TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Plasmid and siRNA Transfection
2.3. RT-qPCR
2.4. Western Blot Analysis
2.5. Chromatin Immunoprecipitation (ChIP) Assay
2.6. Cell Cycle Analysis by Flow Cytometry
2.7. High Content Imaging Analysis
3. Results
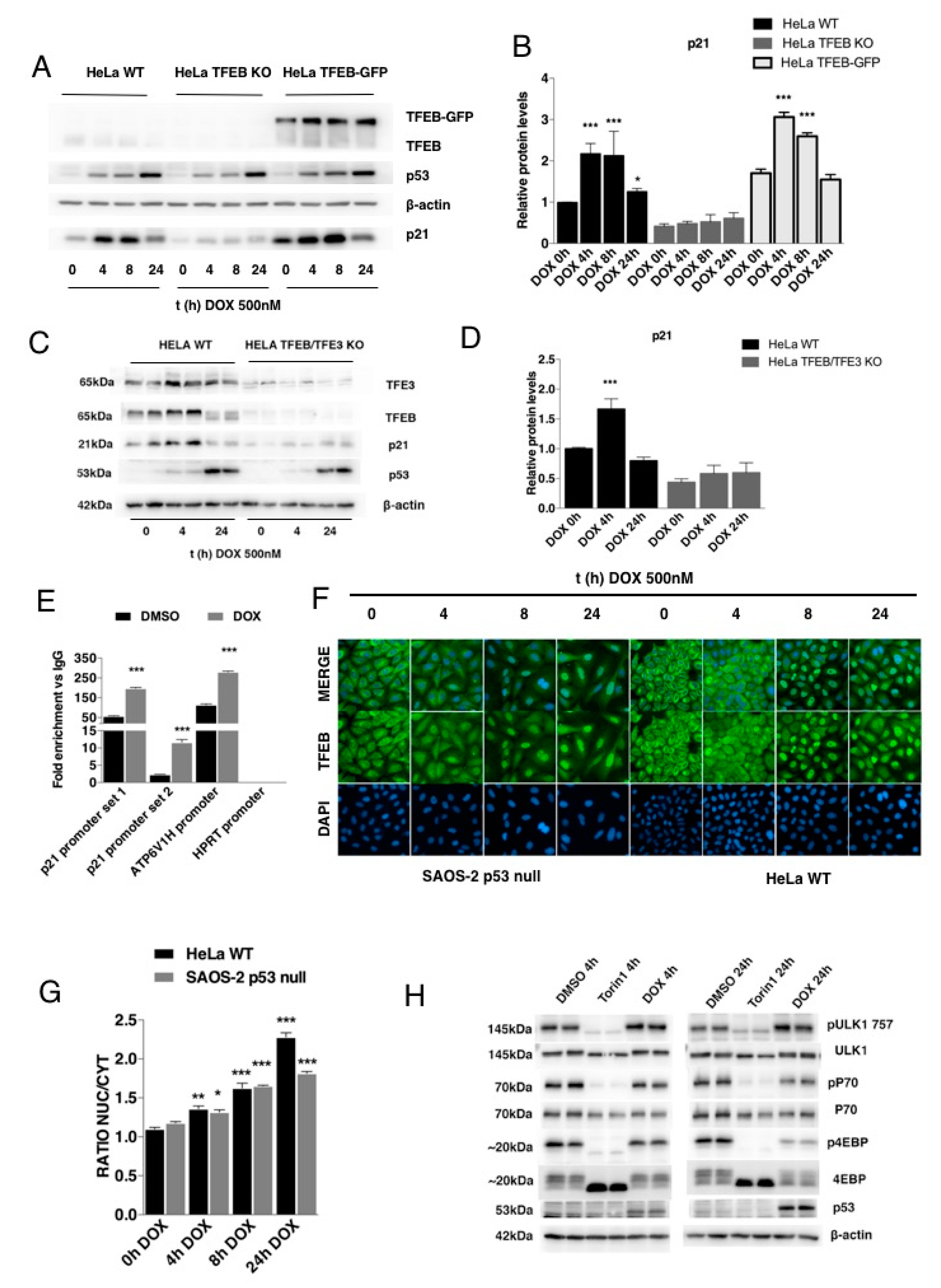
3.1. TFEB Transcriptionally Regulates p21/WAF1 Expression
3.2. TFEB-Mediated Induction of p21 Requires p53
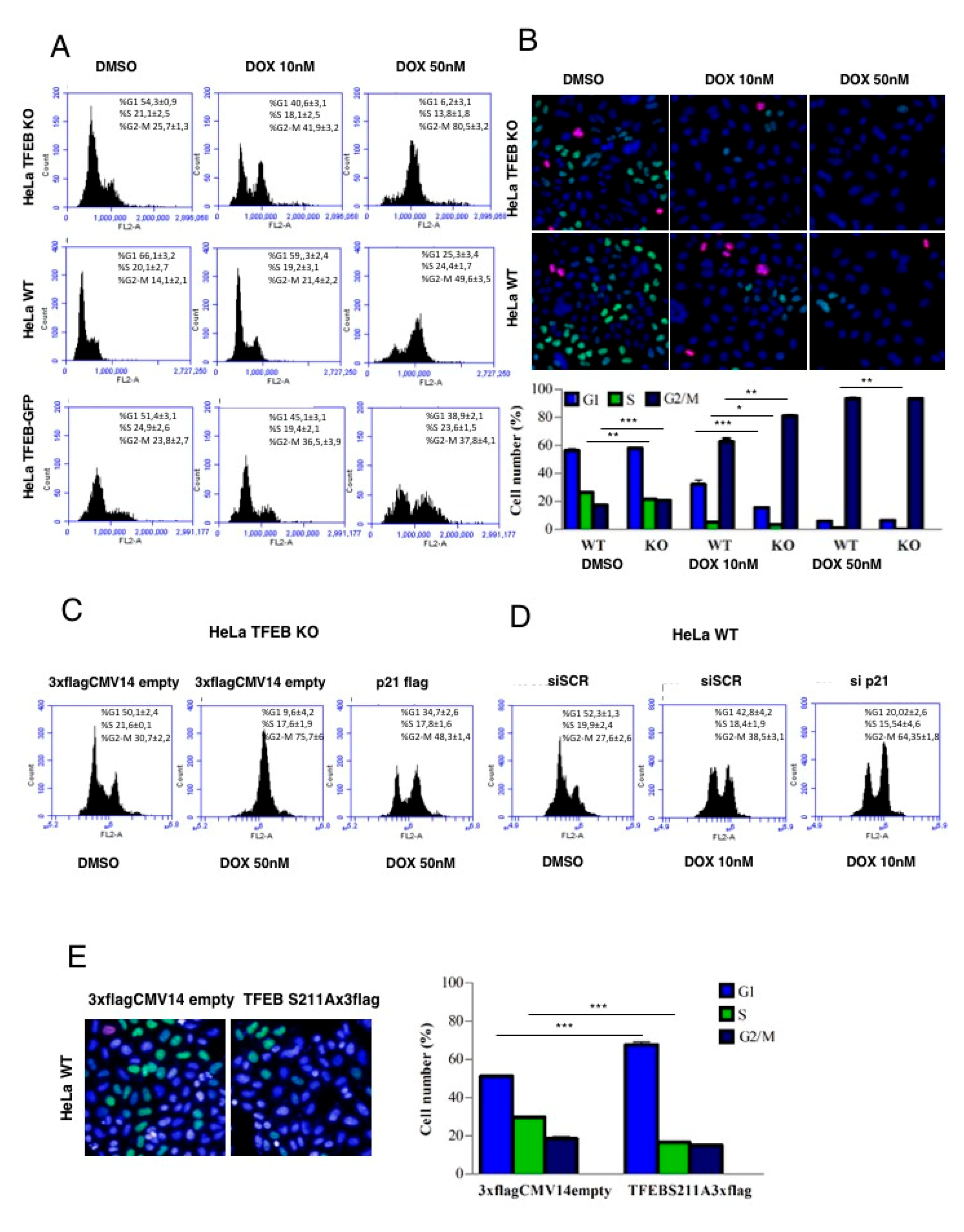
3.3. p21 Modulation in Response to DNA Damage Requires TFEB
3.4. Lack of TFEB Promotes a Faster Arrest in G2/M Phase in Response to DNA Damage
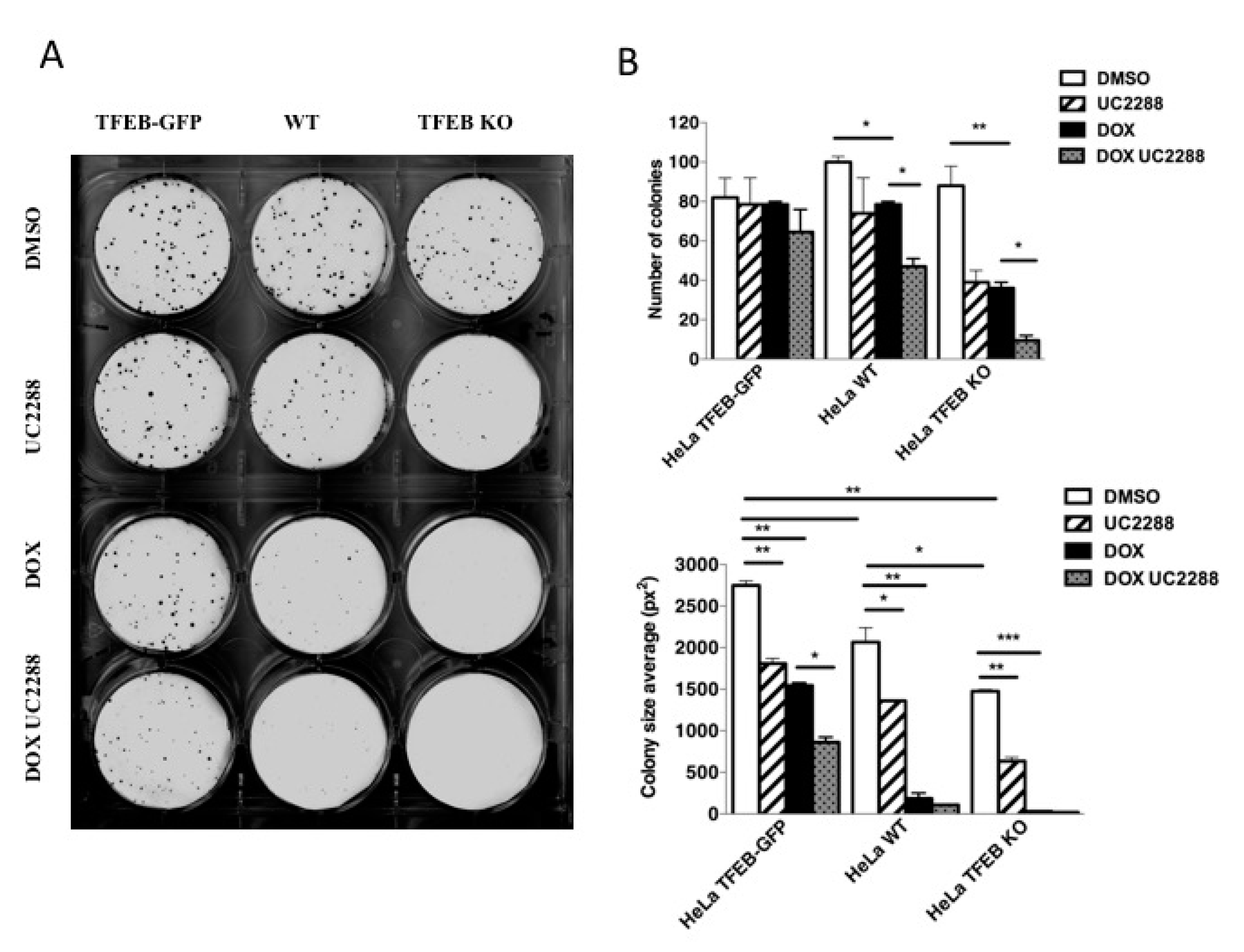
3.5. TFEB Promotes Cancer Cell Survival Upon DNA Damage Through p21 Upregulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Carreira, S.; Goodall, J.; Aksan, I.; La Rocca, S.A.; Galibert, M.-D.; Denat, L.; Larue, L.; Goding, C.R. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 2005, 433, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Singh, A.; Yang, Z.; Garcia, A.; Kong, Y.; Meyskens, F.L. MiTF links Erk1/2 kinase and p21 CIP1/WAF1 activation after UVC radiation in normal human melanocytes and melanoma cells. Mol. Cancer 2010, 9, 214. [Google Scholar] [CrossRef]
- Zhang, X.D. A pair of new statistical parameters for quality control in RNA interference high-throughput screening assays. Genomics 2007, 89, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.H. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell 2003, 4, 425–429. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A two-faced genome guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Wei, C.-Y.; Tan, Q.-X.; Zhu, X.; Qin, Q.-H.; Zhu, F.-B.; Mo, Q.-G.; Yang, W.-P. Expression of CDKN1A/p21 and TGFBR2 in breast cancer and their prognostic significance. Int. J. Clin. Exp. Pathol. 2015, 8, 14619–14629. [Google Scholar]
