Platelet Transforming Growth Factor-β1 Induces Liver Sinusoidal Endothelial Cells to Secrete Interleukin-6

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Proteins
2.3. Materials
2.4. Reagents
2.5. Antibodies
2.6. Isolation and Culture of Liver Sinusoidal Cells, Kupffer Cells or HUVEC
2.7. Preparation of Mouse Activated Platelet Releasate
2.8. Preparation of Platelet Microparticles
2.9. Platelet Releasate Preparation and Fast Protein Liquid Chromatography
2.10. Western Blot
2.11. Immunofluorescence
2.12. Data Analysis
2.13. Statistics
2.14. Study Approval
3. Results
3.1. Activated Platelet Releasate Induces Interleukin-6 Expression and Secretion in Liver Sinusoidal Endothelial Cells
3.2. Platelet-Derived Extracellular Vesicles Are Not Implicated in Interleukin-6 Secretion by Liver Sinusoidal Endothelial Cells
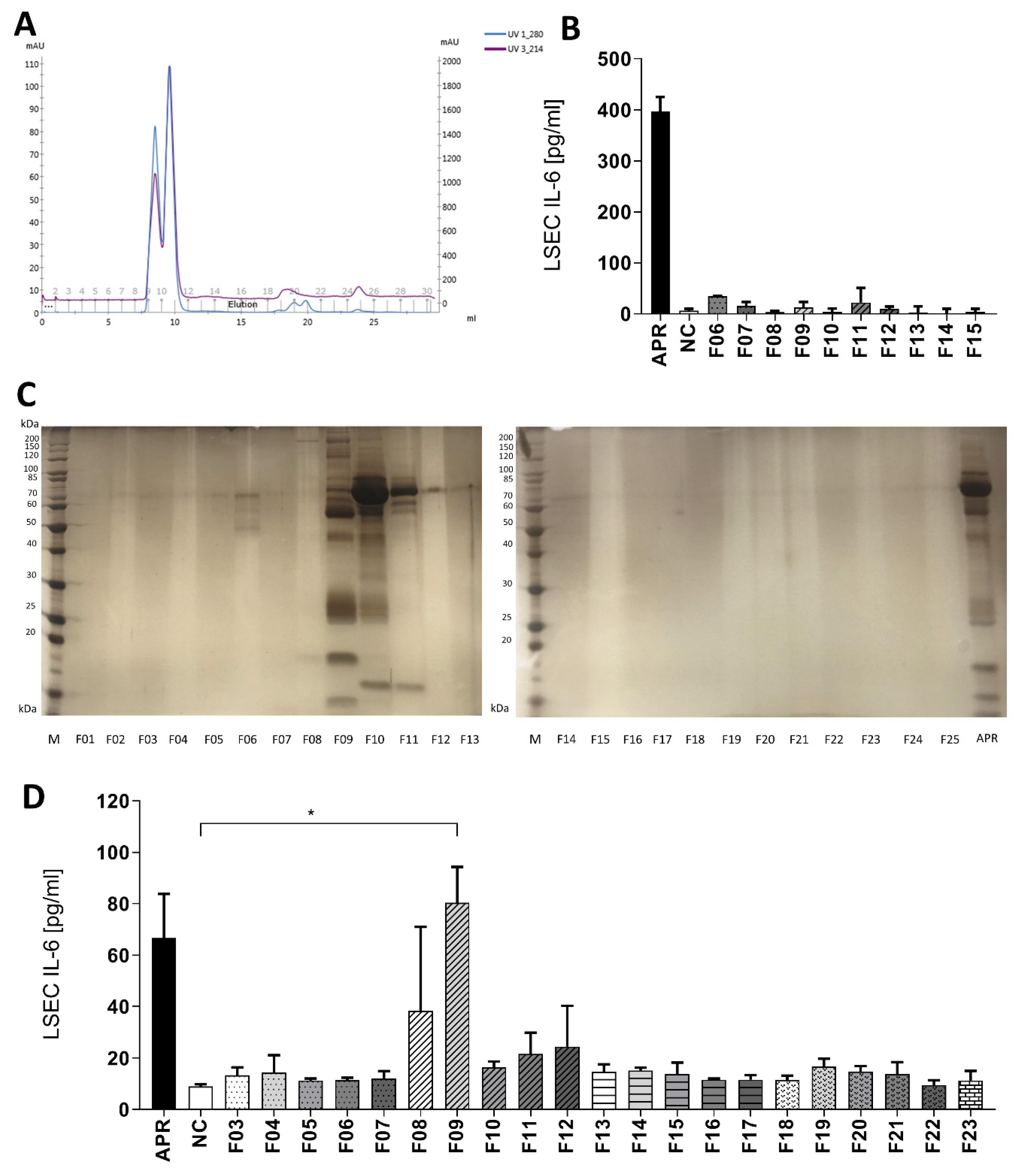
3.3. Identification by Fast Protein Liquid Chromatography of a Single Fraction Inducing Interleukin-6 Secretion in Liver Sinusoidal Endothelial Cells
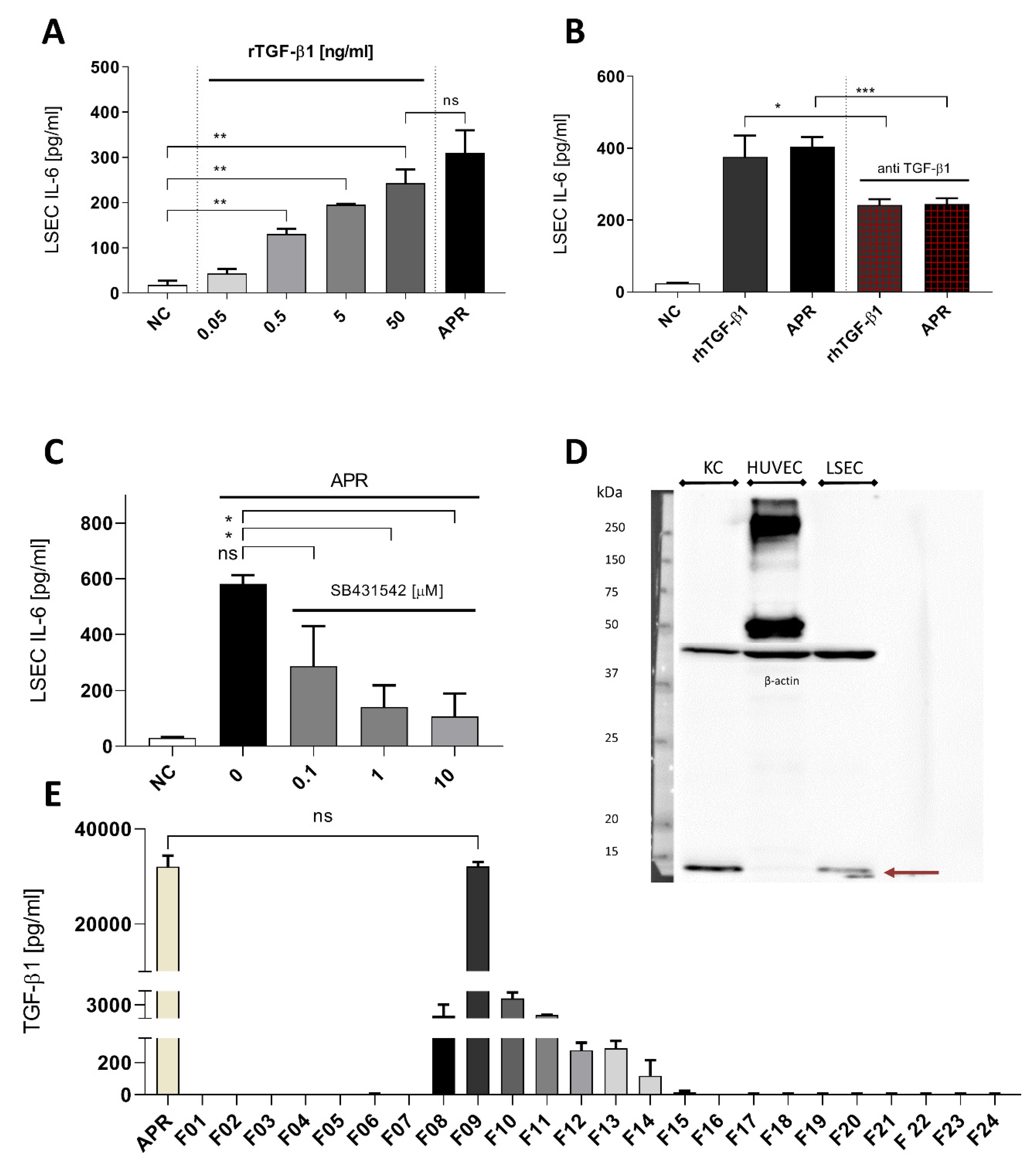
3.4. Von Willebrand Factor Induces Interleukin-6 Secretion by Liver Sinusoidal Endothelial Cells, But Its Absence in Activated Platelet Releasate Has No Impact on Interleukin-6 Secretion
3.5. Transforming Growth Factor β1 Is One of the Principal Activators in Platelet Releasate
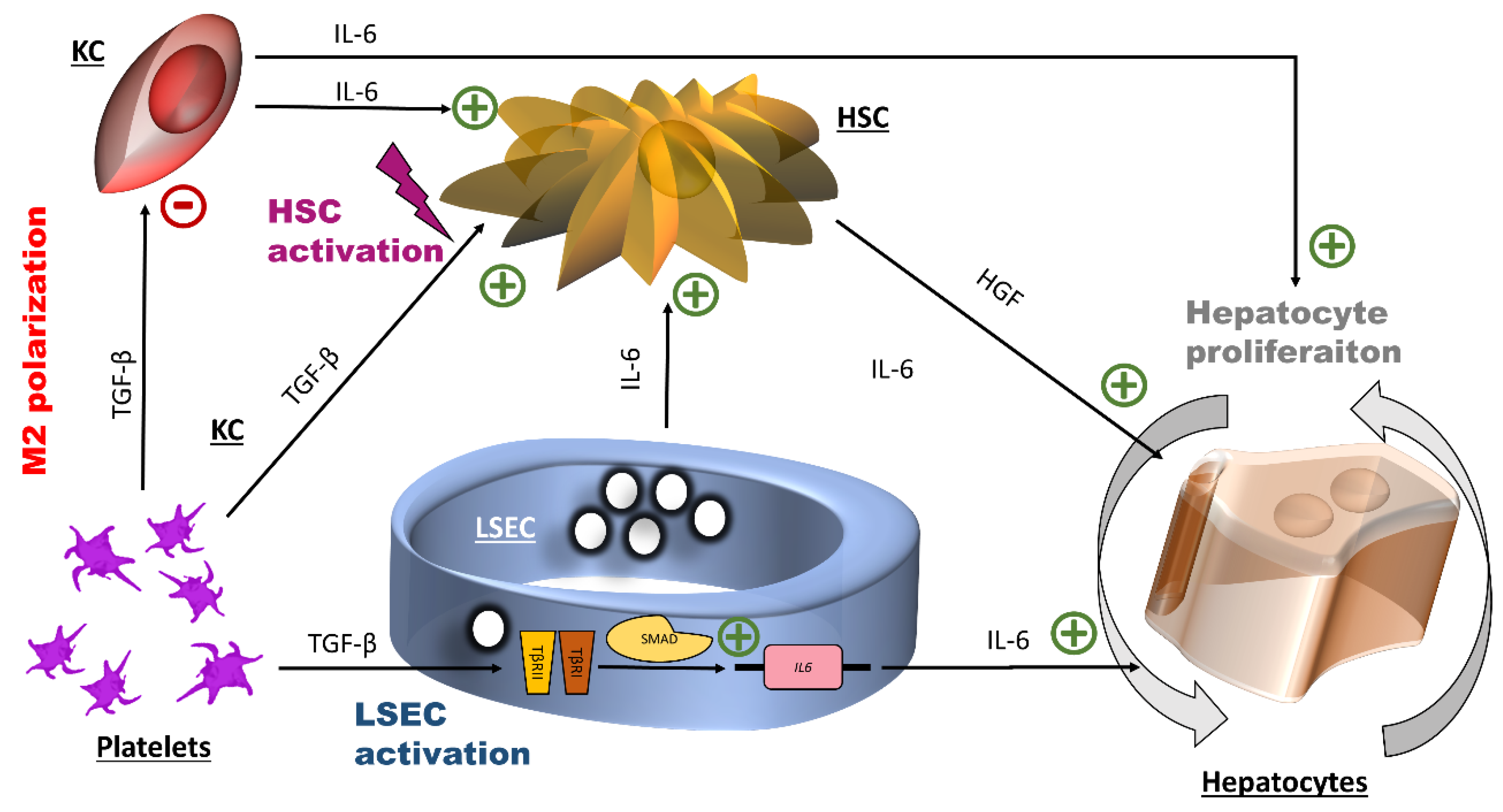
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EV | Extracellular vesicles |
| FBS | Fetal bovine serum |
| HGF | Hepatocyte growth factor |
| HSC | Hepatic stellate cells |
| HUVEC | Human umbilical vein endothelial cells |
| IL-6 | Interleukin-6 |
| KC | Kupffer cells |
| LSEC | Liver sinusoidal endothelial cells |
| vWF | von Willebrand factor |
| PDGF | Platelet-derived growth factor |
| PH | Partial hepatectomy |
| PRP | Platelet rich plasma |
| SMAD | Small mothers against decapentaplegic |
| TGF-β | Transforming growth factor β |
| TβRI | Type I TGF-β receptor |
| TβRII | Type II TGF-β receptor |
| TSP1 | Thrombospondin 1 |
| VEGF | Vascular endothelial growth factor |
References
- Meyer, J.; Lejmi, E.; Fontana, P.; Morel, P.; Gonelle-Gispert, C.; Bühler, L. A focus on the role of platelets in liver regeneration: Do platelet-endothelial cell interactions initiate the regenerative process? J. Hepatol. 2015, 63, 1263–1271. [Google Scholar] [CrossRef]
