The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance

 ,
,  ,
,  ,
,  and
and
Abstract
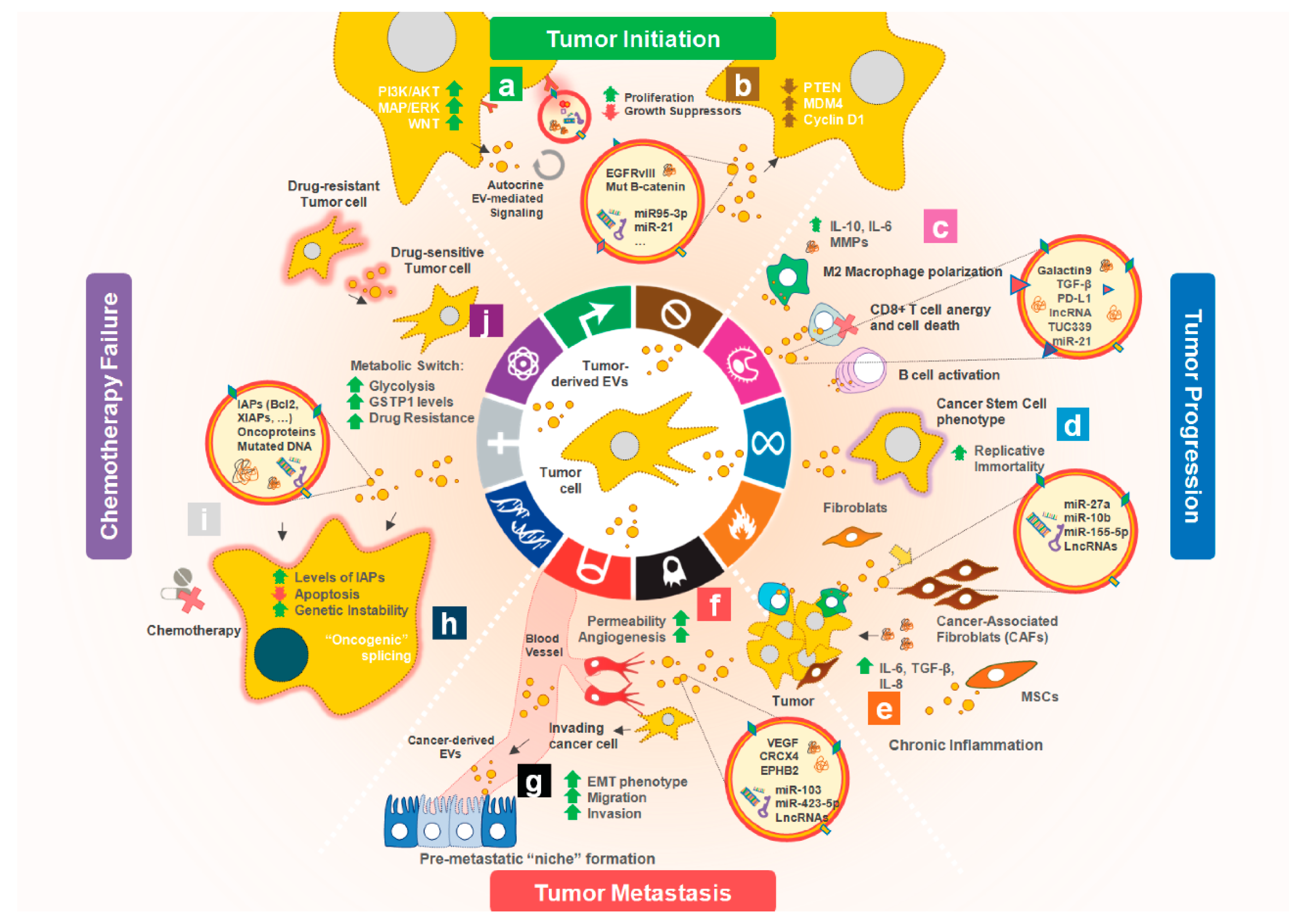
1. Introduction
2. Tumor-Derived EVs affecting Cancer Hallmarks
2.1. Promoting Cell Proliferation and Escape from Apoptosis
2.2. Sustaining Angiogenesis
2.3. Contributing to Cancer Cell Invasion and Metastasis
2.4. Reprogramming Energy Metabolism
2.5. Transferring Mutations
2.6. Modulating the TME: Evading Immune Response and Promoting Inflammation
2.6.1. Impact of Tumor-Derived EVs on Macrophages
2.6.2. Impact of Tumor-Derived EVs on T Lymphocytes
2.6.3. Impact of Tumor-Derived EVs on Fibroblasts
3. Tumor-Derived EVs Affecting Cancer Therapy Resistance
3.1. Intercellular Transfer of Drug Resistant Traits between Cancer Cells
3.1.1. Transferring Drug-Efflux Pumps
3.1.2. Transferring Apoptotic Modulators
3.1.3. Transferring Other Proteins
3.1.4. Transferring microRNAs, mRNAs and lncRNAs
miRNAs
mRNAs
lncRNAs
3.1.5. Transferring Lipids
3.2. Intercellular Transfer of Traits between the Microenvironment and Tumor Cells
3.3. Drug Efflux Mediated by EVs
3.4. Increased Release of EVs by Drug Resistant Cells
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AGAP2-AS1 | AGAP2 Antisense RNA 1 |
| ALIX | ALG-2-interacting protein X |
| Alk | Anaplastic Lymphoma Kinase |
| APAF1 | Apoptotic Peptidase Activating Factor 1 |
| AREG | amphiregulin |
| ARF6 | ADP-ribosylation factor 6 |
| ATP1A1 | ATPase Na+/K+ Transporting Subunit Alpha 1 |
| ATP1B3 | ATPase Na+/K+ Transporting Subunit Beta 3 |
| AXL | AXL Receptor Tyrosine Kinase |
| BAX | BCL2 Associated X, Apoptosis Regulator |
| Bcl-2 | B-cell lymphoma 2 |
| BRCP/ABCG2 | breast cancer resistance protein |
| CADM1 | cell adhesion molecule 1 |
| CAFs | cancer-associated fibroblasts |
| CALN1 | Calneuron 1 |
| CAV1 | Caveolin 1 |
| CCAL | colorectal cancer-associated lncRNA |
| CDKN1B | Cyclin Dependent Kinase Inhibitor 1B |
| CEACAM | carcinoembryonic antigen related cell adhesion molecule |
| CLIC1 | Chloride Intracellular Channel 1 |
| CLIC1 | protein chloride intracellular channel-1 |
| CXCR-4 | C-X-C chemokine receptor type 4 |
| DCK | Deoxycytidine Kinase |
| DNMT1 | DNA methyltranferase 1 |
| DR | drug resistant |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial to Mesenchymal Transition |
| EPACAM | Hepatic and Glial Cell Adhesion Molecule |
| EPHB2 | ephrin type B receptor 2 |
| ERK | extracellular-signal-regulated kinase |
| EVs | Extracellular Vesicles |
| FGF | fibroblast growth factor |
| GLUT-1 | Glucose transporter 1 |
| GSTP1 | Glutathione S-transferase P |
| HER-2 | Human Epidermal growth factor Receptor-type 2 |
| HMGA | high mobility group A |
| HNF1A-AS1 | HNF1A Antisense RNA 1 |
| HOTTIP | HOXA Distal Transcript Antisense RNA |
| HSP70 | Heat Shock Protein 70 |
| IAP | Inhibitors of apoptosis proteins |
| ICAM-1 | intercellular adhesion molecule 1 |
| ING5 | Inhibitor Of Growth Family Member 5 |
| ISEV | International Society for Extracellular Vesicles |
| JNK | c-Jun N-terminal kinase |
| LAMP | lysosomal associated membrane protein |
| LncRNA | long non-coding RNAs |
