Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
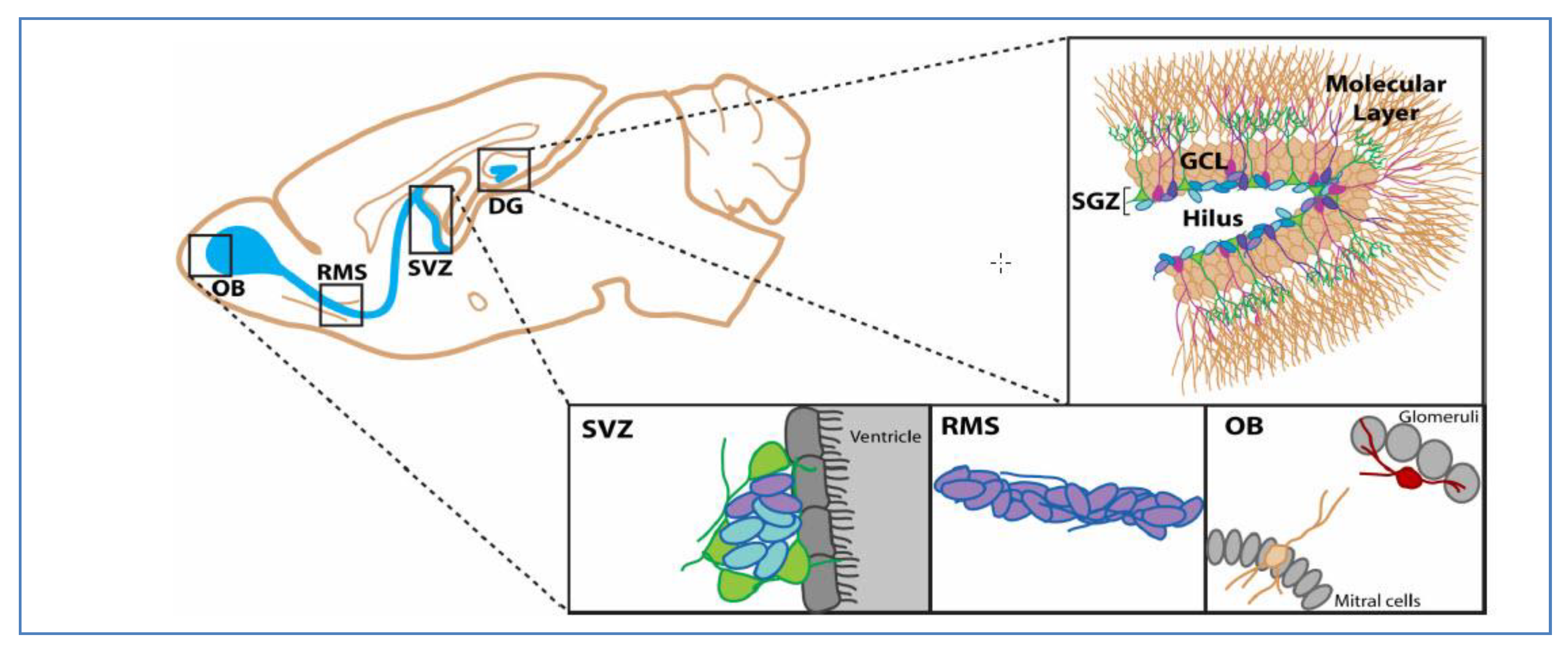
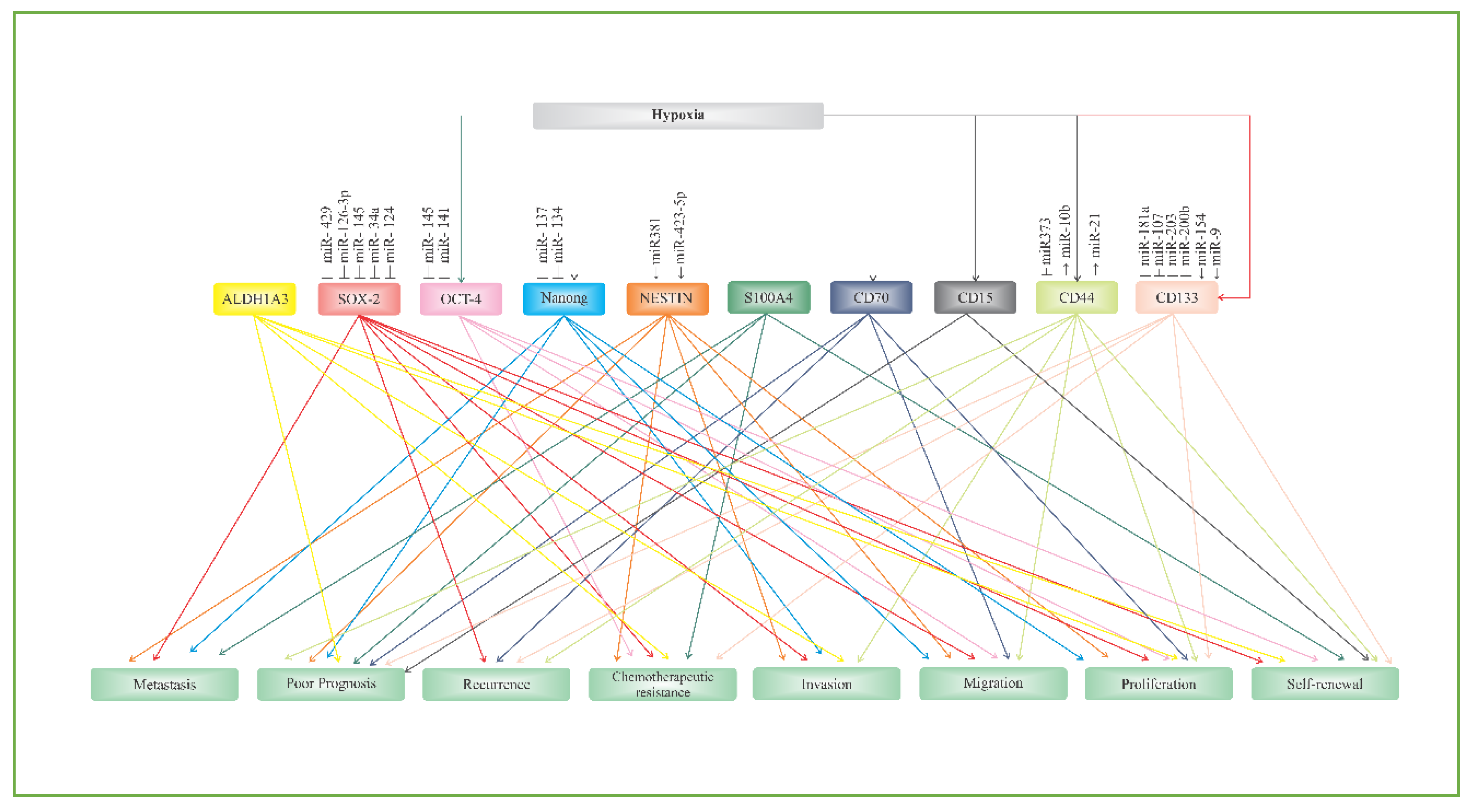
2. Putative Biomarkers of Glioblastoma Stem Cells
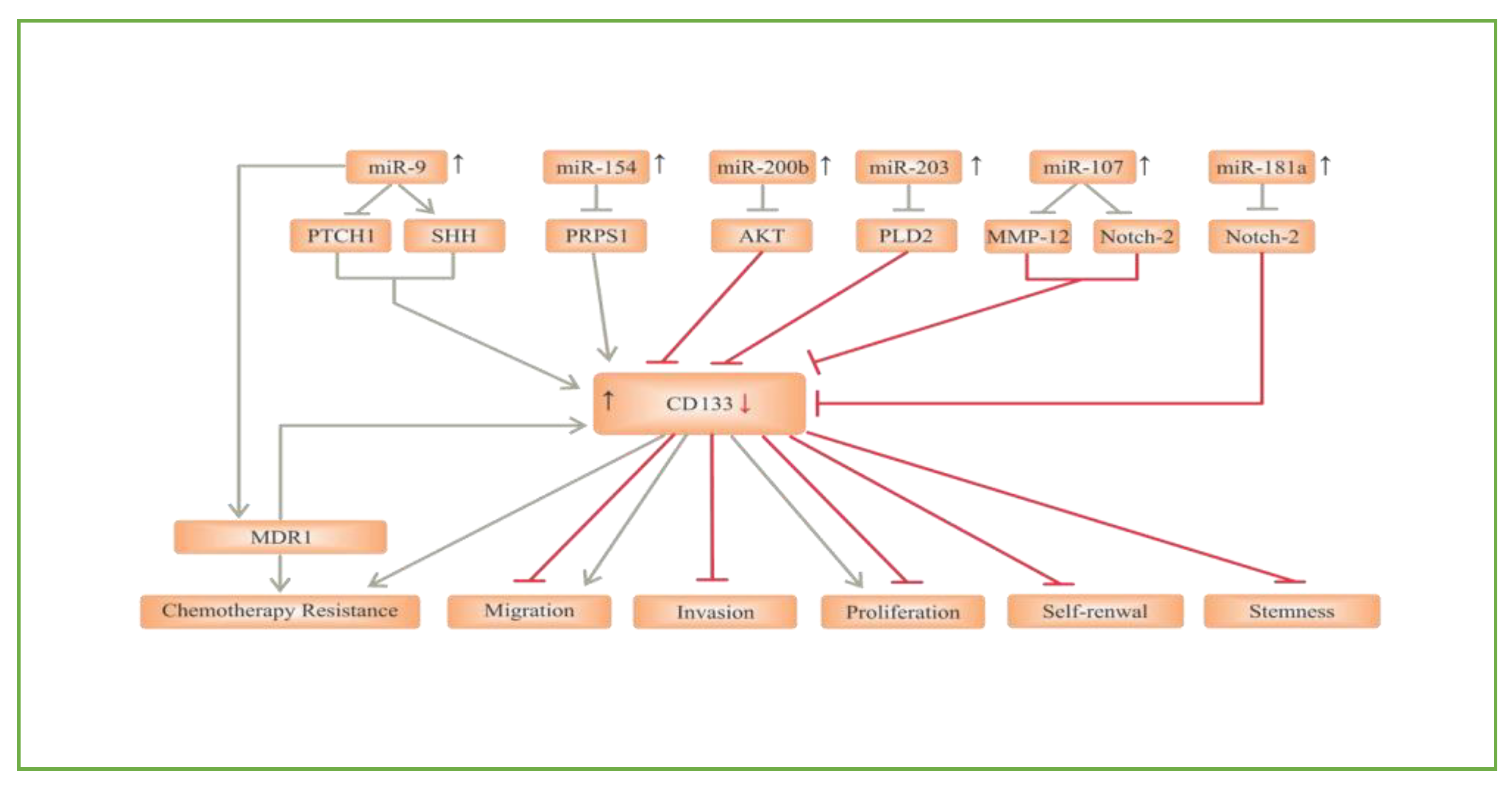
2.1. CD133
2.2. CD44
2.3. CD15
2.4. CD70 (CD27L)
2.5. S100A4
2.6. ALDH1A3
2.7. NANOG
2.8. OCT-4
2.9. SOX-2
2.10. NESTIN
3. Conclusions
Funding
Conflicts of Interest
References
- Rajasekhar, V.K.; Vemuri, M.C. Regulatory Networks in Stem Cells; Humana Press: Totowa, NJ, USA, 2009. [Google Scholar]
- Bonnet, M.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [PubMed]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; D’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 1–17. [Google Scholar]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [PubMed]
- Llaguno, S.R.A.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural Stem Cells and the Origin of Gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar]
- Johnson, M.A.; Ables, J.L.; Eisch, A.J. Cell-intrinsic signals that regulate adult neurogenesis in vivo: Insights from inducible approaches. BMB Rep. 2009, 42, 245–259. [Google Scholar]
- Bent, M.V.D. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: A clinician’s perspective. Acta Neuropathol. 2010, 120, 297–304. [Google Scholar]
- Ellison, D.W.; Kocak, M.; Figarella-Branger, D.; Felice, G.; Godfraind, C.; Pietsch, T.; Frappaz, D.; Massimino, M.; Grill, J.; Boyett, J.M.; et al. Histopathological grading of pediatric ependymoma: Reproducibility and clinical relevance in European trial cohorts. J. Negat. Results Biomed. 2011, 10, 7. [Google Scholar]
- Sturm, D.; Orr, B.A.; Toprak, U.H.; Hovestadt, V.; Jones, D.T.W.; Capper, D.; Sill, M.; Buchhalter, I.; Northcott, P.A.; Leis, I.; et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164, 1060–1072. [Google Scholar]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar]
- Molinaro, A.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [PubMed]
- Pearson, J.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal. Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [PubMed]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2012, 19, 764–772. [Google Scholar] [PubMed]
- Li, B.; McCrudden, C.M.; Yuen, H.F.; Xi, X.; Lyu, P.; Chan, K.W.; Zhang, S.-D.; Kwok, H.F. CD133 in brain tumor: The prognostic factor. Oncotarget 2016, 8, 11144–11159. [Google Scholar]
- Xia, P. CD133 mRNA may be a suitable prognostic marker for human breast cancer. Stem Cell Investig. 2017, 4, 87. [Google Scholar]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2018, 27, 257–269. [Google Scholar]
- Han, M.; Guo, L.; Zhang, Y.; Huang, B.; Chen, A.; Chen, W.; Liu, X.; Sun, S.; Wang, K.; Liu, A.; et al. Clinicopathological and Prognostic Significance of CD133 in Glioma Patients: A Meta-Analysis. Mol. Neurobiol. 2015, 53, 720–727. [Google Scholar]
- Kazama, S.; Kishikawa, J.; Kiyomatsu, T.; Kawai, K.; Nozawa, H.; Ishihara, S.; Watanabe, T. Expression of the stem cell marker CD133 is related to tumor development in colorectal carcinogenesis. Asian J. Surg. 2018, 41, 274–278. [Google Scholar]
- Yoon, C.-H.; Kim, M.-J.; Kim, R.-K.; Lim, E.-J.; Choi, K.-S.; An, S.; Hwang, S.-G.; Kang, S.-G.; Suh, Y.; Park, M.-J.; et al. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene 2012, 31, 4655–4666. [Google Scholar]
- Ulasov, I.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic Hedgehog and Notch Pathways Enhances Sensitivity of CD133+ Glioma Stem Cells to Temozolomide Therapy. Mol. Med. 2010, 17, 103–112. [Google Scholar]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Köhrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [PubMed]
- Kuo, S.-H.; Yang, S.-Y.; You, S.-L.; Lien, H.-C.; Lin, C.; Lin, P.; Huang, C.-S. Polymorphisms of ESR1, UGT1A1, HCN1, MAP3K1 and CYP2B6 are associated with the prognosis of hormone receptor-positive early breast cancer. Oncotarget 2017, 8, 20925–20938. [Google Scholar] [PubMed]