- Meyer, J.; Balaphas, A.; Fontana, P.; Sadoul, K.; Morel, P.; Gonelle-Gispert, C.; Bühler, L. Platelets in liver regeneration. ISBT Sci. Ser. 2017, 12, 455–462. [Google Scholar] [CrossRef]
- Balaphas, A.; Meyer, J.; Sadoul, K.; Fontana, P.; Morel, P.; Gonelle-Gispert, C.; Bühler, L.H. Platelets and Platelet-Derived Extracellular Vesicles in Liver Physiology and Disease. Hepatol. Commun. 2019, 3, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Karimian, G.; Adelmeijer, J.; Giepmans, B.N.G.; Porte, R.J.; Lisman, T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood 2015, 126, 798–806. [Google Scholar] [CrossRef]
- Peng, Q.; Yeh, H.; Wei, L.; Enjyoj, K.; Machaidze, Z.; Csizmad, E.; Schuetz, C.; Lee, K.M.; Deng, S.; Robson, S.C.; et al. Mechanisms of xenogeneic baboon platelet aggregation and phagocytosis by porcine liver sinusoidal endothelial cells. PLoS ONE 2012, 7, e47273. [Google Scholar] [CrossRef]
- Lejmi, E.; Meyer, J.; Sadoul, K.; Fontana, P.; Perroud, E.; Bollinger, A.; Morel, P.; Gonelle-Gispert, C.; Buhler, L.H. Human Platelets Are Internalized By Liver Sinusoidal Endothelial Cells and Enhance Secretion of EGF. Transplantation 2014, 98, 417. [Google Scholar] [CrossRef]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.-A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef]
- Starlinger, P.; Assinger, A.; Haegele, S.; Wanek, D.; Zikeli, S.; Schauer, D.; Birner, P.; Fleischmann, E.; Gruenberger, B.; Brostjan, C.; et al. Evidence for serotonin as a relevant inducer of liver regeneration after liver resection in humans. Hepatol. Baltim. Md 2014, 60, 257–266. [Google Scholar] [CrossRef]
- Matsuo, R.; Ohkohchi, N.; Murata, S.; Ikeda, O.; Nakano, Y.; Watanabe, M.; Hisakura, K.; Myronovych, A.; Kubota, T.; Narimatsu, H.; et al. Platelets Strongly Induce Hepatocyte Proliferation with IGF-1 and HGF In Vitro. J. Surg. Res. 2008, 145, 279–286. [Google Scholar] [CrossRef]
- Meyer, J.; Fontana, P.; Gonelle-Gispert, C.; Bühler, L. Reply to: “The role of platelets in liver regeneration-What do not we know?”. J. Hepatol. 2015, 63, 1538–1539. [Google Scholar] [CrossRef]