| MAPK | mitogen-activated protein kinase |
| MDM4 | MDM4 Regulator of P53 |
| MDR | multidrug resistance |
| MECP2 | Methyl-CpG Binding Protein 2 |
| miRs | microRNAs |
| MLH1 | MutL homolog 1 |
| MMPs | Matrix Metalloproteases |
| mRNAs | Messenger RNA |
| MRP1/ABCC1 | multidrug resistance-associated protein 1 |
| MSC | Mesenchymal Stromal Cells |
| MVs | ectosomes or microparticles, |
| NEU1 | lysosomal sialidase |
| NFATc3 | Nuclear Factor Of Activated T Cells 3 |
| NSCLC | non-small cell lung cancer |
| PAX2 | paired box gene 2 |
| PDCD4 | programmed cell death 4 |
| PDGFR | platelet-derived growth factor |
| PDK | Protein 3-phosphoinositide-dependent protein kinase |
| PD-L1 | Programmed death-ligand 1 |
| P-gp/MDR1/ABCB1 | P-glycoprotein |
| PTPRZ1 | Protein Tyrosine Phosphatase Receptor Type Z1 |
| RAB7A | RAB7A, Member RAS Oncogene Family |
| RBM11 | RNA Binding Motif Protein 11 |
| RhoA | member of the small GTPases family |
| RIG-I | retinoic acid-inducible gene I |
| ROR | Receptor tyrosine kinase-like orphan receptor |
| ROS | reactive oxygen species |
| SBF2-AS1 | SBF2 antisense RNA 1 |
| SNHG14 | small nucleolar RNA host gene 14 |
| SOD2 | Superoxide dismutase 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TERF1 | Telomeric Repeat Binding Factor 1 |
| TERT | Telomerase reverse transcriptase |
| TGFBR1 | Transforming Growth Factor Beta Receptor 1 |
| TGF-β | transforming growth factor beta |
| TGM2 | Transglutaminase 2 |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TME | tumor microenvironment |
| TP53INP1 | Tumor Protein P53 Inducible Nuclear Protein 1 |
| TrpC5 | Short transient receptor potential channel 5 |
| TSG101 | tumor susceptibility gene 101 protein |
| TSGA10 | testis-specific gene antigen |
| TUFT1 | Tuftelin 1 |
| VE-cadherin | vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| VLDLR | Very Low Density Lipoprotein Receptor |
| XIAP | X-linked inhibitor of apoptosis protein |
| XRCC4 | X-ray repair cross-complementing protein 4 |
| ZEB1-AS1 | ZEB1 Antisense RNA 1 |
References
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Roy, S.; Lin, H.Y.; Chou, C.Y.; Huang, C.H.; Small, J.; Sadik, N.; Ayinon, C.M.; Lansbury, E.; Cruz, L.; Yekula, A.; et al. Navigating the Landscape of Tumor Extracellular Vesicle Heterogeneity. Int. J. Mol. Sci. 2019, 20, 1349. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Abdelmohsen, K.; Mustapic, M.; Kapogiannis, D.; Gorospe, M. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA 2017, 8, e1413. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Kramer-Albers, E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013, 7, 182. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Yoshioka, Y.; Hagiwara, K.; Takeshita, F.; Ochiya, T. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J. Biol. Chem. 2012, 287, 1397–1405. [Google Scholar] [CrossRef]
