MMTR/Dmap1 Sets the Stage for Early Lineage Commitment of Embryonic Stem Cells by Crosstalk with PcG Proteins

 , ,
, ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Establishment of MMTR/Dmap1 Full-Length, N-terminal, and C-terminal Half Overexpression and Knockdown ESC Lines
2.3. Total RNA Preparation and RT-qPCR
2.4. Western Blot
2.5. Cell Cycle Analyses Using Flow Cytometry
2.6. In Vivo Differentiation Assay
2.7. Chromatin Immunoprecipitation-Quantitative PCR (ChIP-qPCR) Analysis
2.8. Microarray Data Analysis
2.9. ChIP Data Processing and Analyses
2.10. Bioinformatics Analysis
3. Results
3.1. MMTR/Dmap1 Is Required for Accurate Differentiation of mESCs
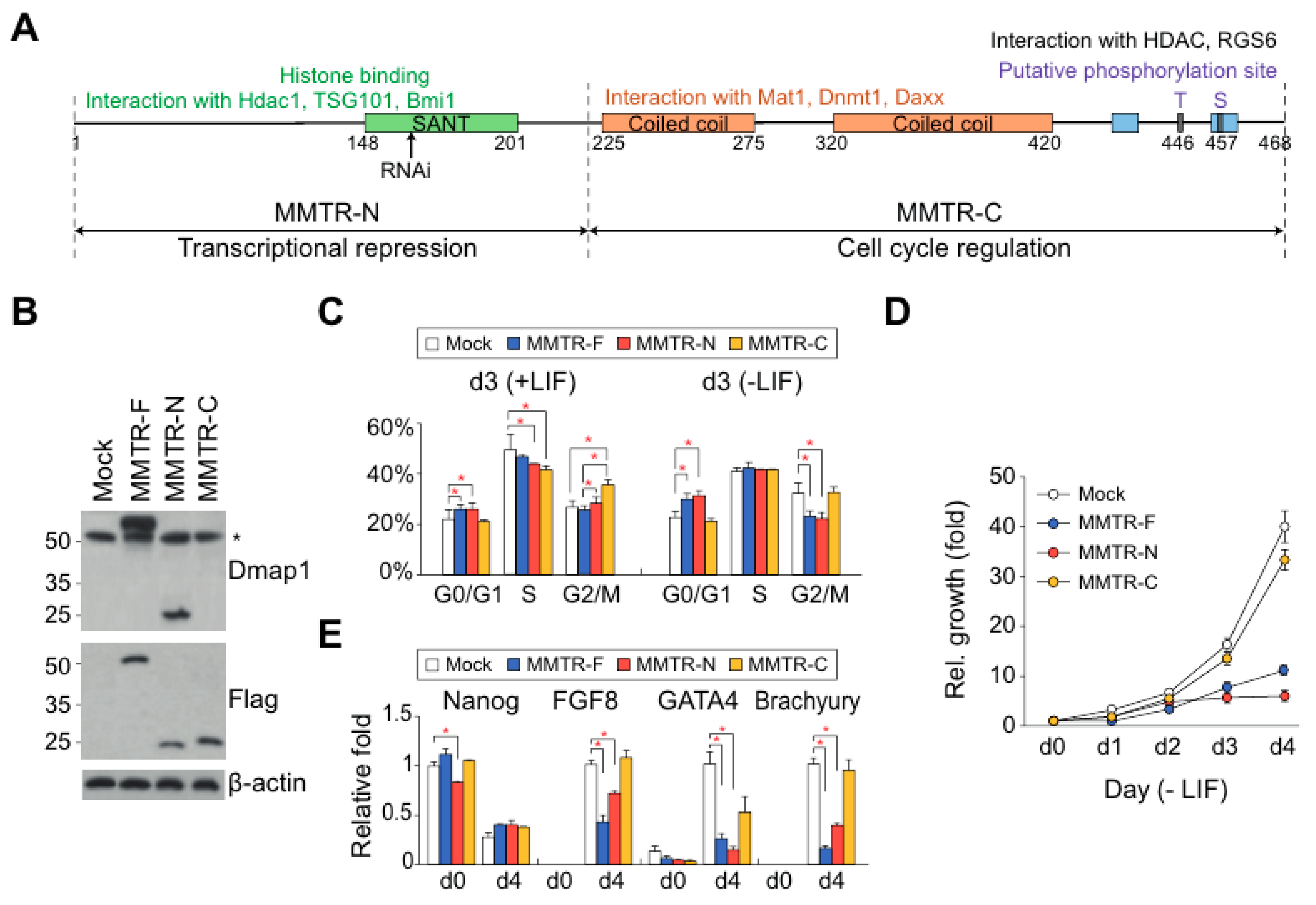
3.2. The Distinct Roles between N- and C-Terminal Regions of MMTR/Dmap1 in Differentiation of mESCs
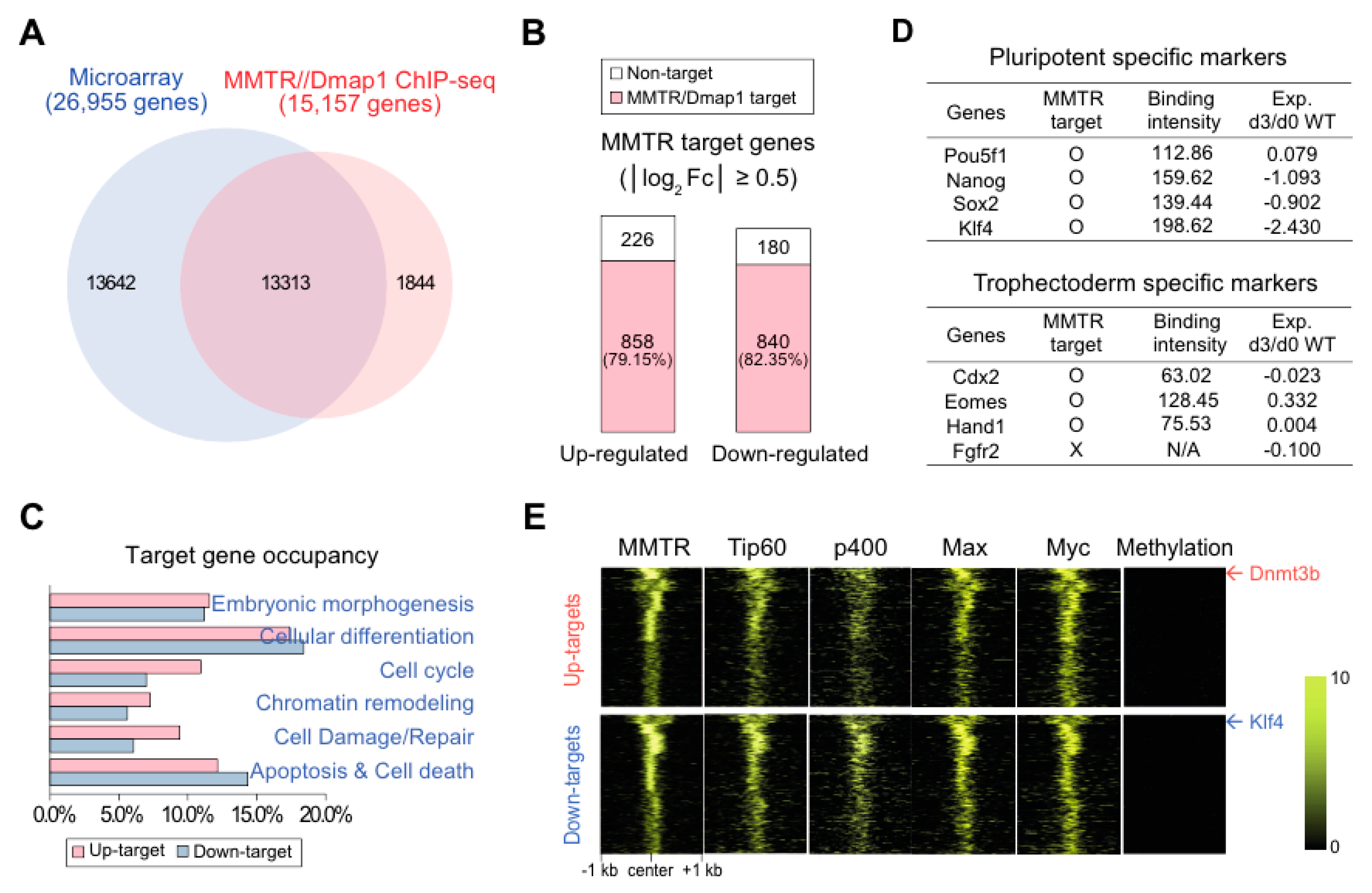
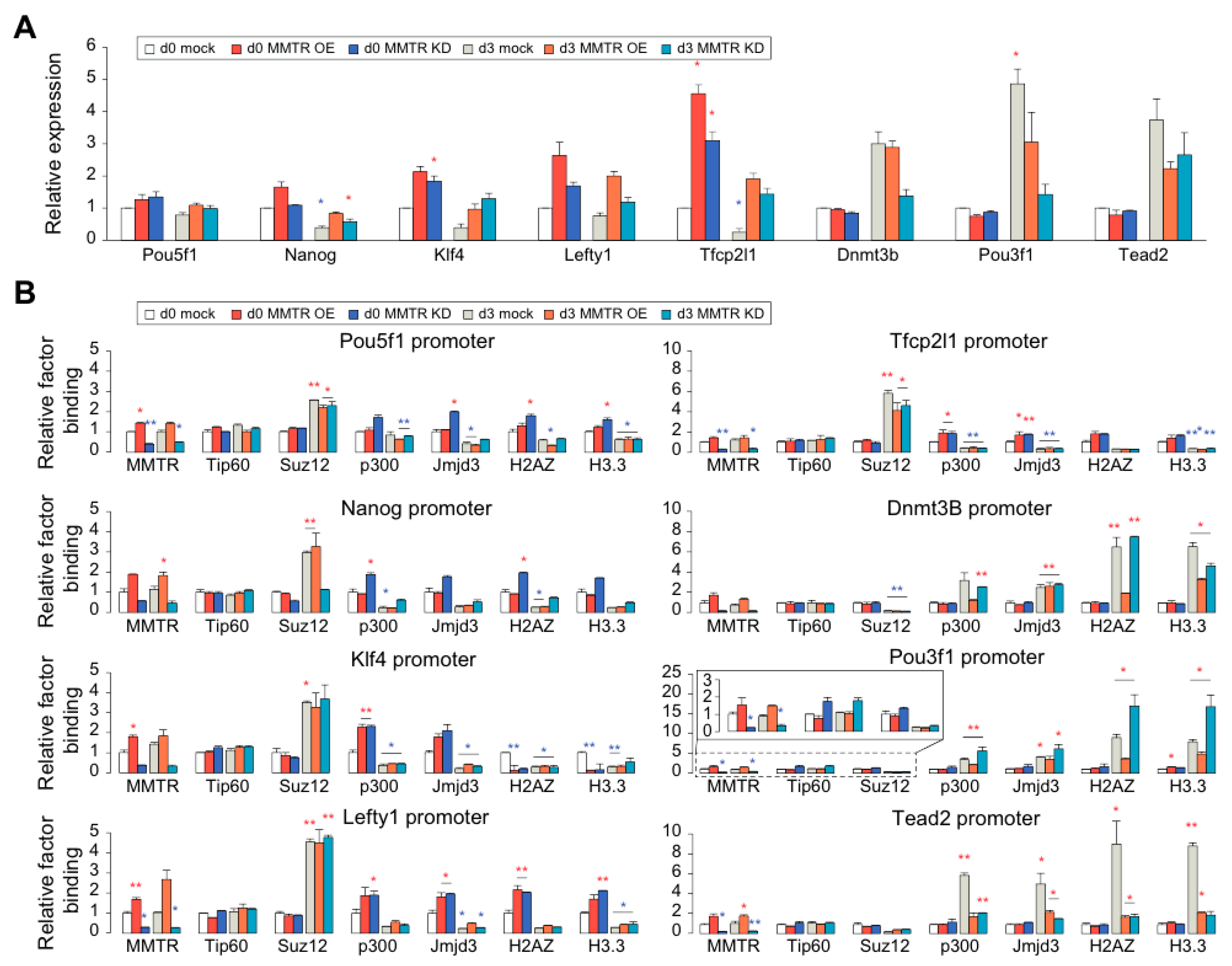
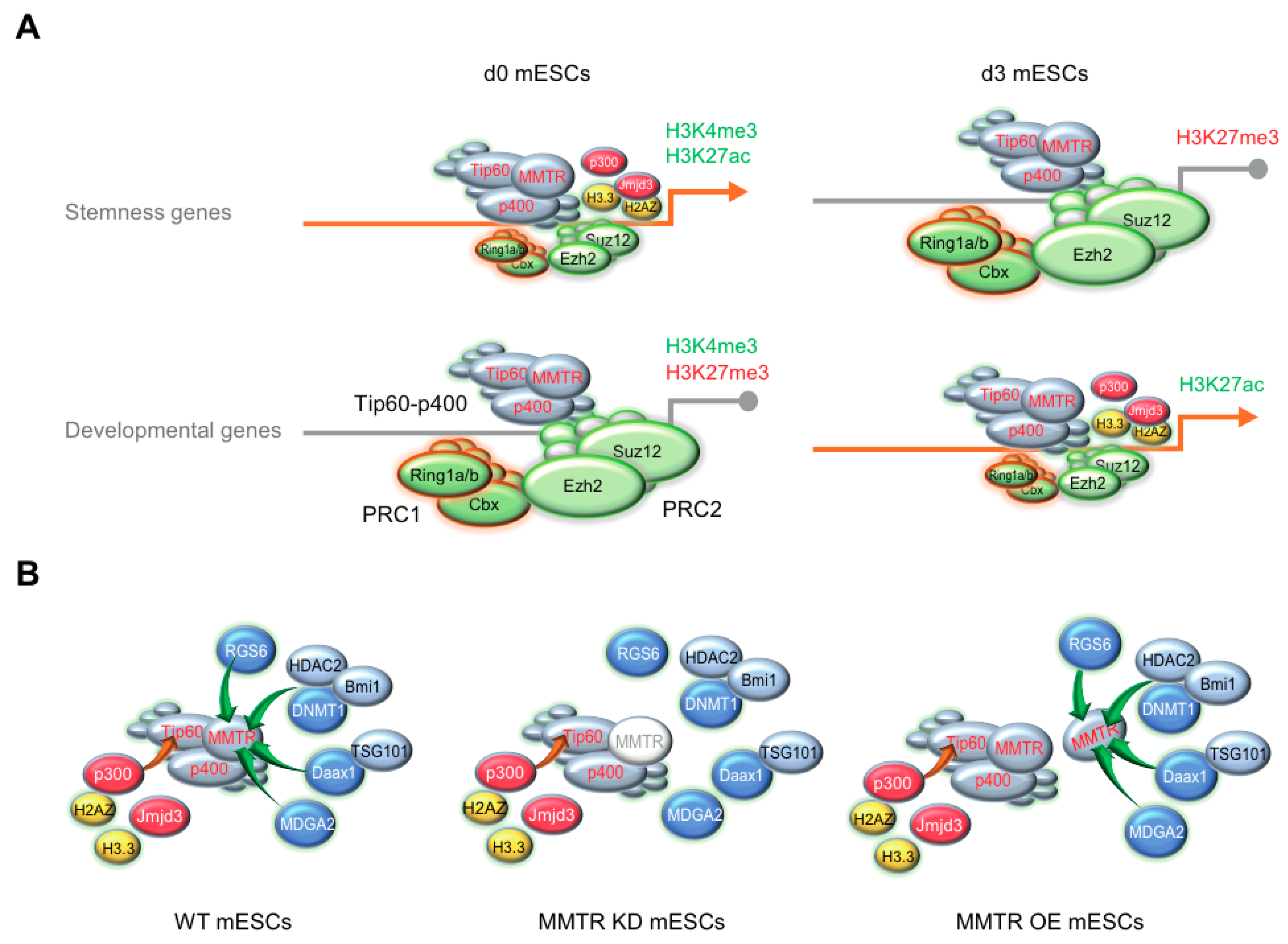
3.3. MMTR/Dmap1 Functions as a Member of the Tip60-p400 Complex for Chromatin Remodeling during Commitment of mESC Early Differentiation
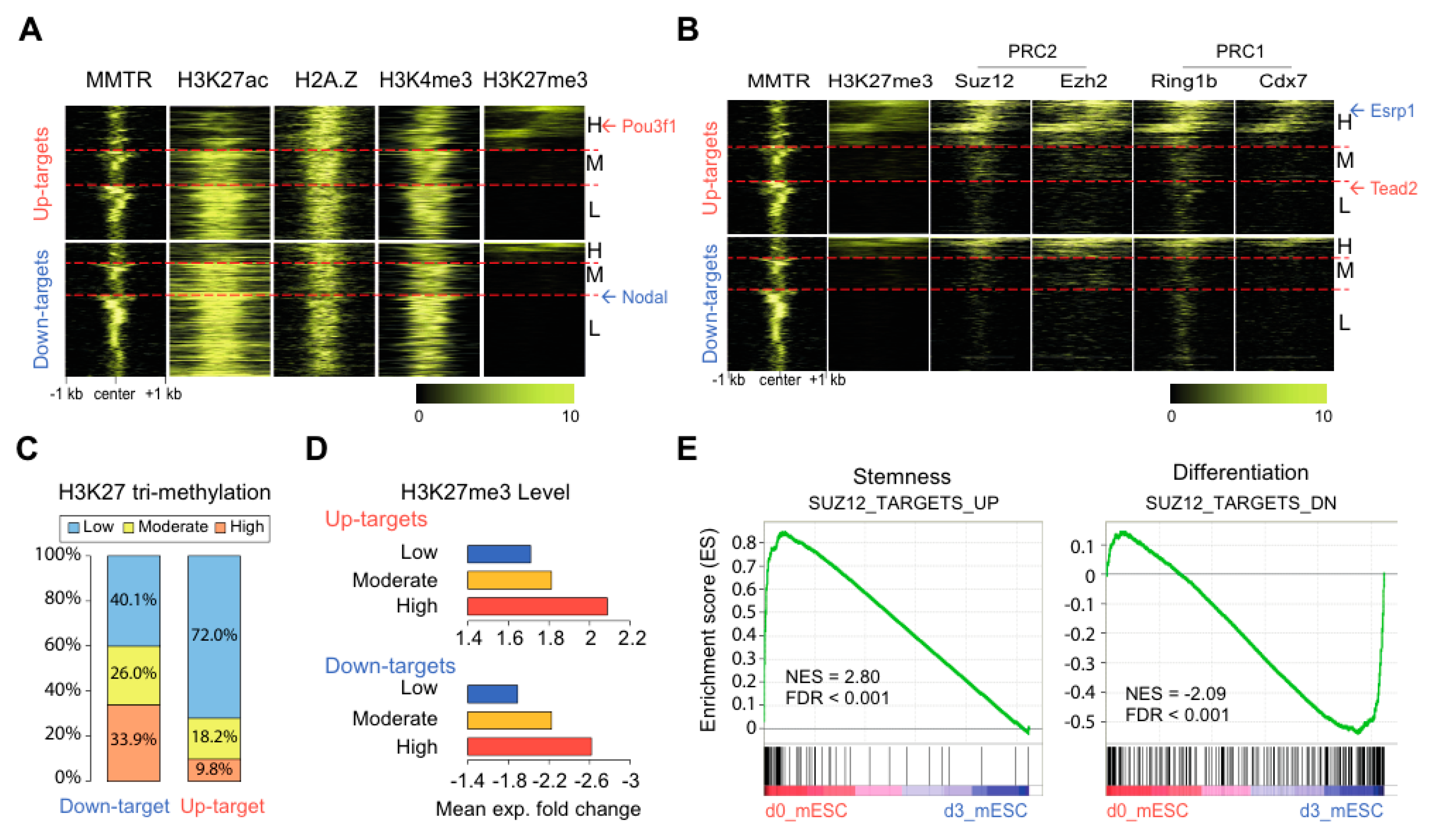
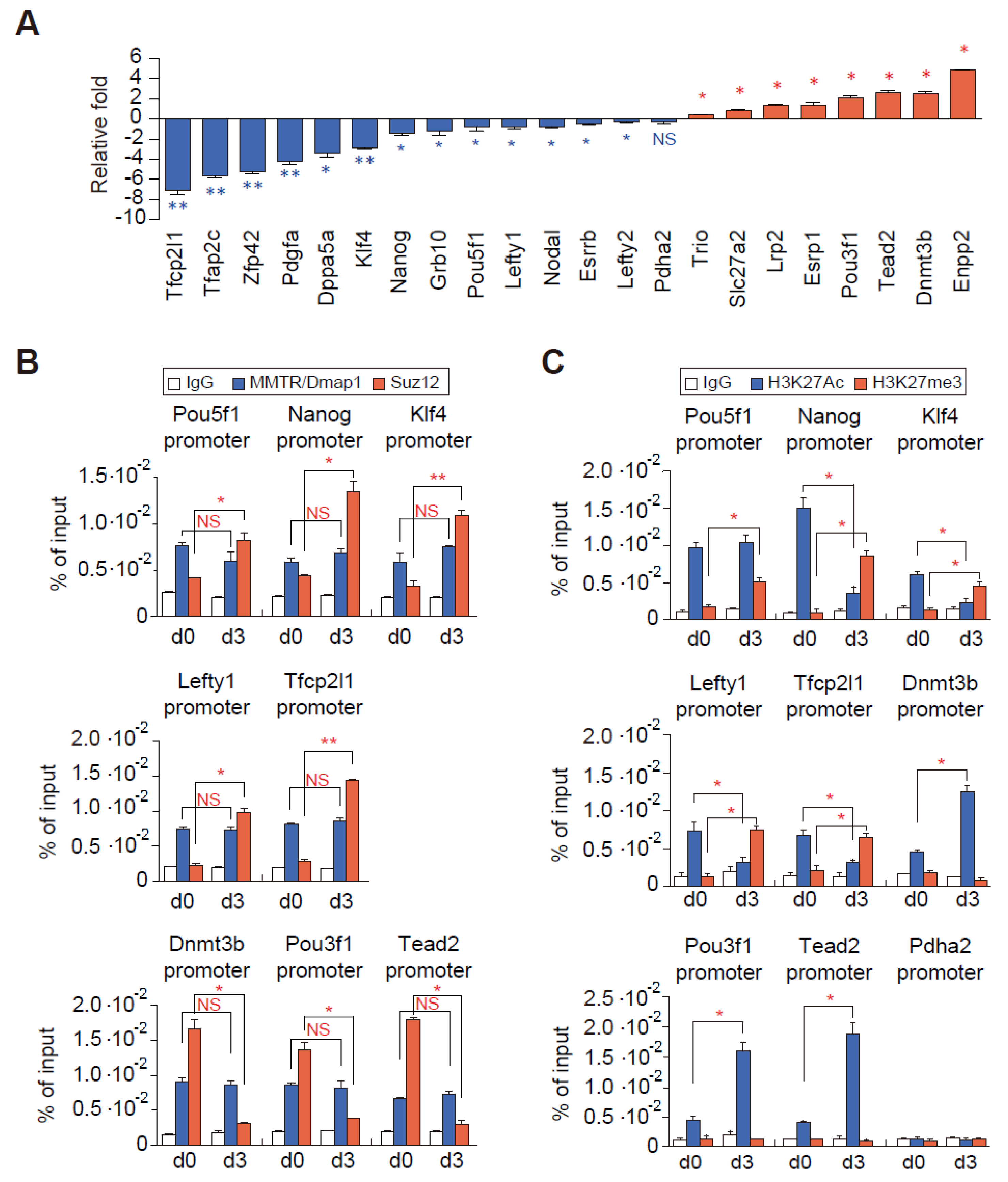
3.4. Suz12 Binding to the Commitment Gene Promoters, but not MMTR/Dmap1, Is Modulated Accompanied by H3K27 Modification during the mESCs Differentiation
