [CrossRef]
- Dragoni, S.; Reforgiato, M.; Zuccolo, E.; Poletto, V.; Lodola, F.; Ruffinatti, F.A.; Bonetti, E.; Guerra, G.; Barosi, G.; Rosti, V.; et al. Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 2015, 43, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; James, A.F.; Foster, R.R.; Hancox, J.C.; Bates, D.O. VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef]
- Zhu, Y.; Lu, Y.; Qu, C.; Miller, M.; Tian, J.; Thakur, D.P.; Zhu, J.; Deng, Z.; Hu, X.; Wu, M.; et al. Identification and optimization of 2-aminobenzimidazole derivatives as novel inhibitors of TRPC4 and TRPC5 channels. Br. J. Pharmacol. 2015, 172, 3495–3509. [Google Scholar] [CrossRef]
- Gaudet, R. TRP channels entering the structural era. J. Physiol. 2008, 586, 3565–3575. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef]
- Cheng, W.; Yang, F.; Takanishi, C.L.; Zheng, J. Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 2007, 129, 191–207. [Google Scholar] [CrossRef]
- O’Leary, C.; McGahon, M.K.; Ashraf, S.; McNaughten, J.; Friedel, T.; Cincola, P.; Barabas, P.; Fernandez, J.A.; Stitt, A.W.; McGeown, J.G.; et al. Involvement of TRPV1 and TRPV4 channels in retinal angiogenesis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3297–3309. [Google Scholar] [CrossRef]
- Cheng, W.; Sun, C.; Zheng, J. Heteromerization of TRP channel subunits: Extending functional diversity. Protein Cell 2010, 1, 802–810. [Google Scholar] [CrossRef]
- Stewart, A.P.; Smith, G.D.; Sandford, R.N.; Edwardson, J.M. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys. J. 2010, 99, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Grimm, C.; Becker, L.; Ricci, A.J.; Heller, S. A novel ion channel formed by interaction of TRPML3 with TRPV5. PLoS ONE 2013, 8, e58174. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, N.M.; Wang, L.; Ranke, H.; Liedtke, W.; Tabuchi, A.; Kuebler, W.M. TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology 2015, 122, 1338–1348. [Google Scholar] [CrossRef]
- Ma, X.; Qiu, S.; Luo, J.; Ma, Y.; Ngai, C.Y.; Shen, B.; Wong, C.O.; Huang, Y.; Yao, X. Functional role of vanilloid transient receptor potential 4-canonical transient receptor potential 1 complex in flow-induced Ca2+ influx. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 851–858. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Carlton-Carew, S.R.E.; Khan, D.M.; Zargaran, A.K.; Jahan, K.S.; Vanessa Ho, W.S.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vasc. Pharmacol. 2017, 96, 53–62. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, K.T.; Wong, C.O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef]
- Cao, E.; Liao, M.; Cheng, Y.; Julius, D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 2013, 504, 113–118. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, T.; Simon, S.A. TRPV1 Receptors and Signal Transduction. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Munns, C.H.; Chung, M.K.; Sanchez, Y.E.; Amzel, L.M.; Caterina, M.J. Role of the outer pore domain in transient receptor potential vanilloid 1 dynamic permeability to large cations. J. Biol. Chem. 2015, 290, 5707–5724. [Google Scholar] [CrossRef] [PubMed]
- Kuzhikandathil, E.V.; Wang, H.; Szabo, T.; Morozova, N.; Blumberg, P.M.; Oxford, G.S. Functional analysis of capsaicin receptor (vanilloid receptor subtype 1) multimerization and agonist responsiveness using a dominant negative mutation. J. Neurosci. 2001, 21, 8697–8706. [Google Scholar] [CrossRef] [PubMed]