- Beldi, G.; Wu, Y.; Sun, X.; Imai, M.; Enjyoji, K.; Csizmadia, E.; Candinas, D.; Erb, L.; Robson, S.C. Regulated catalysis of extracellular nucleotides by vascular CD39/ENTPD1 is required for liver regeneration. Gastroenterology 2008, 135, 1751–1760. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shido, K.; Chavez, D.; Cao, Z.; Ko, J.L.; Rafii, S.; Ding, B.-S. Platelets prime hematopoietic–vascular niche to drive angiocrine-mediated liver regeneration. Signal Transduct. Target. Ther. 2017, 2, sigtrans201644. [Google Scholar] [CrossRef] [PubMed]
- Balaphas, A.; Meyer, J.; Perozzo, R.; Fontana, P.; Berndt, S.; Turzi, A.; Morel, P.; Scapozza, L.; Sadoul, K.; Gonelle-Gispert, C.; et al. Abstracts of the 106th Annual Congress of the Swiss Society of Surgery, held in Berne, Switzerland, 15–17 May 2019. Br. J. Surg. 2019, 106 (Suppl. S4), 5–27. [Google Scholar] [CrossRef]
- Kawasaki, T.; Murata, S.; Takahashi, K.; Nozaki, R.; Ohshiro, Y.; Ikeda, N.; Pak, S.; Myronovych, A.; Hisakura, K.; Fukunaga, K.; et al. Activation of human liver sinusoidal endothelial cell by human platelets induces hepatocyte proliferation. J. Hepatol. 2010, 53, 648–654. [Google Scholar] [CrossRef]
- Nowatari, T.; Murata, S.; Nakayama, K.; Sano, N.; Maruyama, T.; Nozaki, R.; Ikeda, N.; Fukunaga, K.; Ohkohchi, N. Sphingosine 1-phosphate has anti-apoptotic effect on liver sinusoidal endothelial cells and proliferative effect on hepatocytes in a paracrine manner in human. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2015, 45, 1136–1145. [Google Scholar] [CrossRef]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef]
- Fazel Modares, N.; Polz, R.; Haghighi, F.; Lamertz, L.; Behnke, K.; Zhuang, Y.; Kordes, C.; Häussinger, D.; Sorg, U.R.; Pfeffer, K.; et al. IL-6 Trans-signaling Controls Liver Regeneration After Partial Hepatectomy. Hepatol. Baltim. Md 2019. [Google Scholar] [CrossRef]
- Sun, R.; Jaruga, B.; Kulkarni, S.; Sun, H.; Gao, B. IL-6 modulates hepatocyte proliferation via induction of HGF/p21cip1: Regulation by SOCS3. Biochem. Biophys. Res. Commun. 2005, 338, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Kariv, R.; Enden, A.; Zvibel, I.; Rosner, G.; Brill, S.; Shafritz, D.A.; Halpern, Z.; Oren, R. Triiodothyronine and interleukin-6 (IL-6) induce expression of HGF in an immortalized rat hepatic stellate cell line. Liver Int. Off. J. Int. Assoc. Study Liver 2003, 23, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 188–202. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Lacotte, S.; Morel, P.; Gonelle-Gispert, C.; Bühler, L. An optimized method for mouse liver sinusoidal endothelial cell isolation. Exp. Cell Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wijten, P.; van Holten, T.; Woo, L.L.; Bleijerveld, O.B.; Roest, M.; Heck, A.J.R.; Scholten, A. High precision platelet releasate definition by quantitative reversed protein profiling--brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Klass, B.R.; Grobbelaar, A.O.; Rolfe, K.J. Transforming growth factor beta1 signalling, wound healing and repair: A multifunctional cytokine with clinical implications for wound repair, a delicate balance. Postgrad. Med. J. 2009, 85, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Kirschbaum, M.; Jenne, C.N.; Veldhuis, Z.J.; Sjollema, K.A.; Lenting, P.J.; Giepmans, B.N.G.; Porte, R.J.; Kubes, P.; Denis, C.V.; Lisman, T. Transient von Willebrand factor-mediated platelet influx stimulates liver regeneration after partial hepatectomy in mice. Liver Int. Off. J. Int. Assoc. Study Liver 2017. [Google Scholar] [CrossRef]
- Starlinger, P.; Pereyra, D.; Haegele, S.; Braeuer, P.; Oehlberger, L.; Primavesi, F.; Kohler, A.; Offensperger, F.; Reiberger, T.; Ferlitsch, A.; et al. Perioperative von Willebrand factor dynamics are associated with liver regeneration and predict outcome after liver resection. Hepatol. Baltim. Md 2018, 67, 1516–1530. [Google Scholar] [CrossRef]
- Starlinger, P.; Schauer, D.; Alidzanovic, L.; Zikeli, S.; Gebhardt, K.; Luf, F.; Fleischmann, E.; Perisanidis, B.; Gruenberger, B.; Gruenberger, T.; et al. Clinical evidence for thrombospondin-1 as a relevant suppressor of liver regeneration. J. Hepatol. 2013, 58, 1053–1054. [Google Scholar] [CrossRef]
- Li, Y.; Turpin, C.P.; Wang, S. Role of thrombospondin 1 in liver diseases. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2017, 47, 186–193. [Google Scholar] [CrossRef]
- Hu, J.; Srivastava, K.; Wieland, M.; Runge, A.; Mogler, C.; Besemfelder, E.; Terhardt, D.; Vogel, M.J.; Cao, L.; Korn, C.; et al. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science 2014, 343, 416–419. [Google Scholar] [CrossRef]
- Alkozai, E.M.; Nijsten, M.W.; de Jong, K.P.; de Boer, M.T.; Peeters, P.M.J.G.; Slooff, M.J.; Porte, R.J.; Lisman, T. Immediate postoperative low platelet count is associated with delayed liver function recovery after partial liver resection. Ann. Surg. 2010, 251, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Ohkohchi, N.; Matsuo, R.; Ikeda, O.; Myronovych, A.; Hoshi, R. Platelets promote liver regeneration in early period after hepatectomy in mice. World J. Surg. 2007, 31, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Jirik, F.R.; Podor, T.J.; Hirano, T.; Kishimoto, T.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J. Immunol. 1989, 142, 144–147. [Google Scholar] [PubMed]