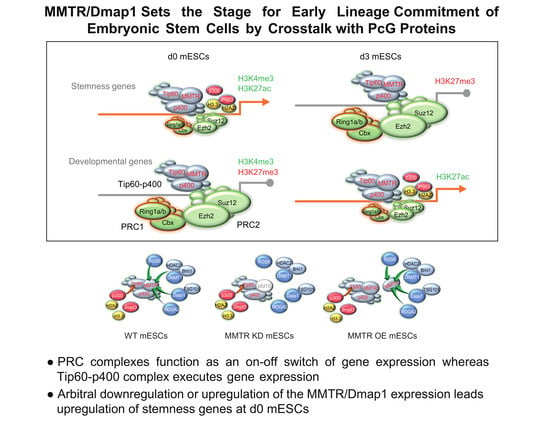
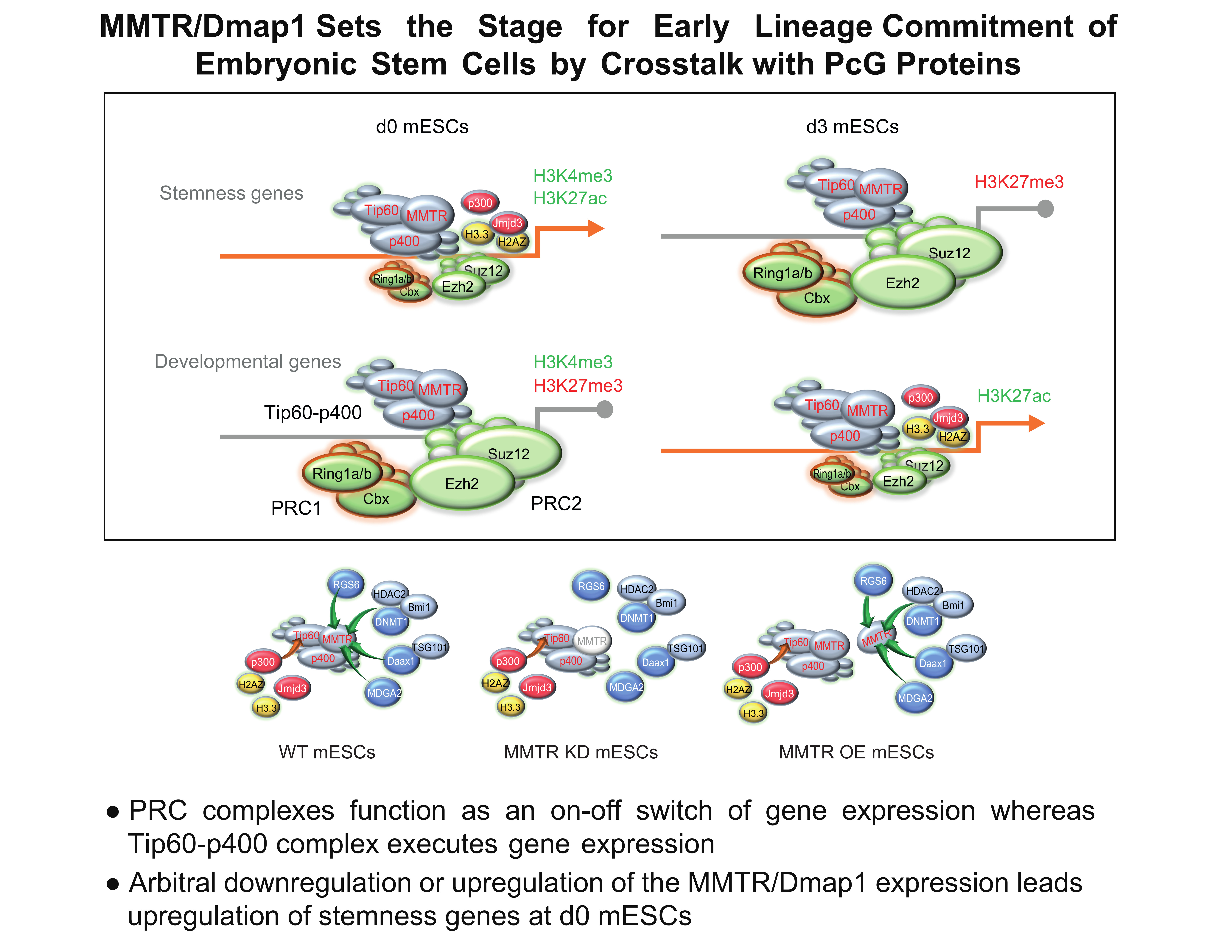
3.5. PRC Complexes Function as an On–Off Switch, whereas the Tip60-p400 Complex Executes Gene Expression via Recruiting Factors Required for Gene Activation During the mESCs Differentiation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Martin, G.R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7634–7638. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Devito, L.; Miere, C.; Codognotto, S. Human embryonic and induced pluripotent stem cells in clinical trials. Br. Med. Bull. 2015, 116, 19–27. [Google Scholar] [CrossRef]
- Keller, G. Embryonic stem cell differentiation: Emergence of a new era in biology and medicine. Genes. Dev. 2005, 19, 1129–1155. [Google Scholar] [CrossRef]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.L.; Zhang, W.; Jiang, J.; et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Kim, J.; Chu, J.; Shen, X.; Wang, J.; Orkin, S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008, 132, 1049–1061. [Google Scholar] [CrossRef]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef]