- Geron, M.; Hazan, A.; Priel, A. Animal toxins providing insights into TRPV1 activation mechanism. Toxins 2017, 9, 326. [Google Scholar] [CrossRef]
- Patacchini, R.; Santicioli, P.; Giuliani, S.; Maggi, C.A. Pharmacological investigation of hydrogen sulfide (H2S) contractile activity in rat detrusor muscle. Eur. J. Pharmacol. 2005, 509, 171–177. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Siemens, J.; Zhou, S.; Piskorowski, R.; Nikai, T.; Lumpkin, E.A.; Basbaum, A.I.; King, D.; Julius, D. Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature 2006, 444, 208–212. [Google Scholar] [CrossRef]
- Vriens, J.; Appendino, G.; Nilius, B. Pharmacology of vanilloid transient receptor potential cation channels. Mol. Pharmacol. 2009, 75, 1262–1279. [Google Scholar] [CrossRef]
- Appendino, G.; Minassi, A.; Pagani, A.; Ech-Chahad, A. The role of natural products in the ligand deorphanization of TRP channels. Curr. Pharm. Des. 2008, 14, 2–17. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; De Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.J.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef]
- McNamara, F.N.; Randall, A.; Gunthorpe, M.J. Effects of piperine, the pungent component of black pepper, at the human vanilloid receptor (TRPV1). Br. J. Pharmacol. 2005, 144, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Blair, N.T.; Clapham, D.E. Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J. Neurosci. 2005, 25, 8924–8937. [Google Scholar] [CrossRef] [PubMed]
- Ahern, G.P.; Wang, X.; Miyares, R.L. Polyamines are potent ligands for the capsaicin receptor TRPV1. J. Biol. Chem. 2006, 281, 8991–8995. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Droogmans, G.; Wissenbach, U.; Janssens, A.; Flockerzi, V.; Nilius, B. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 2004, 430, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Aneiros, E.; Cao, L.; Papakosta, M.; Stevens, E.B.; Phillips, S.; Grimm, C. The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 2011, 30, 994–1002. [Google Scholar] [CrossRef]
- Vennekens, R.; Owsianik, G.; Nilius, B. Vanilloid transient receptor potential cation channels: An overview. Curr. Pharm. Des. 2008, 14, 18–31. [Google Scholar] [CrossRef]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef]
- Su, K.H.; Lee, K.I.; Shyue, S.K.; Chen, H.Y.; Wei, J.; Lee, T.S. Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int. J. Biol. Sci. 2014, 10, 990–996. [Google Scholar] [CrossRef]
- Guo, B.C.; Wei, J.; Su, K.H.; Chiang, A.N.; Zhao, J.F.; Chen, H.Y.; Shyue, S.K.; Lee, T.S. Transient receptor potential vanilloid type 1 is vital for (-)-epigallocatechin-3-gallate mediated activation of endothelial nitric oxide synthase. Mol. Nutr. Food Res. 2015, 59, 646–657. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.B.; Su, K.H.; Kou, Y.R.; Guo, B.C.; Lee, K.I.; Wei, J.; Lee, T.S. Role of transient receptor potential vanilloid 1 in regulating erythropoietin-induced activation of endothelial nitric oxide synthase. Acta Physiol. 2017, 219, 465–477. [Google Scholar] [CrossRef]
- Huang, W.; Wang, H.; Galligan, J.J.; Wang, D.H. Transient receptor potential vanilloid subtype 1 channel mediated neuropeptide secretion and depressor effects: Role of endoplasmic reticulum associated Ca2+ release receptors in rat dorsal root ganglion neurons. J. Hypertens. 2008, 26, 1966–1975. [Google Scholar] [CrossRef]
- Karai, L.J.; Russell, J.T.; Iadarola, M.J.; Olah, Z. Vanilloid receptor 1 regulates multiple calcium compartments and contributes to Ca2+-induced Ca2+ release in sensory neurons. J. Biol. Chem. 2004, 279, 16377–16387. [Google Scholar] [CrossRef] [PubMed]