- Gao, L.; Chen, B.; Li, J.; Yang, F.; Cen, X.; Liao, Z.; Long, X. Wnt/β-catenin signaling pathway inhibits the proliferation and apoptosis of U87 glioma cells via different mechanisms. PLoS ONE 2017, 12, e0181346. [Google Scholar]
- Kahlert, U.D.; Suwala, A.K.; Koch, K.; Natsumeda, M.; Orr, B.A.; Hayashi, M.; Maciaczyk, J.; Eberhart, C.G. Pharmacologic Wnt Inhibition Reduces Proliferation, Survival, and Clonogenicity of Glioblastoma Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 889–900. [Google Scholar]
- Wang, P.; Wan, W.-W.; Xiong, S.-L.; Feng, H.; Wu, N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017, 3, 16105. [Google Scholar]
- Aghajani, M.; Mansoori, B.; Mohammadi, A.; Asadzadeh, Z.; Baradaran, B. New emerging roles of CD133 in cancer stem cell: Signaling pathway and miRNA regulation. J. Cell. Physiol. 2019, 234, 21642–21661. [Google Scholar]
- Huang, S.-X.; Zhao, Z.-Y.; Weng, G.-H.; He, X.-Y.; Wu, C.-J.; Fu, C.-Y.; Sui, Z.-Y.; Ma, Y.-S.; Liu, T. Upregulation of miR-181a suppresses the formation of glioblastoma stem cells by targeting the Notch2 oncogene and correlates with good prognosis in patients with glioblastoma multiforme. Biochem. Biophys. Res. Commun. 2017, 486, 1129–1136. [Google Scholar]
- Chen, L.; Chen, X.-R.; Chen, F.-F.; Liu, Y.; Li, P.; Zhang, R.; Yan, K.; Yi, Y.-J.; Xu, Z.-M.; Jang, X.-D. MicroRNA-107 Inhibits U87 Glioma Stem Cells Growth and Invasion. Cell. Mol. Neurobiol. 2013, 33, 651–657. [Google Scholar]
- Chen, Z.; Li, D.; Cheng, Q.; Ma, Z.; Jiang, B.; Peng, R.; Chen, R.; Cao, Y.; Wan, X. MicroRNA-203 inhibits the proliferation and invasion of U251 glioblastoma cells by directly targeting PLD2. Mol. Med. Rep. 2013, 9, 503–508. [Google Scholar]
- Deng, Y.; Zhu, G.; Luo, H.; Zhao, S. MicroRNA-203 As a Stemness Inhibitor of Glioblastoma Stem Cells. Mol. Cells 2016, 39, 619–624. [Google Scholar]
- Liu, A.; Yu, Q.; Peng, Z.; Huang, Y.; Diao, S.; Cheng, J.; Wang, W.; Hong, M. miR-200b inhibits CD133+ glioma cells by targeting the AKT pathway. Oncol. Lett. 2017, 13, 4701–4707. [Google Scholar] [PubMed]
- Zhao, W.J.; Yang, L.; He, H.J. MiR-200b suppresses proliferation of glioma and its stem cells by targeting CD133. Tumor 2014, 34, 231–237. [Google Scholar]
- Zhao, C.; Ma, Z.; Mou, S.; Yang, Y.; Zhang, Y.; Yao, W. Targeting effect of microRNA on CD133 and its impact analysis on proliferation and invasion of glioma cells. Genet. Mol. Res. 2017, 16, 16. [Google Scholar]
- Yang, L.; Yan, Z.; Wang, Y.; Ma, W.; Li, C. Down-expression of miR-154 suppresses tumourigenesis in CD133+glioblastoma stem cells. Cell Biochem. Funct. 2016, 34, 404–413. [Google Scholar] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Rameshwar, P. High expression of miR-9 in CD133+ glioblastoma cells in chemoresistance to temozolomide. J. Cancer Stem Cell Res. 2015, 3, 1. [Google Scholar]
- Brown, D.V.; Filiz, G.; Daniel, P.M.; Hollande, F.; Dworkin, S.; Amiridis, S.; Kountouri, N.; Ng, W.; Morokoff, A.; Mantamadiotis, T. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS ONE 2017, 12, e0172791. [Google Scholar]
- Barrantes-Freer, A.; Renovanz, M.; Eich, M.; Braukmann, A.; Sprang, B.; Spirin, P.; Pardo, L.A.; Giese, A.; Kim, E. CD133 Expression Is Not Synonymous to Immunoreactivity for AC133 and Fluctuates throughout the Cell Cycle in Glioma Stem-Like Cells. PLoS ONE 2015, 10, e0130519. [Google Scholar]
- Dahlrot, R.H.; Hansen, S.; Jensen, S.S.; Schrøder, H.D.; Hjelmborg, J.; Kristensen, B.W. Clinical value of CD133 and nestin in patients with glioma: A population-based study. Int. J. Clin. Exp. Pathol. 2014, 7, 3739–3751. [Google Scholar]
- Lim, S.; Kim, H.; Ju, S.; Shin, S.; Cho, I.-J.; Park, S.-H.; Grailhe, R.; Lee, C.; Kim, Y.K. Glioblastoma-secreted soluble CD44 activates tau pathology in the brain. Exp. Mol. Med. 2018, 50, 5. [Google Scholar]
- Fakhri, S.; Mehrjardi, A.Z.; Noori, M. Expression of CD44 and CD133 in glioma stem cells. Int. J. Tumor Ther. 2018, 7, 27–33. [Google Scholar]