- Podor, T.J.; Jirik, F.R.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Human endothelial cells produce IL-6. Lack of responses to exogenous IL-6. Ann. N. Y. Acad. Sci. 1989, 557, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Breviario, F.; Proserpio, P.; Biondi, A.; Vecchi, A.; Van Damme, J.; Dejana, E.; Mantovani, A. IL-1 stimulates IL-6 production in endothelial cells. J. Immunol. 1989, 142, 549–553. [Google Scholar] [PubMed]
- Loppnow, H.; Libby, P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell. Immunol. 1989, 122, 493–503. [Google Scholar] [CrossRef]
- Chi, L.; Li, Y.; Stehno-Bittel, L.; Gao, J.; Morrison, D.C.; Stechschulte, D.J.; Dileepan, K.N. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2001, 21, 231–240. [Google Scholar] [CrossRef]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.-P.; Hillan, K.J.; Ferrara, N. Angiogenesis-Independent Endothelial Protection of Liver: Role of VEGFR-1. Science 2003, 299, 890–893. [Google Scholar] [CrossRef]
- Luna, G.; Paez, J.; Cardier, J.E. Expression of the hematopoietic stem cell antigen Sca-1 (LY-6A/E) in liver sinusoidal endothelial cells: Possible function of Sca-1 in endothelial cells. Stem Cells Dev. 2004, 13, 528–535. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, F.; Liu, J.; Xiao, X.; Huang, L.; He, D. Triple labeling with three thymidine analogs reveals a well-orchestrated regulation of hepatocyte proliferation during liver regeneration. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2011, 41, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-Y.; Paris, L.L.; Chihara, R.K.; Tector, A.J.; Burlak, C. Immortalized porcine liver sinusoidal endothelial cells: An in vitro model of xenotransplantation-induced thrombocytopenia. Xenotransplantation 2012, 19, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Balaphas, A.; Meyer, J.; Sadoul, R.; Morel, P.; Gonelle-Gispert, C.; Bühler, L.H. Extracellular vesicles: Future diagnostic and therapeutic tools for liver disease and regeneration. Liver Int. Off. J. Int. Assoc. Study Liver 2019. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Tandon, N.N.; Nakamura, T.; Cone, J.; Fukuhara, S.; Kambayashi, J. High-shear-stress-induced activation of platelets and microparticles enhances expression of cell adhesion molecules in THP-1 and endothelial cells. Atherosclerosis 2001, 158, 277–287. [Google Scholar] [CrossRef]
- Laffont, B.; Corduan, A.; Plé, H.; Duchez, A.-C.; Cloutier, N.; Boilard, E.; Provost, P. Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood 2013, 122, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Wagner, G.P. The origin of platelets enabled the evolution of eutherian placentation. Biol. Lett. 2019, 15, 20190374. [Google Scholar] [CrossRef]
- Zufferey, A.; Fontana, P.; Reny, J.-L.; Nolli, S.; Sanchez, J.-C. Platelet proteomics. Mass Spectrom. Rev. 2012, 31, 331–351. [Google Scholar] [CrossRef]
- Zufferey, A.; Schvartz, D.; Nolli, S.; Reny, J.-L.; Sanchez, J.-C.; Fontana, P. Characterization of the platelet granule proteome: Evidence of the presence of MHC1 in alpha-granules. J. Proteom. 2014, 101, 130–140. [Google Scholar] [CrossRef]
- Lenting, P.J.; Casari, C.; Christophe, O.D.; Denis, C.V. Von Willebrand factor: The old, the new and the unknown. J. Thromb. Haemost. JTH 2012, 10, 2428–2437. [Google Scholar] [CrossRef]
- De Ceunynck, K.; De Meyer, S.F.; Vanhoorelbeke, K. Unwinding the von Willebrand factor strings puzzle. Blood 2013, 121, 270–277. [Google Scholar] [CrossRef]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. Von Willebrand factor biosynthesis, secretion, and clearance: Connecting the far ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Smith, D.M.; Flanders, K.C.; Sporn, M.B. Latent transforming growth factor-beta from human platelets. A high molecular weight complex containing precursor sequences. J. Biol. Chem. 1988, 263, 7646–7654. [Google Scholar]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Laping, N.J.; Grygielko, E.; Mathur, A.; Butter, S.; Bomberger, J.; Tweed, C.; Martin, W.; Fornwald, J.; Lehr, R.; Harling, J.; et al. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol. Pharmacol. 2002, 62, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Brown, C.W.; Matzuk, M.M. Genetic analysis of the mammalian transforming growth factor-beta superfamily. Endocr. Rev. 2002, 23, 787–823. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. Off. Publ. Wound Heal. Soc. Eur. Tissue Repair Soc. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Bissell, D.M.; Wang, S.S.; Jarnagin, W.R.; Roll, F.J. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J. Clin. Investig. 1995, 96, 447–455. [Google Scholar] [CrossRef]