- Lozano, C.; Cordova, C.; Marchant, I.; Zuniga, R.; Ochova, P.; Ramirez-Barrantes, R.; Gonzalez-Arriagada, W.A.; Rodriguez, B.; Olivero, P. Intracellular aggregated TRPV1 is associated with lower survival in breast cancer patients. Breast Cancer 2018, 10, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Pecze, L.; Blum, W.; Henzi, T.; Schwaller, B. Endogenous TRPV1 stimulation leads to the activation of the inositol phospholipid pathway necessary for sustained Ca(2+) oscillations. Biochim. Biophys. Acta 2016, 1863, 2905–2915. [Google Scholar] [CrossRef]
- Pecze, L.; Josvay, K.; Blum, W.; Petrovics, G.; Vizler, C.; Olah, Z.; Schwaller, B. Activation of endogenous TRPV1 fails to induce overstimulation-based cytotoxicity in breast and prostate cancer cells but not in pain-sensing neurons. Biochim. Biophys. Acta 2016, 1863, 2054–2064. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sorgard, M.; Di Marzo, V.; Julius, D.; Hogestatt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef]
- Lo, Y.C.; Hsiao, H.C.; Wu, D.C.; Lin, R.J.; Liang, J.C.; Yeh, J.L.; Chen, I.J. A novel capsaicin derivative VOA induced relaxation in rat mesenteric and aortic arteries: Involvement of CGRP, NO, cGMP, and endothelium-dependent activities. J. Cardiovasc. Pharmacol. 2003, 42, 511–520. [Google Scholar] [CrossRef]
- Poblete, I.M.; Orliac, M.L.; Briones, R.; Adler-Graschinsky, E.; Huidobro-Toro, J.P. Anandamide elicits an acute release of nitric oxide through endothelial TRPV1 receptor activation in the rat arterial mesenteric bed. J. Physiol. 2005, 568, 539–551. [Google Scholar] [CrossRef]
- Yang, D.; Luo, Z.; Ma, S.; Wong, W.T.; Ma, L.; Zhong, J.; He, H.; Zhao, Z.; Cao, T.; Yan, Z.; et al. Activation of TRPV1 by dietary capsaicin improves endothelium-dependent vasorelaxation and prevents hypertension. Cell Metab. 2010, 12, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pu, Y.; Wang, P.; Chen, S.; Zhao, Y.; Liu, C.; Shang, Q.; Zhu, Z.; Liu, D. TRPV1-mediated UCP2 upregulation ameliorates hyperglycemia-induced endothelial dysfunction. Cardiovasc. Diabetol. 2013, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Wang, P.; Ma, L.; Gao, P.; Gong, L.; Li, L.; Li, Q.; Sun, F.; Zhou, X.; He, H.; et al. Ameliorating endothelial mitochondrial dysfunction restores coronary function via transient receptor potential vanilloid 1-mediated protein kinase A/Uncoupling protein 2 pathway. Hypertension 2016, 67, 451–460. [Google Scholar] [CrossRef]
- Peng, W.J.; Liu, Y.; Yu, Y.R.; Fu, Y.Q.; Zhao, Y.; Kuang, H.B.; Huang, Q.R.; He, M.; Luo, D. Rutaecarpine prevented dysfunction of endothelial gap junction induced by Ox-LDL via activation of TRPV1. Eur. J. Pharmacol. 2015, 756, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca 2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Ye, F.; Deng, P.Y.; Li, D.; Luo, D.; Li, N.S.; Deng, S.; Deng, H.W.; Li, Y.J. Involvement of endothelial cell-derived CGRP in heat stress-induced protection of endothelial function. Vasc. Pharmacol. 2007, 46, 238–246. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, Y.W.; Peng, W.J.; Peng, J.; Chen, Q.Q.; Li, D.; Deng, H.W.; Li, Y.J. Transient receptor potential vanilloid 1-mediated expression and secretion of endothelial cell-derived calcitonin gene-related peptide. Regul. Pept. 2008, 150, 66–72. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Capsaicin may have important potential for promoting vascular and metabolic health. Open Heart 2015, 2, e000262. [Google Scholar] [CrossRef]