- Klank, R.L.; Grunke, S.A.D.; Bangasser, B.L.; Forster, C.L.; Price, M.A.; Odde, T.J.; Santacruz, K.S.; Rosenfeld, S.S.; Canoll, P.; Turley, E.A.; et al. Biphasic Dependence of Glioma Survival and Cell Migration on CD44 Expression Level. Cell Rep. 2017, 18, 23–31. [Google Scholar] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 1–15. [Google Scholar]
- Johansson, E.; Grassi, E.S.; Pantazopoulou, V.; Tong, B.; Lindgren, D.; Berg, T.J.; Pietras, E.J.; Axelson, H.; Pietras, A. CD44 Interacts with HIF-2α to Modulate the Hypoxic Phenotype of Perinecrotic and Perivascular Glioma Cells. Cell Rep. 2017, 20, 1641–1653. [Google Scholar] [PubMed]
- Wang, H.-H.; Liao, C.-C.; Chow, N.-H.; Huang, L.L.-H.; Chuang, J.-I.; Wei, K.-C.; Shin, J.-W. Whether CD44 is an applicable marker for glioma stem cells. Am. J. Transl. Res. 2017, 9, 4785–4806. [Google Scholar] [PubMed]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.-H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar]
- Bhat, K.P.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal Differentiation Mediated by NF-ΚB Promotes Radiation Resistance in Glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar]
- Brown, D.V.; Daniel, P.M.; D’Abaco, G.M.; Gogos, A.; Ng, W.; Morokoff, A.; Mantamadiotis, T. Coexpression analysis of CD133 and CD44 identifies Proneural and Mesenchymal subtypes of glioblastoma multiforme. Oncotarget 2015, 6, 6267–6280. [Google Scholar]
- Sasayama, T.; Nishihara, M.; Kondoh, T.; Hosoda, K.; Kohmura, E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RhoC. Int. J. Cancer 2009, 125, 1407–1413. [Google Scholar]
- Bourguignon, L. Hyaluronan-CD44 interaction promotes microRNA signaling and RhoGTPase activation leading to tumor progression. Small GTPases 2012, 3, 53–59. [Google Scholar]
- Wei, F.; Cao, C.; Xu, X.; Wang, J. Diverse functions of miR-373 in cancer. J. Transl. Med. 2015, 13, 162. [Google Scholar]
- Wei, F.; Wang, Q.; Su, Q.; Huang, H.; Luan, J.; Xu, X.; Wang, J. miR-373 Inhibits Glioma Cell U251 Migration and Invasion by Down-Regulating CD44 and TGFBR2. Cell. Mol. Neurobiol. 2016, 36, 1389–1397. [Google Scholar] [PubMed]
- Yamamuro, S.; Okamoto, Y.; Sano, E.; Ochiai, Y.; Ogino, A.; Ohta, T.; Hara, H.; Ueda, T.; Nakayama, T.; Yoshino, A.; et al. Characterization of glioma stem-like cells from human glioblastomas. Int. J. Oncol. 2015, 47, 91–96. [Google Scholar] [PubMed]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [PubMed]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-Associated Neutrophils as a New Prognostic Factor in Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e98259. [Google Scholar]
- Kenney-Herbert, E.; Al-Mayhani, T.; Piccirillo, S.G.M.; Fowler, J.; Spiteri, I.; Jones, P.H.; Watts, C. CD15 Expression Does Not Identify a Phenotypically or Genetically Distinct Glioblastoma Population. STEM CELLS Transl. Med. 2015, 4, 822–831. [Google Scholar]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar]
- William, D.; Walther, M.; Schneider, B.; Linnebacher, M.; Classen, C.F. Temozolomide-induced increase of tumorigenicity can be diminished by targeting of mitochondria in in vitro models of patient individual glioblastoma. PLoS ONE 2018, 13, e0191511. [Google Scholar]
- Lazari, D.; Alexiou, G.A.; Markopoulos, G.S.; Vartholomatos, E.; Hodaj, E.; Chousidis, I.; Leonardos, I.; Galani, V.; Kyritsis, A.P. N-(p-coumaroyl) serotonin inhibits glioblastoma cells growth through triggering S-phase arrest and apoptosis. J. Neuro-Oncology 2017, 28, 373–381. [Google Scholar]
- Vincent, P.; for the H.E.S.S. collaboration; De Angelis, A.; Mansutti, O.H.E.S.S. Science with the New Generation of High Energy Gamma-Ray Experiments; World Scientific Pub Co Pte Ltd: Singapore, 2006; Volume 71, pp. 11–19. [Google Scholar]
- Pratt, D.; Pittaluga, S.; Palisoc, M.; Fetsch, P.; Xi, L.; Raffeld, M.; Gilbert, M.R.; Quezado, M. Expression of CD70 (CD27L) Is Associated With Epithelioid and Sarcomatous Features in IDH-Wild-Type Glioblastoma. J. Neuropathol. Exp. Neurol. 2017, 76, 697–708. [Google Scholar]