- Ikeda, H.; Nagoshi, S.; Ohno, A.; Yanase, M.; Maekawa, H.; Fujiwara, K. Activated rat stellate cells express c-met and respond to hepatocyte growth factor to enhance transforming growth factor beta1 expression and DNA synthesis. Biochem. Biophys. Res. Commun. 1998, 250, 769–775. [Google Scholar] [CrossRef]
- Ichikawa, T.; Zhang, Y.Q.; Kogure, K.; Hasegawa, Y.; Takagi, H.; Mori, M.; Kojima, I. Transforming growth factor beta and activin tonically inhibit DNA synthesis in the rat liver. Hepatol. Baltim. Md 2001, 34, 918–925. [Google Scholar] [CrossRef]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. TGF-beta/Smad signaling in the injured liver. Z. Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- O’Connor-McCourt, M.D.; Wakefield, L.M. Latent transforming growth factor-beta in serum. A specific complex with alpha 2-macroglobulin. J. Biol. Chem. 1987, 262, 14090–14099. [Google Scholar] [PubMed]
- Kang, L.-I.; Mars, W.M.; Michalopoulos, G.K. Signals and cells involved in regulating liver regeneration. Cells 2012, 1, 1261–1292. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Park, J.-I.; Lee, M.-G.; Cho, K.; Park, B.-J.; Chae, K.-S.; Byun, D.-S.; Ryu, B.-K.; Park, Y.-K.; Chi, S.-G. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene 2003, 22, 4314–4332. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Kwon, S.J.; Lee, J.-H.; Jeon, S.S.; Jang, K.T.; Choi, H.Y.; Lee, H.M.; Kim, W.-J.; Kim, S.J.; Kim, I.Y. Induction of Interleukin-6 Expression by Bone Morphogenetic Protein-6 in Macrophages Requires Both SMAD and p38 Signaling Pathways. J. Biol. Chem. 2010, 285, 39401–39408. [Google Scholar] [CrossRef]
- Qi, Y.; Ge, J.; Ma, C.; Wu, N.; Cui, X.; Liu, Z. Activin A regulates activation of mouse neutrophils by Smad3 signalling. Open Biol. 2017, 7. [Google Scholar] [CrossRef]
- Crist, A.M.; Zhou, X.; Garai, J.; Lee, A.R.; Thoele, J.; Ullmer, C.; Klein, C.; Zabaleta, J.; Meadows, S.M. Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation 2019, 139, 2049–2063. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Gene Name | Protein | Peptide | MW (kDa) | Identity (%) |