- Bratz, I.N.; Dick, G.M.; Tune, J.D.; Edwards, J.M.; Neeb, Z.P.; Dincer, U.D.; Sturek, M. Impaired capsaicin-induced relaxation of coronary arteries in a porcine model of the metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2489–H2496. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, L.; Xu, H.; Liu, S.; Zhu, F.; Yan, F.; Shen, S.; Zhu, M. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 2017, 260, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.X.; Jing, Y.; Liu, Y.L.; Xu, Z.M.; Yuan, F.; Wang, M.L.; Geng, Z.; Tian, H.L. Inhibition of transient receptor potential vanilloid 1 attenuates blood-brain barrier disruption after traumatic brain injury in mice. J. Neurotrauma 2019, 36, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Golech, S.A.; McCarron, R.M.; Chen, Y.; Bembry, J.; Lenz, F.; Mechoulam, R.; Shohami, E.; Spatz, M. Human brain endothelium: Coexpression and function of vanilloid and endocannabinoid receptors. Brain Res. Mol. Brain Res. 2004, 132, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hind, W.H.; Tufarelli, C.; Neophytou, M.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharmacol. 2015, 172, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Min, J.K.; Han, K.Y.; Kim, E.C.; Kim, Y.M.; Lee, S.W.; Kim, O.H.; Kim, K.W.; Gho, Y.S.; Kwon, Y.G. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004, 64, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Doucette, C.D.; Hilchie, A.L.; Liwski, R.; Hoskin, D.W. Piperine, a dietary phytochemical, inhibits angiogenesis. J. Nutr. Biochem. 2013, 24, 231–239. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca(2+) signaling and the resistance to anticancer treatments: Partners in crime. Int. J. Mol. Sci. 2018, 19, E217. [Google Scholar] [CrossRef]
- Wu, T.T.; Peters, A.A.; Tan, P.T.; Roberts-Thomson, S.J.; Monteith, G.R. Consequences of activating the calcium-permeable ion channel TRPV1 in breast cancer cells with regulated TRPV1 expression. Cell Calcium 2014, 56, 59–67. [Google Scholar] [CrossRef]
- Ramirez-Barrantes, R.; Cordova, C.; Gatica, S.; Rodriguez, B.; Lozano, C.; Marchant, I.; Echeverria, C.; Simon, F.; Olivero, P. Transient receptor potential vanilloid 1 expression mediates capsaicin-induced cell death. Front. Physiol. 2018, 9, 682. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. A Review on Potential Involvement of TRPV1 Channels in Ischemia-Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 38–45. [Google Scholar] [CrossRef]
- Wang, L.; Wang, D.H. TRPV1 gene knockout impairs postischemic recovery in isolated perfused heart in mice. Circulation 2005, 112, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Wang, D.H. N-oleoyldopamine, a novel endogenous capsaicin-like lipid, protects the heart against ischemia-reperfusion injury via activation of TRPV1. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H728–H735. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Wang, D.H. Protease-activated receptor 2-mediated protection of myocardial ischemia-reperfusion injury: Role of transient receptor potential vanilloid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1681–R1690. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Amadesi, S.; Nie, J.; Vergnolle, N.; Cottrell, G.S.; Grady, E.F.; Trevisani, M.; Manni, C.; Geppetti, P.; McRoberts, J.A.; Ennes, H.; et al. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J. Neurosci. 2004, 24, 4300–4312. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; Di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Balasubramanian, A.; Marrelli, S.P. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R149–R156. [Google Scholar] [CrossRef]