- Jin, L.; Ge, H.; Yang, C.; Long, Y.; Chang, Y.; Mu, L.; Sayour, E.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70 as a novel target of CAR-T-cell therapy for gliomas. J. Clin. Oncol. 2017, 35, 148. [Google Scholar]
- Ge, H.; Mu, L.; Jin, L.; Yang, C.; Chang, Y.E.; Long, Y.; DeLeon, G.; Deleyrolle, L.; Mitchell, D.A.; Kubilis, P.S.; et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int. J. Cancer 2017, 141, 1434–1444. [Google Scholar] [PubMed]
- Al Sayed, M.F.; Jaeger-Ruckstuhl, C.A.; Hilmenyuk, T.; Claus, C.; Bourquin, J.-P.; Bornhauser, B.C.; Radpour, R.; Riether, C.; Ochsenbein, A.F. CD70 reverse signaling enhances NK cell function and immunosurveillance in CD27-expressing B-cell malignancies. Blood 2017, 130, 297–309. [Google Scholar] [PubMed]
- Wang, Q.J.; Yu, Z.; Hanada, K.-I.; Patel, K.; Kleiner, D.; Restifo, N.P.; Yang, J.C. Preclinical Evaluation of Chimeric Antigen Receptors Targeting CD70-Expressing Cancers. Clin. Cancer Res. 2016, 23, 2267–2276. [Google Scholar] [PubMed]
- Chahlavi, A.; Rayman, P.; Richmond, A.L.; Biswas, K.; Zhang, R.; Vogelbaum, M.; Tannenbaum, C.; Barnett, G.; Finke, J.H. Glioblastomas Induce T-Lymphocyte Death by Two Distinct Pathways Involving Gangliosides and CD70. Cancer Res. 2005, 65, 5428–5438. [Google Scholar] [PubMed]
- Kitajima, S.; Lee, K.L.; Fujioka, M.; Sun, W.; You, J.; Chia, G.S.; Wanibuchi, H.; Tomita, S.; Araki, M.; Kato, H.; et al. Hypoxia-inducible factor-2 alpha up-regulates CD70 under hypoxia and enhances anchorage-independent growth and aggressiveness in cancer cells. Oncotarget 2018, 9, 19123–19135. [Google Scholar]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar]
- Chow, K.-H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.A.; Neilson, E.G.; et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar]
- Cheng, L.-H.; Hung, K.-F.; Huang, T.-F.; Hsieh, H.-P.; Wang, S.-Y.; Huang, C.-Y.; Lo, J.-F. Attenuation of cancer-initiating cells stemness properties by abrogating S100A4 calcium binding ability in head and neck cancers. Oncotarget 2016, 7, 78946–78957. [Google Scholar]
- Decaestecker, C.; De Clercq, S.; Salmon, I. S100A4, a key factor in glioblastoma biology. Transl. Cancer Res. 2018, 7, S71–S73. [Google Scholar]
- Pankratova, S.; Klingelhofer, J.; Dmytriyeva, O.; Owczarek, S.; Renziehausen, A.; Syed, N.; Porter, A.E.; Dexter, D.T.; Kiryushko, D. The S100A4 Protein Signals through the ErbB4 Receptor to Promote Neuronal Survival. Theranostics 2018, 8, 3977–3990. [Google Scholar] [PubMed]
- Ambartsumian, N.; Klingelhöfer, J.; Grigorian, M. The Multifaceted S100A4 Protein in Cancer and Inflammation. In Calcium-Binding Proteins of the EF-Hand Superfamily; Humana Press: New York, NY, USA, 2019; pp. 339–365. [Google Scholar]
- Dmytriyeva, O.; Pankratova, S.; Owczarek, S.; Sonn, K.; Soroka, V.; Ridley, C.M.; Marsolais, A.; López-Hoyos, M.; Ambartsumian, N.; Lukanidin, E.; et al. The metastasis-promoting S100A4 protein confers neuroprotection in brain injury. Nat. Commun. 2012, 3, 1197. [Google Scholar]
- He, Z.; Yu, L.; Luo, S.; Li, M.; Li, J.; Li, Q.; Sun, Y.; Wang, C. miR-296 inhibits the metastasis and epithelial-mesenchymal transition of colorectal cancer by targeting S100A4. BMC Cancer 2017, 17, 140. [Google Scholar]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; De Groot, J. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2013, 20, 187–198. [Google Scholar] [PubMed]
- Xu, Y.; Wang, J.; Xu, Y.; Xiao, H.; Li, J.; Wang, Z. Screening critical genes associated with malignant glioma using bioinformatics analysis. Mol. Med. Rep. 2017, 16, 6580–6589. [Google Scholar]
- Aguilar-Morante, D.; Morales-Garcia, J.A.; Santos, Á.; Perez-Castillo, A. CCAAT/Enhancer binding protein β induces motility and invasion of glioblastoma cells through transcriptional regulation of the calcium binding protein S100A4. Oncotarget 2015, 6, 4369–4384. [Google Scholar]