- Puhka, M.; Takatalo, M.; Nordberg, M.E.; Valkonen, S.; Nandania, J.; Aatonen, M.; Yliperttula, M.; Laitinen, S.; Velagapudi, V.; Mirtti, T.; et al. Metabolomic Profiling of Extracellular Vesicles and Alternative Normalization Methods Reveal Enriched Metabolites and Strategies to Study Prostate Cancer-Related Changes. Theranostics 2017, 7, 3824–3841. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, M.H.; Caires, H.R.; Abols, A.; Xavier, C.P.R.; Line, A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drug Resist. Update 2019, 47, 100647. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef]
- Deregibus, M.C.; Cantaluppi, V.; Calogero, R.; Lo Iacono, M.; Tetta, C.; Biancone, L.; Bruno, S.; Bussolati, B.; Camussi, G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007, 110, 2440–2448. [Google Scholar] [CrossRef]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Aalberts, M.; van Dissel-Emiliani, F.M.; van Adrichem, N.P.; van Wijnen, M.; Wauben, M.H.; Stout, T.A.; Stoorvogel, W. Identification of distinct populations of prostasomes that differentially express prostate stem cell antigen, annexin A1, and GLIPR2 in humans. Biol. Reprod. 2012, 86, 82. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, P.A.; Zhou, H.; Pisitkun, T.; Wang, N.S.; Star, R.A.; Knepper, M.A.; Yuen, P.S. Isolation and purification of exosomes in urine. Methods Mol. Biol. 2010, 641, 89–99. [Google Scholar]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filen, J.J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Lasser, C.; O’Neil, S.E.; Ekerljung, L.; Ekstrom, K.; Sjostrand, M.; Lotvall, J. RNA-containing exosomes in human nasal secretions. Am. J. Rhinol. Allergy 2011, 25, 89–93. [Google Scholar] [CrossRef]
- Palanisamy, V.; Sharma, S.; Deshpande, A.; Zhou, H.; Gimzewski, J.; Wong, D.T. Nanostructural and transcriptomic analyses of human saliva derived exosomes. PLoS ONE 2010, 5, e8577. [Google Scholar] [CrossRef]
- Koga, Y.; Yasunaga, M.; Moriya, Y.; Akasu, T.; Fujita, S.; Yamamoto, S.; Matsumura, Y. Exosome can prevent RNase from degrading microRNA in feces. J. Gastrointest. Oncol. 2011, 2, 215–222. [Google Scholar]
- Armstrong, D.; Wildman, D.E. Extracellular Vesicles and the Promise of Continuous Liquid Biopsies. J. Pathol. Transl. Med. 2018, 52, 1–8. [Google Scholar] [CrossRef]
- Yokoi, A.; Yoshioka, Y.; Yamamoto, Y.; Ishikawa, M.; Ikeda, S.I.; Kato, T.; Kiyono, T.; Takeshita, F.; Kajiyama, H.; Kikkawa, F.; et al. Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 2017, 8, 14470. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular Vesicle-Mediated Cell(-)Cell Communication in the Nervous System: Focus on Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Perut, F.; Roncuzzi, L.; Baldini, N. The Emerging Roles of Extracellular Vesicles in Osteosarcoma. Front. Oncol. 2019, 9, 1342. [Google Scholar] [CrossRef]
- Graves, L.E.; Ariztia, E.V.; Navari, J.R.; Matzel, H.J.; Stack, M.S.; Fishman, D.A. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 2004, 64, 7045–7049. [Google Scholar] [CrossRef]
- Menck, K.; Bleckmann, A.; Wachter, A.; Hennies, B.; Ries, L.; Schulz, M.; Balkenhol, M.; Pukrop, T.; Schatlo, B.; Rost, U.; et al. Characterisation of tumour-derived microvesicles in cancer patients’ blood and correlation with clinical outcome. J. Extracell. Vesicles 2017, 6, 1340745. [Google Scholar] [CrossRef]