- Tomoyose, K.; Okada, Y.; Sumioka, T.; Miyajima, M.; Flanders, K.C.; Shirai, K.; Morii, T.; Reinach, P.S.; Yamanaka, O.; Saika, S. Suppression of in vivo neovascularization by the loss of TRPV1 in mouse cornea. J. Ophthalmol. 2015, 2015, 706404. [Google Scholar] [CrossRef]
- Su, L.; Zhang, Y.; He, K.; Wei, S.; Pei, H.; Wang, Q.; Yang, D.; Yang, Y. Activation of transient receptor potential vanilloid 1 accelerates re-endothelialization and inhibits neointimal formation after vascular injury. J. Vasc. Surg. 2017, 65, 197–205. [Google Scholar] [CrossRef]
- Buccheri, D.; Piraino, D.; Andolina, G.; Cortese, B. Understanding and managing in-stent restenosis: A review of clinical data, from pathogenesis to treatment. J. Thorac. Dis. 2016, 8, E1150–E1162. [Google Scholar] [CrossRef]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef]
- Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Pharmacological actions of multi-target-directed evodiamine. Molecules 2013, 18, 1826–1843. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.C.; Kou, Y.R.; Shyue, S.K.; Su, K.H.; Wei, J.; Cheng, L.C.; Yu, Y.B.; Pan, C.C.; Lee, T.S. Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 2011, 91, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen sulfide and endothelial dysfunction: Relationship with nitric oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive store-operated Ca(2+) entry leads to enhanced nitric oxide production and proliferation in infantile hemangioma-derived endothelial colony-forming cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Ching, L.C.; Chen, C.Y.; Su, K.H.; Hou, H.H.; Shyue, S.K.; Kou, Y.R.; Lee, T.S. Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol. Med. 2012, 18, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeon, H.L.; Park, S.J.; Shin, J.Y. Effect of statins, metformin, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers on age-related macular degeneration. Yonsei Med. J. 2019, 60, 679–686. [Google Scholar] [CrossRef]
- Nishimoto-Hazuku, A.; Hirase, T.; Ide, N.; Ikeda, Y.; Node, K. Simvastatin stimulates vascular endothelial growth factor production by hypoxia-inducible factor-1alpha upregulation in endothelial cells. J. Cardiovasc. Pharmacol. 2008, 51, 267–273. [Google Scholar] [CrossRef]
- Settergren, M.; Bohm, F.; Ryden, L.; Pernow, J. Cholesterol lowering is more important than pleiotropic effects of statins for endothelial function in patients with dysglycaemia and coronary artery disease. Eur. Heart J. 2008, 29, 1753–1760. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhao, J.F.; Lin, S.J.; Shyue, S.K.; Guo, B.C.; Lu, T.M.; Lee, T.S. Asymmetric dimethylarginine limits the efficacy of simvastatin activating endothelial nitric oxide synthase. J. Am. Heart Assoc. 2016, 5, e003327. [Google Scholar] [CrossRef]
- Lipinski, M.J.; Cauthen, C.A.; Biondi-Zoccai, G.G.; Abbate, A.; Vrtovec, B.; Khan, B.V.; Vetrovec, G.W. Meta-analysis of randomized controlled trials of statins versus placebo in patients with heart failure. Am. J. Cardiol. 2009, 104, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Su, K.H.; Lin, S.J.; Wei, J.; Lee, K.I.; Zhao, J.F.; Shyue, S.K.; Lee, T.S. The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. 2014, 212, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, H.J.; Kim, G.C.; Choi, J.H.; Hong, J.W. Plasma cupping induces VEGF expression in skin cells through nitric oxide-mediated activation of hypoxia inducible factor 1. Sci. Rep. 2019, 9, 3821. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Zhou, J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha). Curr. Med. Chem. 2003, 10, 845–855. [Google Scholar] [CrossRef]