- Feng, H.; Liu, Y.; Bian, X.; Zhou, F.; Liu, Y. ALDH1A3 affects colon cancer in vitro proliferation and invasion depending on CXCR4 status. Br. J. Cancer 2017, 118, 224–232. [Google Scholar]
- Wu, W.; Schecker, J.; Würstle, S.; Schneider, F.; Schönfelder, M.; Schlegel, J. Aldehyde dehydrogenase 1A3 (ALDH1A3) is regulated by autophagy in human glioblastoma cells. Cancer Lett. 2018, 417, 112–123. [Google Scholar]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Fedele, M. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar]
- Cheng, P.; Wang, J.; Waghmare, I.; Sartini, S.; Coviello, V.; Zhang, Z.; Kim, S.-H.; Mohyeldin, A.; Pavlyukov, M.S.; Minata, M.; et al. FOXD1-ALDH1A3 Signaling Is a Determinant for the Self-Renewal and Tumorigenicity of Mesenchymal Glioma Stem Cells. Cancer Res. 2016, 76, 7219–7230. [Google Scholar] [PubMed]
- Zhang, W.; Liu, Y.; Hu, H.; Huang, H.; Bao, Z.; Yang, P.; Wang, Y.; You, G.; Yan, W.; Jiang, T.; et al. ALDH1A3: A Marker of Mesenchymal Phenotype in Gliomas Associated with Cell Invasion. PLoS ONE 2015, 10, e0142856. [Google Scholar]
- Li, M.; Xiao, A.; Floyd, D.; Olmez, I.; Lee, J.; Godlewski, J.; Bronisz, A.; Bhat, K.P.; Sulman, E.P.; Nakano, I.; et al. CDK4/6 Inhibition Is More Active against the Glioblastoma Proneural Subtype. Oncotarget 2017, 8, 55319–55331. [Google Scholar] [PubMed]
- Li, G.; Li, Y.; Liu, X.; Wang, Z.; Zhang, C.; Wu, F.; Jiang, H.; Zhang, W.; Bao, Z.; Wang, Y.; et al. ALDH1A3 induces mesenchymal differentiation and serves as a predictor for survival in glioblastoma. Cell Death Dis. 2018, 9, 1190. [Google Scholar] [PubMed]
- Sullivan, K.E.; Rojas, K.; Cerione, R.A.; Nakano, I.; Wilson, K.F. The stem cell/cancer stem cell marker ALDH1A3 regulates the expression of the survival factor tissue transglutaminase, in mesenchymal glioma stem cells. Oncotarget 2017, 8, 22325–22343. [Google Scholar]
- Huang, G.; Ho, B.; Conroy, J.; Liu, S.; Qiang, H.; Golubovskaya, V.M. The microarray gene profiling analysis of glioblastoma cancer cells reveals genes affected by FAK inhibitor Y15 and combination of Y15 and temozolomide. Anti-Cancer Agents Med. Chem. 2014, 14, 9–17. [Google Scholar]
- Hattermann, K.; Flüh, C.; Engel, D.; Mehdorn, H.M.; Synowitz, M.; Mentlein, R.; Held-Feindt, J. Stem cell markers in glioma progression and recurrence. Int. J. Oncol. 2016, 49, 1899–1910. [Google Scholar]
- Runtu, F.; Ichwan, S.; I. Wanandi, S.; Hardiany, N.S. Evaluating the potential of NANOG as a glioma malignancy biomarker. J. Physics: Conf. Ser. 2018, 1073, 032057. [Google Scholar]
- Liang, C.; Zhao, T.; Ge, H.; Xu, Y.; Ren, S.; Yue, C.; Li, G.; Wu, J. The clinicopathological and prognostic value of Nanog in human gastrointestinal luminal cancer: A meta-analysis. Int. J. Surg. 2018, 53, 193–200. [Google Scholar]
- Yang, Y.; Niu, C.-S.; Cheng, C.-D. Pin1-Nanog expression in human glioma is correlated with advanced tumor progression. Oncol. Rep. 2013, 30, 560–566. [Google Scholar]
- Kakiuchi, S.; Minami, Y.; Miyata, Y.; Mizutani, Y.; Goto, H.; Kawamoto, S.; Yakushijin, K.; Kurata, K.; Matsuoka, H.; Minami, H. NANOG Expression as a Responsive Biomarker during Treatment with Hedgehog Signal Inhibitor in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 486. [Google Scholar]
- Liu, Y.; Liu, D.L.; Dong, L.L.; Wen, D.; Shi, D.M.; Zhou, J.; Fan, J.; Wu, W.Z. MiR-612 Suppresses Stem Cell-like Property of Hepatocellular Carcinoma Cells by Modulating Sp1/Nanog Signaling. Cell Death Dis. 2016, 7, e2377. [Google Scholar] [PubMed]
- Elsir, T.; Edqvist, P.-H.; Carlsson, J.; Ribom, D.; Bergqvist, M.; Ekman, S.; Popova, S.; Alafuzoff, I.; Ponten, F.; Nistér, M.; et al. A study of embryonic stem cell-related proteins in human astrocytomas: Identification of Nanog as a predictor of survival. Int. J. Cancer 2013, 134, 1123–1131. [Google Scholar] [PubMed]