- Hallal, S.; Russell, B.P.; Wei, H.; Lee, M.Y.T.; Toon, C.W.; Sy, J.; Shivalingam, B.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles from Neurosurgical Aspirates Identifies Chaperonin Containing TCP1 Subunit 6A as a Potential Glioblastoma Biomarker with Prognostic Significance. Proteomics 2019, 19, e1800157. [Google Scholar] [CrossRef]
- Sedlarikova, L.; Bollova, B.; Radova, L.; Brozova, L.; Jarkovsky, J.; Almasi, M.; Penka, M.; Kuglik, P.; Sandecka, V.; Stork, M.; et al. Circulating exosomal long noncoding RNA PRINS-First findings in monoclonal gammopathies. Hematol. Oncol. 2018, 36, 786–791. [Google Scholar] [CrossRef]
- Yadav, D.K.; Bai, X.; Yadav, R.K.; Singh, A.; Li, G.; Ma, T.; Chen, W.; Liang, T. Liquid biopsy in pancreatic cancer: The beginning of a new era. Oncotarget 2018, 9, 26900–26933. [Google Scholar] [CrossRef]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef]
- Claridge, B.; Kastaniegaard, K.; Stensballe, A.; Greening, D.W. Post-translational and transcriptional dynamics—Regulating extracellular vesicle biology. Expert Rev. Proteom. 2019, 16, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Leonardi, T.; Huang, B.; Iraci, N.; Vega, B.; Pluchino, S. Extracellular vesicles and their synthetic analogues in aging and age-associated brain diseases. Biogerontology 2015, 16, 147–185. [Google Scholar] [CrossRef] [PubMed]
- Baek, R.; Sondergaard, E.K.; Varming, K.; Jorgensen, M.M. The impact of various preanalytical treatments on the phenotype of small extracellular vesicles in blood analyzed by protein microarray. J. Immunol. Methods 2016, 438, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bjorge, I.M.; Kim, S.Y.; Mano, J.F.; Kalionis, B.; Chrzanowski, W. Extracellular vesicles, exosomes and shedding vesicles in regenerative medicine—A new paradigm for tissue repair. Biomater. Sci. 2017, 6, 60–78. [Google Scholar] [CrossRef]
- Syn, N.L.; Wang, L.; Chow, E.K.; Lim, C.T.; Goh, B.C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef]
- Zhupanyn, P.; Ewe, A.; Buch, T.; Malek, A.; Rademacher, P.; Muller, C.; Reinert, A.; Jaimes, Y.; Aigner, A. Extracellular vesicle (ECV)-modified polyethylenimine (PEI) complexes for enhanced siRNA delivery in vitro and in vivo. J. Control. Release 2020, 319, 63–76. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.L.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135e10. [Google Scholar] [CrossRef]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef]
- Vella, L.J.; Behren, A.; Coleman, B.; Greening, D.W.; Hill, A.F.; Cebon, J. Intercellular Resistance to BRAF Inhibition Can Be Mediated by Extracellular Vesicle-Associated PDGFRbeta. Neoplasia 2017, 19, 932–940. [Google Scholar] [CrossRef]
- Jin, H.; Liu, P.; Wu, Y.; Meng, X.; Wu, M.; Han, J.; Tan, X. Exosomal zinc transporter ZIP4 promotes cancer growth and is a novel diagnostic biomarker for pancreatic cancer. Cancer Sci. 2018, 109, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.X.; Liao, J.; Xie, M.; Gao, Z.K.; Wang, X.H.; Zhang, Y.; Shang, M.H.; Yin, L.H.; Pu, Y.P.; Liu, R. miR-93-5p Transferred by Exosomes Promotes the Proliferation of Esophageal Cancer Cells via Intercellular Communication by Targeting PTEN. Biomed. Environ. Sci. 2018, 31, 171–185. [Google Scholar] [PubMed]