- Campbell, W.B.; Fleming, I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflug. Arch. 2010, 459, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Cheranov, S.Y.; Karpurapu, M.; Wang, D.; Zhang, B.; Venema, R.C.; Rao, G.N. An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 2008, 111, 5581–5591. [Google Scholar] [CrossRef] [PubMed]
- Romashko, M.; Schragenheim, J.; Abraham, N.G.; McClung, J.A. epoxyeicosatrienoic acid as therapy for diabetic and ischemic cardiomyopathy. Trends Pharmacol. Sci. 2016, 37, 945–962. [Google Scholar] [CrossRef]
- Park, S.K.; Herrnreiter, A.; Pfister, S.L.; Gauthier, K.M.; Falck, B.A.; Falck, J.R.; Campbell, W.B. GPR40 is a low-affinity epoxyeicosatrienoic acid receptor in vascular cells. J. Biol. Chem. 2018, 293, 10675–10691. [Google Scholar] [CrossRef]
- Clement, Y. Can green tea do that? A literature review of the clinical evidence. Prev. Med. 2009, 49, 83–87. [Google Scholar] [CrossRef]
- Bai, Q.; Lyu, Z.; Yang, X.; Pan, Z.; Lou, J.; Dong, T. Epigallocatechin-3-gallate promotes angiogenesis via up-regulation of Nfr2 signaling pathway in a mouse model of ischemic stroke. Behav. Brain Res. 2017, 321, 79–86. [Google Scholar] [CrossRef]
- Deka, A.; Vita, J.A. Tea and cardiovascular disease. Pharmacol. Res. 2011, 64, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Warming Up to New Possibilities with the Capsaicin Receptor TRPV1: mTOR, AMPK, and Erythropoietin. Curr. Neurovasc. Res. 2017, 14, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Kimakova, P.; Solar, P.; Solarova, Z.; Komel, R.; Debeljak, N. Erythropoietin and its angiogenic activity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Maltaneri, R.E.; Schiappacasse, A.; Chamorro, M.E.; Nesse, A.B.; Vittori, D.C. Aquaporin-1 plays a key role in erythropoietin-induced endothelial cell migration. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118569. [Google Scholar] [CrossRef] [PubMed]
- Hache, G.; Garrigue, P.; Bennis, Y.; Stalin, J.; Moyon, A.; Cerami, A.; Brines, M.; Blot-Chabaud, M.; Sabatier, F.; Dignat-George, F.; et al. ARA290, a specific agonist of erythropoietin/CD131 heteroreceptor, improves circulating endothelial progenitors’ angiogenic potential and homing ability. Shock 2016, 46, 390–397. [Google Scholar] [CrossRef]
- Garrigue, P.; Hache, G.; Bennis, Y.; Brige, P.; Stalin, J.; Pellegrini, L.; Velly, L.; Orlandi, F.; Castaldi, E.; Dignat-George, F.; et al. Erythropoietin pretreatment of transplanted endothelial colony-forming cells enhances recovery in a cerebral ischemia model by increasing their homing ability: A SPECT/CT study. J. Nucl. Med. 2016, 57, 1798–1804. [Google Scholar] [CrossRef][Green Version]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. Cell Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef]
- Mergler, S.; Valtink, M.; Taetz, K.; Sahlmuller, M.; Fels, G.; Reinach, P.S.; Engelmann, K.; Pleyer, U. Characterization of transient receptor potential vanilloid channel 4 (TRPV4) in human corneal endothelial cells. Exp. Eye Res. 2011, 93, 710–719. [Google Scholar] [CrossRef]
- Mergler, S.; Valtink, M.; Coulson-Thomas, V.J.; Lindemann, D.; Reinach, P.S.; Engelmann, K.; Pleyer, U. TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 2010, 90, 758–770. [Google Scholar] [CrossRef]