- Masui, S.; Nakatake, Y.; Toyooka, Y.; Shimosato, D.; Yagi, R.; Takahashi, K.; Okochi, H.; Okuda, A.; Matoba, R.; Sharov, A.A.; et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat. Cell Biol. 2007, 9, 625–635. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Mannervik, M.; Nibu, Y.; Zhang, H.; Levine, M. Transcriptional coregulators in development. Science 1999, 284, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef]
- Young, R.A. Control of the embryonic stem cell state. Cell 2011, 144, 940–954. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Rountree, M.R.; Bachman, K.E.; Baylin, S.B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet. 2000, 25, 269–277. [Google Scholar] [CrossRef]
- Xin, H.; Yoon, H.G.; Singh, P.B.; Wong, J.; Qin, J. Components of a pathway maintaining histone modification and heterochromatin protein 1 binding at the pericentric heterochromatin in Mammalian cells. J. Biol. Chem. 2004, 279, 9539–9546. [Google Scholar] [CrossRef]
- Cai, Y.; Jin, J.; Florens, L.; Swanson, S.K.; Kusch, T.; Li, B.; Workman, J.L.; Washburn, M.P.; Conaway, R.C.; Conaway, J.W. The mammalian YL1 protein is a shared subunit of the TRRAP/TIP60 histone acetyltransferase and SRCAP complexes. J. Biol. Chem. 2005, 280, 13665–13670. [Google Scholar] [CrossRef]