- Chen, D.; Wu, X.; Xia, M.; Wu, F.; Ding, J.; Jiao, Y.; Zhan, Q.; An, F. Upregulated exosomic miR23b3p plays regulatory roles in the progression of pancreatic cancer. Oncol. Rep. 2017, 38, 2182–2188. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Ren, Z.J.; Tang, J.H.; Yu, Q. Exosomal MicroRNA MiR-1246 Promotes Cell Proliferation, Invasion and Drug Resistance by Targeting CCNG2 in Breast Cancer. Cell Physiol. Biochem. 2017, 44, 1741–1748. [Google Scholar] [CrossRef]
- Kitdumrongthum, S.; Metheetrairut, C.; Charoensawan, V.; Ounjai, P.; Janpipatkul, K.; Panvongsa, W.; Weerachayaphorn, J.; Piyachaturawat, P.; Chairoungdua, A. Dysregulated microRNA expression profiles in cholangiocarcinoma cell-derived exosomes. Life Sci. 2018, 210, 65–75. [Google Scholar] [CrossRef]
- Soldevilla, B.; Rodriguez, M.; San Millan, C.; Garcia, V.; Fernandez-Perianez, R.; Gil-Calderon, B.; Martin, P.; Garcia-Grande, A.; Silva, J.; Bonilla, F.; et al. Tumor-derived exosomes are enriched in DeltaNp73, which promotes oncogenic potential in acceptor cells and correlates with patient survival. Hum. Mol. Genet. 2014, 23, 467–478. [Google Scholar] [CrossRef]
- Teng, Y.; Ren, Y.; Hu, X.; Mu, J.; Samykutty, A.; Zhuang, X.; Deng, Z.; Kumar, A.; Zhang, L.; Merchant, M.L.; et al. MVP-mediated exosomal sorting of miR-193a promotes colon cancer progression. Nat. Commun. 2017, 8, 14448. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, T.; Chen, Y.; Xiao, J. Exosome-mediated miR-200b promotes colorectal cancer proliferation upon TGF-beta1 exposure. Biomed. Pharmacother. 2018, 106, 1135–1143. [Google Scholar] [CrossRef]
- Guo, K.; Yao, J.; Yu, Q.; Li, Z.; Huang, H.; Cheng, J.; Wang, Z.; Zhu, Y. The expression pattern of long non-coding RNA PVT1 in tumor tissues and in extracellular vesicles of colorectal cancer correlates with cancer progression. Tumour. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Lee, J.C.; Zhao, J.T.; Gundara, J.; Serpell, J.; Bach, L.A.; Sidhu, S. Papillary thyroid cancer-derived exosomes contain miRNA-146b and miRNA-222. J. Surg. Res. 2015, 196, 39–48. [Google Scholar] [CrossRef]
- Cappellesso, R.; Tinazzi, A.; Giurici, T.; Simonato, F.; Guzzardo, V.; Ventura, L.; Crescenzi, M.; Chiarelli, S.; Fassina, A. Programmed cell death 4 and microRNA 21 inverse expression is maintained in cells and exosomes from ovarian serous carcinoma effusions. Cancer Cytopathol. 2014, 122, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, X.M.; Tao, H.M.; Xiong, W.; Jie, S.H.; Li, H.Y. Regulation of the expression of zinc finger protein genes by microRNAs enriched within acute lymphoblastic leukemia-derived microvesicles. Genet. Mol. Res. 2015, 14, 11884–11895. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liang, W.; Fu, M.; Huang, Z.H.; Li, X.; Zhang, W.; Zhang, P.; Qian, H.; Jiang, P.C.; Xu, W.R.; et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J. Cancer Res. Clin. Oncol. 2017, 143, 991–1004. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhou, M.W.; Bai, L.; Han, R.Y.; Lv, K.; Wang, Z. Extracellular vesicles promote esophageal cancer progression by delivering lncZEB1-AS1 between cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2662–2670. [Google Scholar]