- Weiss, R.H.; Borowsky, A.D.; Seligson, D.; Lin, P.-Y.; Dillard-Telm, L.; Belldegrun, A.S.; Figlin, R.A.; Pantuck, A.D. p21 is a prognostic marker for renal cell carcinoma: Implications for novel therapeutic approaches. J. Urol. 2007, 177, 63–69. [Google Scholar] [CrossRef]
- Cronin, J.C.; Wunderlich, J.; Loftus, S.K.; Prickett, T.D.; Wei, X.; Ridd, K.; Vemula, S.; Burrell, A.S.; Agrawal, N.S.; Lin, J.C.; et al. Frequent mutations in the MITF pathway in melanoma. Pigment Cell Melanoma Res. 2009, 22, 435–444. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Ladanyi, M. Distinctive neoplasms characterised by specific chromosomal translocations comprise a significant proportion of paediatric renal cell carcinomas. Pathology 2003, 35, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Lu, Y.J.; Sidhar, S.K.; Parker, C.; Gill, S.; Smedley, D.; Hamoudi, R.; Linehan, W.M.; Shipley, J.; Cooper, C.S. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene 1997, 15, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.J.; Hsi, B.-L.; Arroyo, J.D.; Vargas, S.O.; Yeh, Y.A.; Motyckova, G.; Valencia, P.; Perez-Atayde, A.R.; Argani, P.; Ladanyi, M.; et al. Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc. Natl. Acad. Sci. USA 2003, 100, 6051–6056. [Google Scholar] [CrossRef]