|---|---|---|---|---|---|
| 1 | TGFB1 | Transforming growth factor β-1 | 5 | 44 | 89.7 |
| 2 | TGFBI | Transforming growth factor, β-induced | 8 | 75 | 90.6 |
| 3 | SERPINA3 | Alpha-1-antichymotrypsin | 22 | 48 | na |
| 4 | SERPINC1 | Antithrombin-III | 4 | 53 | 87.3 |
| 5 | C6 | Complement factor 6 | 18 | 105 | na |
| 6 | CFB | Complement factor B | 22 | 86 | 83.6 |
| 7 | FN1 | Fibronectin | 62 | 263 | 88.2 |
| 8 | NID1 | Nidogen-1 | 26 | 136 | 85 |
| 9 | THBS1 | Thrombospondin-1 | 50 | 129 | 94.8 |
| 10 | VWF | Von Willebrand factor | 29 | 309 | 83.2 |
| 11 | CALU | Calumenin | 3 | 37 | 98.1 |
| 12 | CP | Ceruloplasmin | 49 | 122 | 83.1 |
| 13 | SPARC | Basement-membrane protein 40 | 4 | 35 | 92.4 |
| 14 | TTR | Transthyretin | 7 | 16 | 81.6 |
| 15 | APLP2 | Amyloid-like protein 2 | 8 | 87 | 85.5 |
| 16 | FSTL1 | Follistatin-related protein 1 | 2 | 35 | 91.9 |
| 17 | MAN2A1 | Alpha-mannosidase 2 | 2 | 131 | 80.4 |
| 18 | SERPINA4 | Kallistatin | 2 | 49 | na |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaphas, A.; Meyer, J.; Perozzo, R.; Zeisser-Labouebe, M.; Berndt, S.; Turzi, A.; Fontana, P.; Scapozza, L.; Gonelle-Gispert, C.; Bühler, L.H. Platelet Transforming Growth Factor-β1 Induces Liver Sinusoidal Endothelial Cells to Secrete Interleukin-6. Cells 2020, 9, 1311. https://doi.org/10.3390/cells9051311
Balaphas A, Meyer J, Perozzo R, Zeisser-Labouebe M, Berndt S, Turzi A, Fontana P, Scapozza L, Gonelle-Gispert C, Bühler LH. Platelet Transforming Growth Factor-β1 Induces Liver Sinusoidal Endothelial Cells to Secrete Interleukin-6. Cells. 2020; 9(5):1311. https://doi.org/10.3390/cells9051311
Chicago/Turabian StyleBalaphas, Alexandre, Jeremy Meyer, Remo Perozzo, Magali Zeisser-Labouebe, Sarah Berndt, Antoine Turzi, Pierre Fontana, Leonardo Scapozza, Carmen Gonelle-Gispert, and Leo H. Bühler. 2020. "Platelet Transforming Growth Factor-β1 Induces Liver Sinusoidal Endothelial Cells to Secrete Interleukin-6" Cells 9, no. 5: 1311. https://doi.org/10.3390/cells9051311
APA StyleBalaphas, A., Meyer, J., Perozzo, R., Zeisser-Labouebe, M., Berndt, S., Turzi, A., Fontana, P., Scapozza, L., Gonelle-Gispert, C., & Bühler, L. H. (2020). Platelet Transforming Growth Factor-β1 Induces Liver Sinusoidal Endothelial Cells to Secrete Interleukin-6. Cells, 9(5), 1311. https://doi.org/10.3390/cells9051311