- Ihori, H.; Nozawa, T.; Sobajima, M.; Shida, T.; Fukui, Y.; Fujii, N.; Inoue, H. Waon therapy attenuates cardiac hypertrophy and promotes myocardial capillary growth in hypertensive rats: A comparative study with fluvastatin. Heart Vessel. 2016, 31, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Sobajima, M.; Nozawa, T.; Shida, T.; Ohori, T.; Suzuki, T.; Matsuki, A.; Inoue, H. Repeated sauna therapy attenuates ventricular remodeling after myocardial infarction in rats by increasing coronary vascularity of noninfarcted myocardium. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H548–H554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.S.; Kim, Y.K.; Cho, K.H.; Chung, J.H. Infrared exposure induces an angiogenic switch in human skin that is partially mediated by heat. Br. J. Dermatol. 2006, 155, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Calcium signaling in endothelial colony forming cells in health and disease. Adv. Exp. Med. Biol 2020, 1131, 1013–1030. [Google Scholar] [CrossRef]
- Moccia, F.; Guerra, G. Ca(2+) signalling in endothelial progenitor cells: Friend or foe? J. Cell Physiol. 2016, 231, 314–327. [Google Scholar] [CrossRef]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vasc. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef]
- Griffin, M.F.; Butler, P.E.; Seifalian, A.M.; Kalaskar, D.M. Control of stem cell fate by engineering their micro and nanoenvironment. World J. Stem Cells 2015, 7, 37–50. [Google Scholar] [CrossRef]
- Puhl, S.L. Cannabinoid-sensitive receptors in cardiac physiology and ischaemia. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118462. [Google Scholar] [CrossRef]
- Fenno, L.; Yizhar, O.; Deisseroth, K. The development and application of optogenetics. Annu. Rev. Neurosci. 2011, 34, 389–412. [Google Scholar] [CrossRef]
- Antognazza, M.R.; Martino, N.; Ghezzi, D.; Feyen, P.; Colombo, E.; Endeman, D.; Benfenati, F.; Lanzani, G. Shedding light on living cells. Adv. Mater. 2015, 27, 7662–7669. [Google Scholar] [CrossRef]
- Di Maria, F.; Lodola, F.; Zucchetti, E.; Benfenati, F.; Lanzani, G. The evolution of artificial light actuators in living systems: From planar to nanostructured interfaces. Chem. Soc. Rev. 2018, 47, 4757–4780. [Google Scholar] [CrossRef] [PubMed]
- Martino, N.; Bossio, C.; Vaquero Morata, S.; Lanzani, G.; Antognazza, M.R. Optical control of living cells electrical activity by conjugated polymers. J. Vis. Exp. 2016, e53494. [Google Scholar] [CrossRef] [PubMed]
- Feyen, P.; Colombo, E.; Endeman, D.; Nova, M.; Laudato, L.; Martino, N.; Antognazza, M.R.; Lanzani, G.; Benfenati, F.; Ghezzi, D. Light-evoked hyperpolarization and silencing of neurons by conjugated polymers. Sci. Rep. 2016, 6, 22718. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, S.; Bossio, C.; Bellani, S.; Martino, N.; Zucchetti, E.; Lanzani, G.; Antognazza, M.R. Conjugated polymers for the optical control of the electrical activity of living cells. J. Mater. Chem. B 2016, 4, 5272–5283. [Google Scholar] [CrossRef] [PubMed]
- Zucchetti, E.; Zangoli, M.; Bargigia, I.; Bossio, C.; Di Maria, F.; Barbarella, G.; D’Andrea, C.; Lanzani, G.; Antognazza, M.R. Poly(3-hexylthiophene) nanoparticles for biophotonics: Study of the mutual interaction with living cells. J. Mater. Chem. B 2017, 5, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Martino, N.; Tullii, G.; Lanzani, G.; Antognazza, M.R. Conjugated polymers mediate effective activation of the Mammalian Ion Channel Transient Receptor Potential Vanilloid 1. Sci. Rep. 2017, 7, 8477. [Google Scholar] [CrossRef]