- Kuiper, R.P.; Schepens, M.; Thijssen, J.; van Asseldonk, M.; van den Berg, E.; Bridge, J.; Schuuring, E.; Schoenmakers, E.F.P.M.; van Kessel, A.G. Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum. Mol. Genet. 2003, 12, 1661–1669. [Google Scholar] [CrossRef]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Argani, P.; Healey, J.H.; Ueda, T.; Yoshikawa, H.; Meloni-Ehrig, A.; et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 2001, 20, 48–57. [Google Scholar] [CrossRef]
- Sidhar, S.K.; Clark, J.; Gill, S.; Hamoudi, R.; Crew, A.J.; Gwilliam, R.; Ross, M.; Linehan, W.M.; Birdsall, S.; Shipley, J.; et al. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum. Mol. Genet. 1996, 5, 1333–1338. [Google Scholar] [CrossRef]
- Lilleby, W.; Vlatkovic, L.; Meza-Zepeda, L.A.; Revheim, M.-E.; Hovig, E. Translocational renal cell carcinoma (t(6;11)(p21;q12) with transcription factor EB (TFEB) amplification and an integrated precision approach: A case report. J. Med. Case Rep. 2015, 9, 281. [Google Scholar] [CrossRef][Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef]
- Brady, O.A.; Jeong, E.; Martina, J.A.; Pirooznia, M.; Tunc, I.; Puertollano, R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife 2018, 7. [Google Scholar] [CrossRef]
- Doronzo, G.; Astanina, E.; Corà, D.; Chiabotto, G.; Comunanza, V.; Noghero, A.; Neri, F.; Puliafito, A.; Primo, L.; Spampanato, C.; et al. TFEB controls vascular development by regulating the proliferation of endothelial cells. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Soldati, C.; Bithell, A.; Johnston, C.; Wong, K.-Y.; Stanton, L.W.; Buckley, N.J. Dysregulation of REST-regulated coding and non-coding RNAs in a cellular model of Huntington’s disease. J. Neurochem. 2013, 124, 418–430. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Nezich, C.L.; Wang, C.; Fogel, A.I.; Youle, R.J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 2015, 210, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Hemesath, T.J.; Steingrímsson, E.; McGill, G.; Hansen, M.J.; Vaught, J.