- Kogure, T.; Yan, I.K.; Lin, W.L.; Patel, T. Extracellular Vesicle-Mediated Transfer of a Novel Long Noncoding RNA TUC339: A Mechanism of Intercellular Signaling in Human Hepatocellular Cancer. Genes Cancer 2013, 4, 261–272. [Google Scholar] [CrossRef]
- Lang, H.L.; Hu, G.W.; Zhang, B.; Kuang, W.; Chen, Y.; Wu, L.; Xu, G.H. Glioma cells enhance angiogenesis and inhibit endothelial cell apoptosis through the release of exosomes that contain long non-coding RNA CCAT2. Oncol. Rep. 2017, 38, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.L.; Hu, G.W.; Chen, Y.; Liu, Y.; Tu, W.; Lu, Y.M.; Wu, L.; Xu, G.H. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 959–972. [Google Scholar]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef]
- Giusti, I.; Delle Monache, S.; Di Francesco, M.; Sanita, P.; D’Ascenzo, S.; Gravina, G.L.; Festuccia, C.; Dolo, V. From glioblastoma to endothelial cells through extracellular vesicles: Messages for angiogenesis. Tumour. Biol. 2016, 37, 12743–12753. [Google Scholar] [CrossRef]
- Sato, S.; Vasaikar, S.; Eskaros, A.; Kim, Y.; Lewis, J.S.; Zhang, B.; Zijlstra, A.; Weaver, A.M. EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Huang, A.; Dong, J.; Li, S.; Wang, C.; Ding, H.; Li, H.; Su, X.; Ge, X.; Sun, L.; Bai, C.; et al. Exosomal transfer of vasorin expressed in hepatocellular carcinoma cells promotes migration of human umbilical vein endothelial cells. Int. J. Biol. Sci. 2015, 11, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.H.; Zhang, Z.J.; Shang, L.R.; Luo, Y.W.; Lin, Y.F.; Yuan, Y.; Zhuang, S.M. Hepatoma cell-secreted exosomal microRNA-103 increases vascular permeability and promotes metastasis by targeting junction proteins. Hepatology 2018, 68, 1459–1475. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hong, J.; Hong, M.; Wang, Y.; Yu, T.; Zang, S.; Wu, Q. piRNA-823 delivered by multiple myeloma-derived extracellular vesicles promoted tumorigenesis through re-educating endothelial cells in the tumor environment. Oncogene 2019, 38, 5227–5238. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef]
- Lawson, J.; Dickman, C.; MacLellan, S.; Towle, R.; Jabalee, J.; Lam, S.; Garnis, C. Selective secretion of microRNAs from lung cancer cells via extracellular vesicles promotes CAMK1D-mediated tube formation in endothelial cells. Oncotarget 2017, 8, 83913–83924. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, L.; Chen, C.; Ming, P.; Huang, Q.; Li, C.; Cao, D.; Xu, X.; Ge, W. The extracellular vesicles secreted by lung cancer cells in radiation therapy promote endothelial cell angiogenesis by transferring miR-23a. PeerJ 2017, 5, e3627. [Google Scholar] [CrossRef]