- Cai, Y.; Jin, J.; Tomomori-Sato, C.; Sato, S.; Sorokina, I.; Parmely, T.J.; Conaway, R.C.; Conaway, J.W. Identification of new subunits of the multiprotein mammalian TRRAP/TIP60-containing histone acetyltransferase complex. J. Biol. Chem. 2003, 278, 42733–42736. [Google Scholar] [CrossRef]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Cote, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, T.; Negishi, M.; Nakamura, S.; Oguro, H.; Satoh, K.; Ichinose, M.; Iwama, A. Depletion of Dnmt1-associated protein 1 triggers DNA damage and compromises the proliferative capacity of hematopoietic stem cells. Int. J. Hematol. 2010, 91, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Sapountzi, V.; Logan, I.R.; Robson, C.N. Cellular functions of TIP60. Int. J. Biochem. Cell Biol. 2006, 38, 1496–1509. [Google Scholar] [CrossRef] [PubMed]
- Penicud, K.; Behrens, A. DMAP1 is an essential regulator of ATM activity and function. Oncogene 2014, 33, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takenobu, H.; Ohira, M.; Nakazawa, A.; Yoshida, S.; Akita, N.; Shimozato, O.; Iwama, A.; Nakagawara, A.; Kamijo, T. Novel 1p tumour suppressor Dnmt1-associated protein 1 regulates MYCN/ataxia telangiectasia mutated/p53 pathway. Eur. J. Cancer 2014, 50, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Kokosar, M.; Benrick, A.; Perfilyev, A.; Fornes, R.; Nilsson, E.; Maliqueo, M.; Behre, C.J.; Sazonova, A.; Ohlsson, C.; Ling, C.; et al. Epigenetic and Transcriptional Alterations in Human Adipose Tissue of Polycystic Ovary Syndrome. Sci. Rep. 2016, 6, 22883. [Google Scholar] [CrossRef]