- Niu, C.; Li, D.-X.; Liu, Y.-H.; Fu, X.M.; Tang, S.-F. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol. Rep. 2011, 26, 593–601. [Google Scholar]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.A.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar]
- Soni, P.; Qayoom, S.; Husain, N.; Kumar, P.; Chandra, A.; Ojha, B.K.; Gupta, R.K. CD24 and Nanog expression in Stem Cells in Glioblastoma: Correlation with Response to Chemoradiation and Overall Survival. Asian Pac. J. Cancer Prev. 2017, 18, 2215–2219. [Google Scholar]
- Bien-Möller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem Cells Int. 2018, 2018, 1–19. [Google Scholar]
- Fawzy, M.S.; I. Badran, D.; Al Ageeli, E.; Al-Qahtani, S.A.M.; Alghamdi, S.A.; Helal, G.M.; Toraih, E.A. Longevity-Related Gene Transcriptomic Signature in Glioblastoma Multiforme. Oxidative Med. Cell. Longev. 2018, 2018, 1–12. [Google Scholar]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.-N.; Borges, I.; I. Altaba, A.R. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar]
- Bier, A.; Giladi, N.; Kronfeld, N.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Poisson, L.; Decarvalho, A.C.; Slavin, S.; et al. MicroRNA-137 is downregulated in glioblastoma and inhibits the stemness of glioma stem cells by targeting RTVP-1. Oncotarget 2013, 4, 665–676. [Google Scholar]
- Gong, S.; Li, Q.; Jeter, C.; Fan, Q.; Tang, D.G.; Liu, B. Regulation of NANOG in cancer cells. Mol. Carcinog. 2015, 54, 679–687. [Google Scholar] [PubMed]
- Niu, C.-S.; Yang, Y.; Cheng, C.-D. MiR-134 regulates the proliferation and invasion of glioblastoma cells by reducing Nanog expression. Int. J. Oncol. 2013, 42, 1533–1540. [Google Scholar] [PubMed]
- Petersen, J.K.; Jensen, P.; Sørensen, M.D.; Kristensen, B.W. Expression and Prognostic Value of Oct-4 in Astrocytic Brain Tumors. PLoS ONE 2016, 11, e0169129. [Google Scholar]
- Wu, Y.; Sun, B.; Shi, W.; Ni, L.; Chen, J.; Cai, G.; Shi, J. OCT4 is up-regulated by DNA hypomethylation of promoter in recurrent gliomas. Neoplasma 2016, 63, 378–384. [Google Scholar] [PubMed]
- Kijewska, M.; Kocyk, M.; Kloss, M.; Stepniak, K.; Korwek, Z.; Polakowska, R.; Dabrowski, M.; Gieryng, A.; Wojtas, B.; Ciechomska, I.; et al. The embryonic type of SPP1 transcriptional regulation is re-activated in glioblastoma. Oncotarget 2016, 8, 16340–16355. [Google Scholar]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8, 32960–32976. [Google Scholar]
- Li, W.; Zhou, Y.; Zhang, X.; Yang, Y.; Dan, S.; Su, T.; She, S.; Dong, W.; Zhao, Q.; Jia, J.; et al. Dual inhibiting OCT4 and AKT potently suppresses the propagation of human cancer cells. Sci. Rep. 2017, 7, 46246. [Google Scholar]
- Lin, Y.; Yang, Y.; Li, W.; Chen, Q.; Li, J.; Pan, X.; Zhou, L.; Liu, C.; Chen, C.; He, J.; et al. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 2012, 48, 627–640. [Google Scholar]
- Jones, N.M.; Rowe, M.R.; Shepherd, P.R.; McConnell, M.J. Targeted inhibition of dominant PI3-kinase catalytic isoforms increase expression of stem cell genes in glioblastoma cancer stem cell models. Int. J. Oncol. 2016, 49, 207–216. [Google Scholar]
- Srivastava, C.; Irshad, K.; Dikshit, B.; Chattopadhyay, P.; Sarkar, C.; Gupta, D.K.; Sinha, S.; Chosdol, K. FAT1 modulates EMT and stemness genes expression in hypoxic glioblastoma. Int. J. Cancer 2017, 142, 805–812. [Google Scholar]
- Bhagat, M.; Palanichamy, J.K.; Ramalingam, P.; Mudassir, M.; Irshad, K.; Chosdol, K.; Sarkar, C.; Seth, P.; Goswami, S.; Sinha, S.; et al. HIF-2α Mediates a Marked Increase in Migration and Stemness Characteristics in a Subset of Glioma Cells under Hypoxia by Activating an Oct-4/Sox-2-Mena (INV) Axis. Int. J. Biochem. Cell Biol. 2016, 74, 60–71. [Google Scholar] [PubMed]
- Balça-Silva, J.; Carmo, A.D.; Tão, H.; Rebelo, O.; Barbosa, M.; Neto, V.D.M.; Sarmento-Ribeiro, A.B.; Lopes, M.C.; Moreira, J.N. Nucleolin is expressed in patient-derived samples and glioblastoma cells, enabling improved intracellular drug delivery and cytotoxicity. Exp. Cell Res. 2018, 370, 68–77. [Google Scholar] [PubMed]