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A.; Fisher, D.E. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994, 8, 2770–2780. [Google Scholar] [CrossRef] [PubMed]
- Laptenko, O.; Beckerman, R.; Freulich, E.; Prives, C. p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. USA 2011, 108, 10385–10390. [Google Scholar] [CrossRef] [PubMed]
- Saramäki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Kracikova, M.; Akiri, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef]
- Lukin, D.J.; Carvajal, L.A.; Liu, W.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes cell survival due to the reversibility of its cell-cycle checkpoints. Mol. Cancer Res. 2015, 13, 16–28. [Google Scholar] [CrossRef]
- Wenzel, E.S.; Singh, A.T.K. Cell-cycle checkpoints and aneuploidy on the path to cancer. In Vivo 2018, 32, 1–5. [Google Scholar] [CrossRef]
- Wettersten, H.I.; Hee Hwang, S.; Li, C.; Shiu, E.Y.; Wecksler, A.T.; Hammock, B.D.; Weiss, R.H. A novel p21 attenuator which is structurally related to sorafenib. Cancer Biol. Ther. 2013, 14, 278–285. [Google Scholar] [CrossRef]
- Kauffman, E.C.; Ricketts, C.J.; Rais-Bahrami, S.; Yang, Y.; Merino, M.J.; Bottaro, D.P.; Srinivasan, R.; Linehan, W.M. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 2014, 11, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, L.; Su, Q.; Fan, X.; Wang, Y.; Gao, S.; Wang, H.; Chen, H.; Chan, C.B.; Liu, Z. Phosphorylation of MITF by AKT affects its downstream targets and causes TP53-dependent cell senescence. Int. J. Biochem. Cell Biol. 2016, 80, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Yoshida, H. ASPL-TFE3 oncoprotein regulates cell cycle progression and induces cellular senescence by Up-Regulating p21. Neoplasia 2016, 18, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Medendorp, K.; van Groningen, J.J.M.; Vreede, L.; Hetterschijt, L.; Brugmans, L.; van den Hurk, W.H.; van Kessel, A.G. The renal cell carcinoma-associated oncogenic fusion protein PRCCTFE3 provokes p21 WAF1/CIP1-mediated cell cycle delay. Exp. Cell Res. 2009, 315, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Slade, L.; Biswas, D.; Ihionu, F.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. A lysosome independent role for TFEB in activating DNA repair and inhibiting apoptosis in breast cancer cells. Biochem. J. 2020, 477, 137–160. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisonero-Vaquero, S.; Soldati, C.; Cesana, M.; Ballabio, A.; Medina, D.L. TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response. Cells 2020, 9, 1186. https://doi.org/10.3390/cells9051186
Pisonero-Vaquero S, Soldati C, Cesana M, Ballabio A, Medina DL. TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response. Cells. 2020; 9(5):1186. https://doi.org/10.3390/cells9051186
Chicago/Turabian StylePisonero-Vaquero, Sandra, Chiara Soldati, Marcella Cesana, Andrea Ballabio, and Diego Luis Medina. 2020. "TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response" Cells 9, no. 5: 1186. https://doi.org/10.3390/cells9051186
APA StylePisonero-Vaquero, S., Soldati, C., Cesana, M., Ballabio, A., & Medina, D. L. (2020). TFEB Modulates p21/WAF1/CIP1 during the DNA Damage Response. Cells, 9(5), 1186. https://doi.org/10.3390/cells9051186