- Kim, S.J.; Shin, J.H.; Kim, J.; Kim, S.H.; Chae, J.H.; Park, E.J.; Seong, R.H.; Hong, S.H.; Park, S.D.; Jeong, S.; et al. Isolation of developmentally regulated novel genes based on sequence identity and gene expression pattern. Mol. Cells 1999, 9, 207–218. [Google Scholar]
- Kang, B.G.; Shin, J.H.; Yi, J.K.; Kang, H.C.; Lee, J.J.; Heo, H.S.; Chae, J.H.; Shin, I.; Kim, C.G. Corepressor MMTR/DMAP1 is involved in both histone deacetylase 1- and TFIIH-mediated transcriptional repression. Mol. Cell Biol. 2007, 27, 3578–3588. [Google Scholar] [CrossRef][Green Version]
- Shin, J.H.; Kang, H.C.; Park, Y.Y.; Ha, D.H.; Choi, Y.H.; Eum, H.Y.; Kang, B.G.; Chae, J.H.; Shin, I.; Lee, J.H.; et al. Corepressor MMTR/DMAP1 is an intrinsic negative regulator of CAK kinase to regulate cell cycle progression. Biochem. Biophys. Res. Commun. 2010, 402, 110–115. [Google Scholar] [CrossRef]
- Dvir, A.; Conaway, J.W.; Conaway, R.C. Mechanism of transcription initiation and promoter escape by RNA polymerase II. Curr Opin Genet. Dev. 2001, 11, 209–214. [Google Scholar] [CrossRef]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Huff, J.T.; Panning, B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 2008, 134, 162–174. [Google Scholar] [CrossRef]