- Tullii, G.; Giona, F.; Lodola, F.; Bonfadini, S.; Bossio, C.; Varo, S.; Desii, A.; Criante, L.; Sala, C.; Pasini, M.; et al. High-aspect-ratio semiconducting polymer pillars for 3D cell cultures. ACS Appl. Mater. Interfaces 2019, 11, 28125–28137. [Google Scholar] [CrossRef]
- Ghezzi, D.; Antognazza, M.R.; Maccarone, R.; Bellani, S.; Lanzarini, E.; Martino, N.; Mete, M.; Pertile, G.; Bisti, S.; Lanzani, G.; et al. A polymer optoelectronic interface restores light sensitivity in blind rat retinas. Nat. Photonics 2013, 7, 400–406. [Google Scholar] [CrossRef]
- Maya-Vetencourt, J.F.; Ghezzi, D.; Antognazza, M.R.; Colombo, E.; Mete, M.; Feyen, P.; Desii, A.; Buschiazzo, A.; Di Paolo, M.; Di Marco, S.; et al. A fully organic retinal prosthesis restores vision in a rat model of degenerative blindness. Nat. Mater. 2017, 16, 681–689. [Google Scholar] [CrossRef]
- Tortiglione, C.; Antognazza, M.R.; Tino, A.; Bossio, C.; Marchesano, V.; Bauduin, A.; Zangoli, M.; Morata, S.V.; Lanzani, G. Semiconducting polymers are light nanotransducers in eyeless animals. Sci. Adv. 2017, 3, e1601699. [Google Scholar] [CrossRef]
- Gautam, V.; Rand, D.; Hanein, Y.; Narayan, K.S. A polymer optoelectronic interface provides visual cues to a blind retina. Adv. Mater. 2014, 26, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Martino, N.; Feyen, P.; Porro, M.; Bossio, C.; Zucchetti, E.; Ghezzi, D.; Benfenati, F.; Lanzani, G.; Antognazza, M.R. Photothermal cellular stimulation in functional bio-polymer interfaces. Sci. Rep. 2015, 5, 8911. [Google Scholar] [CrossRef] [PubMed]
- Antognazza, M.R.; Abdel Aziz, I.; Lodola, F. Use of exogenous and endogenous photomediators as efficient ROS modulation tools: Results and perspectives for therapeutic purposes. Oxidative Med. Cell. Longev. 2019, 2019, 2867516. [Google Scholar] [CrossRef] [PubMed]
- Bossio, C.; Aziz, I.A.; Tullii, G.; Zucchetti, E.; Debellis, D.; Zangoli, M.; Di Maria, F.; Lanzani, G.; Antognazza, M.R. Photocatalytic activity of polymer nanoparticles modulates intracellular calcium dynamics and reactive oxygen species in HEK-293 cells. Front. Bioeng. Biotech. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Tullii, G.; Desii, A.; Bossio, C.; Bellani, S.; Colombo, M.; Martino, N.; Antognazza, M.R.; Lanzani, G. Bimodal functioning of a mesoporous, light sensitive polymer/electrolyte interface. Org. Electron. 2017, 46, 88–98. [Google Scholar] [CrossRef]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-mediated wound healing: H2O2 entry through AQP3 determines extracellular Ca(2+) influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca2+ signalling in endothelial progenitor cells: A novel means to improve cell-based therapy and impair tumour vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. https://doi.org/10.3390/cells9061341
Negri S, Faris P, Rosti V, Antognazza MR, Lodola F, Moccia F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells. 2020; 9(6):1341. https://doi.org/10.3390/cells9061341
Chicago/Turabian StyleNegri, Sharon, Pawan Faris, Vittorio Rosti, Maria Rosa Antognazza, Francesco Lodola, and Francesco Moccia. 2020. "Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis" Cells 9, no. 6: 1341. https://doi.org/10.3390/cells9061341
APA StyleNegri, S., Faris, P., Rosti, V., Antognazza, M. R., Lodola, F., & Moccia, F. (2020). Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells, 9(6), 1341. https://doi.org/10.3390/cells9061341