- Li, D.; Yang, Z.-K.; Bu, J.-Y.; Xu, C.-Y.; Sun, H.; Tang, J.-B.; Lin, P.; Cheng, W.; Huang, N.; Cui, R.-J.; et al. OCT4B modulates OCT4A expression as ceRNA in tumor cells. Oncol. Rep. 2015, 33, 2622–2630. [Google Scholar] [PubMed]
- Speranza, M.C.; Frattini, V.; Pisati, F.; Kapetis, D.; Porrati, P.; Eoli, M.; Pellegatta, S.; Finocchiaro, G. NEDD9, a novel target of miR-145, increases the invasiveness of glioblastoma. Oncotarget 2012, 3, 723–734. [Google Scholar]
- Yang, Y.-P.; Chien, Y.; Chiou, G.-Y.; Cherng, J.-Y.; Wang, M.-L.; Lo, W.-L.; Chang, Y.-L.; Huang, P.-I.; Chen, Y.-W.; Shih, Y.-H.; et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomater. 2012, 33, 1462–1476. [Google Scholar]
- Gao, X.; Zhu, X.; Sun, Y.; Liu, J. MicroRNA-141 inhibits the self-renewal of glioblastoma stem cells via Jagged1. Mol. Med. Rep. 2017, 16, 167–173. [Google Scholar]
- Garcia, I.; Aldaregia, J.; Vicentic, J.M.; Aldaz, P.; Moreno-Cugnon, L.; Torres-Bayona, S.; Carrasco-Garcia, E.; Garros-Regulez, L.; Egaña, L.; Rubio, A.; et al. Oncogenic activity of SOX1 in glioblastoma. Sci. Rep. 2017, 7, 46575. [Google Scholar]
- Dong, H.; Hao, X.; Cui, B.; Guo, M. MiR-429 suppresses glioblastoma multiforme by targeting SOX2. Cell Biochem. Funct. 2017, 35, 260–268. [Google Scholar]
- Sarlak, G.; Vincent, B. The Roles of the Stem Cell-Controlling Sox2 Transcription Factor: From Neuroectoderm Development to Alzheimer’s Disease? Mol. Neurobiol. 2015, 53, 1679–1698. [Google Scholar]
- Gopal, K.; Gupta, N.; Zhang, H.; Alshareef, A.; Alqahtani, H.; Bigras, G.; Lewis, J.; Douglas, N.; Kneteman, N.; Lavasanifar, A.; et al. Oxidative stress induces the acquisition of cancer stem-like phenotype in breast cancer detectable by using a Sox2 regulatory region-2 (SRR2) reporter. Oncotarget 2015, 7, 3111–3127. [Google Scholar]
- Ren, Z.-H.; Zhang, C.-P.; Ji, T. Expression of SOX2 in oral squamous cell carcinoma and the association with lymph node metastasis. Oncol. Lett. 2016, 11, 1973–1979. [Google Scholar] [PubMed]
- Vasquez, J.C.; Huttner, A.; Zhang, L.; Marks, A.; Chan, A.; Baehring, J.M.; Kahle, K.T.; Dhodapkar, K.M. SOX2 immunity and tissue resident memory in children and young adults with glioma. J. Neuro-Oncology 2017, 134, 41–53. [Google Scholar]
- Garros-Regulez, L.; Aldaz, P.; Arrizabalaga, O.; Moncho-Amor, V.; Carrasco-Garcia, E.; Manterola, L.; Moreno-Cugnon, L.; Barrena, C.; Villanua, J.; Ruiz, I.; et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets 2016, 20, 393–405. [Google Scholar] [PubMed]
- Song, W.-S.; Yang, Y.-P.; Huang, C.-S.; Lu, K.-H.; Liu, W.-H.; Wu, W.-W.; Lee, Y.-Y.; Lo, W.-L.; Lee, S.-D.; Chen, Y.-W.; et al. Sox2, a stemness gene, regulates tumor-initiating and drug-resistant properties in CD133-positive glioblastoma stem cells. J. Chin. Med. Assoc. 2016, 79, 538–545. [Google Scholar] [PubMed]
- Bulstrode, H.; Johnstone, E.; Marqués-Torrejón, M. Ángeles; Ferguson, K.M.; Bressan, R.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genome Res. 2017, 31, 757–773. [Google Scholar]
- Luo, W.; Yan, D.; Song, Z.; Zhu, X.; Liu, X.; Li, X.; Zhao, S. miR-126-3p sensitizes glioblastoma cells to temozolomide by inactivating Wnt/β-catenin signaling via targeting SOX2. Life Sci. 2019, 226, 98–106. [Google Scholar]
- Qian, C.; Wang, B.; Zou, Y.; Zhang, Y.; Hu, X.; Sun, W.; Xiao, H.; Liu, H.; Shi, L. MicroRNA 145 enhances chemosensitivity of glioblastoma stem cells to demethoxycurcumin. Cancer Manag. Res. 2019, 11, 6829–6840. [Google Scholar]
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Neto, V.D.M. MicroRNAs, Hypoxia and the Stem-Like State as Contributors to Cancer Aggressiveness. Front. Genet. 2019, 10, 125. [Google Scholar]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar]
- Park, D.; Xiang, A.P.; Mao, F.F.; Zhang, L.; Di, C.-G.; Liu, X.-M.; Shao, Y.; Ma, B.-F.; Lee, J.-H.; Ha, K.-S.; et al. Nestin Is Required for the Proper Self-Renewal of Neural Stem Cells. STEM CELLS 2010, 28, 2162–2171. [Google Scholar] [PubMed]