- Mohan, K.N.; Ding, F.; Chaillet, J.R. Distinct roles of DMAP1 in mouse development. Mol. Cell. Biol. 2011, 31, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Rohwedel, J.; Horak, V.; Hebrok, M.; Fuchtbauer, E.M.; Wobus, A.M. M-twist expression inhibits mouse embryonic stem cell-derived myogenic differentiation in vitro. Exp. Cell Res. 1995, 220, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Trono, D. Lentiviral vectors: Turning a deadly foe into a therapeutic agent. Gene Ther. 2000, 7, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Park, J.; Lee, J.J.; Ha, D.H.; Kim, J.; Kim, C.G.; Hwang, J.; Kim, C.G. Staufen1-mediated mRNA decay induces Requiem mRNA decay through binding of Staufen1 to the Requiem 3’UTR. Nucleic Acids Res. 2014, 42, 6999–7011. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Kim, J.S.; Son, S.H.; Lim, C.S.; Eum, H.Y.; Ha, D.H.; Park, M.A.; Baek, E.J.; Ryu, B.Y.; Kang, H.C.; et al. Mbd2-CP2c loop drives adult-type globin gene expression and definitive erythropoiesis. Nucleic Acids Res. 2018, 46, 4933–4949. [Google Scholar] [CrossRef]
- Gene Expression Omnibus. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145561 (accessed on 13 February 2020).
- Jiang, P.; Singh, M. CCAT: Combinatorial Code Analysis Tool for transcriptional regulation. Nucleic Acids Res. 2014, 42, 2833–2847. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Nishikawa, S.; Jakt, L.M.; Era, T. Embryonic stem-cell culture as a tool for developmental cell biology. Nat. Rev. Mol. Cell Biol. 2007, 8, 502–507. [Google Scholar] [CrossRef]
- Mansergh, F.C.; Daly, C.S.; Hurley, A.L.; Wride, M.A.; Hunter, S.M.; Evans, M.J. Gene expression profiles during early differentiation of mouse embryonic stem cells. BMC Dev. Biol. 2009, 9, 5. [Google Scholar] [CrossRef]
- Lee, G.E.; Kim, J.H.; Taylor, M.; Muller, M.T. DNA methyltransferase 1-associated protein (DMAP1) is a co-repressor that stimulates DNA methylation globally and locally at sites of double strand break repair. J. Biol. Chem. 2010, 285, 37630–37640. [Google Scholar] [CrossRef] [PubMed]
- Kusch, T.; Florens, L.; Macdonald, W.H.; Swanson, S.K.; Glaser, R.L.; Yates, J.R., 3rd; Abmayr, S.M.; Washburn, M.P.; Workman, J.L. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 2004, 306, 2084–2087. [Google Scholar] [CrossRef] [PubMed]
- Surface, L.E.; Thornton, S.R.; Boyer, L.A. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 2010, 7, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2011, 35, 323–332. [Google Scholar] [CrossRef]
- Leeb, M.; Pasini, D.; Novatchkova, M.; Jaritz, M.; Helin, K.; Wutz, A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010, 24, 265–276. [Google Scholar] [CrossRef]
- Ku, M.; Koche, R.P.; Rheinbay, E.; Mendenhall, E.M.; Endoh, M.; Mikkelsen, T.S.; Presser, A.; Nusbaum, C.; Xie, X.; Chi, A.S.; et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008, 4, e1000242. [Google Scholar] [CrossRef]
- Tolhuis, B.; de Wit, E.; Muijrers, I.; Teunissen, H.; Talhout, W.; van Steensel, B.; van Lohuizen, M. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat. Genet. 2006, 38, 694–699. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Jensen, M.R.; Lazzerini Denchi, E.; Helin, K. Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. Embo. J. 2004, 23, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Nagai, Y.; Deng, G.; Ohtani, T.; Zhu, Z.; Zhou, Z.; Zhang, H.; Ji, M.Q.; Lough, J.W.; Samanta, A.; et al. Dynamic interactions between TIP60 and p300 regulate FOXP3 function through a structural switch defined by a single lysine on TIP60. Cell Rep. 2014, 7, 1471–1480. [Google Scholar] [CrossRef]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef]
- Jin, C.; Felsenfeld, G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007, 21, 1519–1529. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Markoulaki, S.; Levine, S.S.; Hanna, J.; Lodato, M.A.; Sha, K.; Young, R.A.; Jaenisch, R.; Boyer, L.A. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell 2008, 135, 649–661. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Huff, J.T.; Panning, B. Chromatin regulation Tip(60)s the balance in embryonic stem cell self-renewal. Cell Cycle 2008, 7, 3302–3306. [Google Scholar] [CrossRef]
- Negishi, M.; Saraya, A.; Miyagi, S.; Nagao, K.; Inagaki, Y.; Nishikawa, M.; Tajima, S.; Koseki, H.; Tsuda, H.; Takasaki, Y.; et al. Bmi1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem. Biophys. Res. Commun. 2007, 353, 992–998. [Google Scholar] [CrossRef]
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Son, J.; Shen, S.S.; Reinberg, D.; Bonasio, R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Rojas, C.; Hernandez, A.J.; Sarma, K.; Lee, J.T. Regulatory interactions between RNA and polycomb repressive complex 2. Mol. Cell 2014, 55, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.; Liang, H.; Lim, Y.S.; Chan, Y.S.; Yeo, J.C.; Tan, C.P.; Gao, B.; Le, B.; Tan, Z.Y.; Low, K.Y.; et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015, 162, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; van Lohuizen, M. Stem cell regulation by polycomb repressors: Postponing commitment. Curr. Opin. Cell Biol. 2008, 20, 201–207. [Google Scholar] [CrossRef]
- Chamberlain, S.J.; Yee, D.; Magnuson, T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 2008, 26, 1496–1505. [Google Scholar] [CrossRef]
- Eskeland, R.; Freyer, E.; Leeb, M.; Wutz, A.; Bickmore, W.A. Histone acetylation and the maintenance of chromatin compaction by Polycomb repressive complexes. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 71–78. [Google Scholar] [CrossRef]