- Dusart, P.; Fagerberg, L.; Perisic, L.; Civelek, M.; Struck, E.; Hedin, U.; Uhlén, M.; Trégouët, D.A.; Renné, T.; Odeberg, J.; et al. A systems-approach reveals human nestin is an endothelial-enriched, angiogenesis-independent intermediate filament protein. Sci. Rep. 2018, 8, 14668. [Google Scholar] [PubMed]
- Nowak, A.; Grzegrzółka, J.; Paprocka, M.; Piotrowska, A.; Rys, J.; Matkowski, R.; Dziegiel, P. Nestin-positive microvessel density is an independent prognostic factor in breast cancer. Int. J. Oncol. 2017, 51, 668–676. [Google Scholar]
- Nowak, A.; Grzegrzółka, J.; Kmiecik, A.; Piotrowska, A.; Matkowski, R.; Dzięgiel, P. Role of nestin expression in angiogenesis and breast cancer progression. Int. J. Oncol. 2017, 52, 527–535. [Google Scholar] [PubMed]
- Niu, C.; He, H.; Li, M.W. Correlation between glioblastoma stem-like cells and tumor vascularization. Oncol. Rep. 2011, 27, 45–50. [Google Scholar]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of Nestin Suppresses Stem Cell Phenotype of Glioblastomas through the Alteration of Post-Translational Modification of Heat Shock Protein HSPA8/HSC71. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [PubMed]
- Naito, Z.; Ishiwata, T.; Teduka, K.; Yamamoto, T.; Kawahara, K.; Matsuda, Y. Neuroepithelial stem cell marker nestin regulates the migration, invasion and growth of human gliomas. Oncol. Rep. 2011, 26, 91–99. [Google Scholar]
- Lu, W.J.; Lan, F.; He, Q.; Lee, A.; Tang, C.Z.; Dong, L.; Lan, B.; Ma, X.; Wu, J.C.; Shen, L. Inducible expression of stem cell associated intermediate filament nestin reveals an important role in glioblastoma carcinogenesis. Int. J. Cancer 2010, 128, 343–351. [Google Scholar]
- Strojnik, T.; Røsland, G.V.; Sakariassen, P.O.; Kavalar, R.; Lah, T. Neural stem cell markers, nestin and musashi proteins, in the progression of human glioma: Correlation of nestin with prognosis of patient survival. Surg. Neurol. 2007, 68, 133–143. [Google Scholar]
- Arai, H.; Ikota, H.; Sugawara, K.-I.; Nobusawa, S.; Hirato, J.; Nakazato, Y. Nestin expression in brain tumors: Its utility for pathological diagnosis and correlation with the prognosis of high-grade gliomas. Brain Tumor Pathol. 2012, 29, 160–167. [Google Scholar]
- Lv, D.; Lu, L.; Hu, Z.; Fei, Z.; Liu, M.; Wei, L.; Xu, J. Nestin Expression Is Associated with Poor Clinicopathological Features and Prognosis in Glioma Patients: An Association Study and Meta-analysis. Mol. Neurobiol. 2016, 54, 727–735. [Google Scholar] [PubMed]
- Hambardzumyan, L.; Becher, O.J.; Holland, E.C. Cancer stem cells and survival pathways. Cell Cycle 2008, 7, 1371–1378. [Google Scholar] [PubMed]
- Wang, Z.; Yang, J.; Xu, G.; Wang, W.; Liu, C.; Yang, H.; Yu, Z.; Lei, Q.; Xiao, L.; Xiong, J.; et al. Targeting miR-381-NEFL axis sensitizes glioblastoma cells to temozolomide by regulating stemness factors and multidrug resistance factors. Oncotarget 2014, 6, 3147–3164. [Google Scholar]
- Li, S.; Zeng, A.; Hu, Q.; Yan, W.; Liu, Y.; You, Y. miR-423-5p contributes to a malignant phenotype and temozolomide chemoresistance in glioblastomas. Neuro-Oncology 2016, 19, 55–65. [Google Scholar] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassn Mesrati, M.; Behrooz, A.B.; Y. Abuhamad, A.; Syahir, A. Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer. Cells 2020, 9, 1236. https://doi.org/10.3390/cells9051236
Hassn Mesrati M, Behrooz AB, Y. Abuhamad A, Syahir A. Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer. Cells. 2020; 9(5):1236. https://doi.org/10.3390/cells9051236
Chicago/Turabian StyleHassn Mesrati, Malak, Amir Barzegar Behrooz, Asmaa Y. Abuhamad, and Amir Syahir. 2020. "Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer" Cells 9, no. 5: 1236. https://doi.org/10.3390/cells9051236
APA StyleHassn Mesrati, M., Behrooz, A. B., Y. Abuhamad, A., & Syahir, A. (2020). Understanding Glioblastoma Biomarkers: Knocking a Mountain with a Hammer. Cells, 9(5), 1236. https://doi.org/10.3390/cells9051236