- Tamburri, S.; Lavarone, E.; Fernandez-Perez, D.; Conway, E.; Zanotti, M.; Manganaro, D.; Pasini, D. Histone H2AK119 Mono-Ubiquitination Is Essential for Polycomb-Mediated Transcriptional Repression. Mol. Cell 2020, 77, 840–856 e845. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Fursova, N.A.; Kelley, J.R.; Huseyin, M.K.; Feldmann, A.; Klose, R.J. PRC1 Catalytic Activity Is Central to Polycomb System Function. Mol. Cell 2020, 77, 857–874. [Google Scholar] [CrossRef]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schubeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Huang, B.; Zhang, B.; Xiang, Y.; Du, Z.; Xu, Q.; Li, Y.; Wang, Q.; Ma, J.; Peng, X.; et al. Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 2016, 63, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zheng, H.; Kawamura, Y.K.; Zhang, K.; Gassler, J.; Powell, S.; Xu, Q.; Lin, Z.; Xu, K.; Zhou, Q.; et al. Polycomb Group Proteins Regulate Chromatin Architecture in Mouse Oocytes and Early Embryos. Mol. Cell 2020, 77, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; Xie, Y.; Verhoeven, E.; Vermeulen, M.; Lancini, C.; Gargiulo, G.; Hulsman, D.; Mann, M.; Knoblich, J.A.; van Lohuizen, M. The chromodomain helicase Chd4 is required for Polycomb-mediated inhibition of astroglial differentiation. EMBO J. 2013, 32, 1598–1612. [Google Scholar] [CrossRef]
- Reynolds, N.; Salmon-Divon, M.; Dvinge, H.; Hynes-Allen, A.; Balasooriya, G.; Leaford, D.; Behrens, A.; Bertone, P.; Hendrich, B. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J. 2012, 31, 593–605. [Google Scholar] [CrossRef]
- Latos, P.A.; Helliwell, C.; Mosaku, O.; Dudzinska, D.A.; Stubbs, B.; Berdasco, M.; Esteller, M.; Hendrich, B. NuRD-dependent DNA methylation prevents ES cells from accessing a trophectoderm fate. Biol. Open 2012, 1, 341–352. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Gruenewald-Schneider, U.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Smith, C.; Harrison, D.J.; Andrews, R.; Bird, A.P. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 2010, 6, e1001134. [Google Scholar] [CrossRef]
- Colbran, L.L.; Chen, L.; Capra, J.A. Sequence Characteristics Distinguish Transcribed Enhancers from Promoters and Predict Their Breadth of Activity. Genetics 2019, 211, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Steinhaus, R.; Gonzalez, T.; Seelow, D.; Robinson, P.N. Pervasive and CpG-dependent promoter-like characteristics of transcribed enhancers. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Dowell, K.G.; Simons, A.K.; Bai, H.; Kell, B.; Wang, Z.Z.; Yun, K.; Hibbs, M.A. Novel insights into embryonic stem cell self-renewal revealed through comparative human and mouse systems biology networks. Stem Cells 2014, 32, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fisher, R.A. RGS6 interacts with DMAP1 and DNMT1 and inhibits DMAP1 transcriptional repressor activity. J. Biol. Chem. 2004, 279, 14120–14128. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Sugiyama, K.; Yamamoto, T.; Oritani, K.; Shimoda, K.; Matsuda, T. Physical and functional interactions between Daxx and TSG101. Biochem. Biophys. Res. Commun. 2004, 316, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liang, Q.; Li, X.; Tsoi, H.; Zhang, J.; Wang, H.; Go, M.Y.; Chiu, P.W.; Ng, E.K.; Sung, J.J.; et al. MDGA2 is a novel tumour suppressor cooperating with DMAP1 in gastric cancer and is associated with disease outcome. Gut 2016, 65, 1619–1631. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.J.; Son, S.H.; Lim, C.S.; Kim, M.Y.; Lee, S.W.; Lee, S.; Jeon, J.; Ha, D.H.; Jung, N.R.; Han, S.Y.; et al. MMTR/Dmap1 Sets the Stage for Early Lineage Commitment of Embryonic Stem Cells by Crosstalk with PcG Proteins. Cells 2020, 9, 1190. https://doi.org/10.3390/cells9051190
Lee YJ, Son SH, Lim CS, Kim MY, Lee SW, Lee S, Jeon J, Ha DH, Jung NR, Han SY, et al. MMTR/Dmap1 Sets the Stage for Early Lineage Commitment of Embryonic Stem Cells by Crosstalk with PcG Proteins. Cells. 2020; 9(5):1190. https://doi.org/10.3390/cells9051190
Chicago/Turabian StyleLee, Young Jin, Seung Han Son, Chang Su Lim, Min Young Kim, Si Woo Lee, Sangwon Lee, Jinseon Jeon, Dae Hyun Ha, Na Rae Jung, Su Youne Han, and et al. 2020. "MMTR/Dmap1 Sets the Stage for Early Lineage Commitment of Embryonic Stem Cells by Crosstalk with PcG Proteins" Cells 9, no. 5: 1190. https://doi.org/10.3390/cells9051190
APA StyleLee, Y. J., Son, S. H., Lim, C. S., Kim, M. Y., Lee, S. W., Lee, S., Jeon, J., Ha, D. H., Jung, N. R., Han, S. Y., Do, B.-R., Na, I., Uversky, V. N., & Kim, C. G. (2020). MMTR/Dmap1 Sets the Stage for Early Lineage Commitment of Embryonic Stem Cells by Crosstalk with PcG Proteins. Cells, 9(5), 1190. https://doi.org/10.3390/cells9051190