- Masoumi-Dehghi, S.; Babashah, S.; Sadeghizadeh, M. MicroRNA-141-3p-containing small extracellular vesicles derived from epithelial ovarian cancer cells promote endothelial cell angiogenesis through activating the JAK/STAT3 and NF-kappaB signaling pathways. J. Cell Commun. Signal. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Dickman, C.T.; Lawson, J.; Jabalee, J.; MacLellan, S.A.; LePard, N.E.; Bennewith, K.L.; Garnis, C. Selective extracellular vesicle exclusion of miR-142-3p by oral cancer cells promotes both internal and extracellular malignant phenotypes. Oncotarget 2017, 8, 15252–15266. [Google Scholar] [CrossRef]
- Bao, L.; You, B.; Shi, S.; Shan, Y.; Zhang, Q.; Yue, H.; Zhang, J.; Zhang, W.; Shi, Y.; Liu, Y.; et al. Metastasis-associated miR-23a from nasopharyngeal carcinoma-derived exosomes mediates angiogenesis by repressing a novel target gene TSGA10. Oncogene 2018, 37, 2873–2889. [Google Scholar] [CrossRef]
- Campos, A.; Salomon, C.; Bustos, R.; Diaz, J.; Martinez, S.; Silva, V.; Reyes, C.; Diaz-Valdivia, N.; Varas-Godoy, M.; Lobos-Gonzalez, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine (Lond.) 2018, 13, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Yamamoto, H.; Kishida, S.; Kishida, M.; Awada, C.; Takao, T.; Kikuchi, A. Wnt5b-associated exosomes promote cancer cell migration and proliferation. Cancer Sci. 2017, 108, 42–52. [Google Scholar] [CrossRef]
- Higginbotham, J.N.; Demory Beckler, M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Feng, J.; Pan, H.; Jiang, Y.; Duan, Y.; Fa, Z. Exosomes derived from HCC cells with different invasion characteristics mediated EMT through TGF-beta/Smad signaling pathway. Oncol. Targets Ther. 2019, 12, 6897–6905. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, Y.; Xu, Y.; Wang, J.; Zhang, C.; Du, Y.; Wang, L.; Li, L.; Wang, B.; Shen, J.; et al. Horizontal transfer of exosomal CXCR4 promotes murine hepatocarcinoma cell migration, invasion and lymphangiogenesis. Gene 2018, 676, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Zhang, Q.; Lou, Y.; Yang, J.; Nie, G.; Chen, Q.; Chen, Y.; Zhang, J.; Wang, J.; Wei, T.; et al. Primary tumor-derived exosomes facilitate metastasis by regulating adhesion of circulating tumor cells via SMAD3 in liver cancer. Oncogene 2018, 37, 6105–6118. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Wang, X.; Zhao, Y.; Hu, R.; Qin, L. Exosomal miR-93 promotes proliferation and invasion in hepatocellular carcinoma by directly inhibiting TIMP2/TP53INP1/CDKN1A. Biochem. Biophys. Res. Commun. 2018, 502, 515–521. [Google Scholar] [CrossRef]
- Yang, H.; Fu, H.; Wang, B.; Zhang, X.; Mao, J.; Li, X.; Wang, M.; Sun, Z.; Qian, H.; Xu, W. Exosomal miR-423-5p targets SUFU to promote cancer growth and metastasis and serves as a novel marker for gastric cancer. Mol. Carcinog. 2018, 57, 1223–1236. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L.; Bucay, N.; Sekhon, K.; Majid, S.; Shahryari, V.; Dahiya, R.; Tanaka, Y.; Saini, S. microRNA-1246 Is an Exosomal Biomarker for Aggressive Prostate Cancer. Cancer Res. 2018, 78, 1833–1844. [Google Scholar] [CrossRef]
- Cai, Q.; Zhu, A.; Gong, L. Exosomes of glioma cells deliver miR-148a to promote proliferation and metastasis of glioblastoma via targeting CADM1. Bull. Cancer 2018, 105, 643–651. [Google Scholar] [CrossRef]
- Yoshimura, A.; Sawada, K.; Nakamura, K.; Kinose, Y.; Nakatsuka, E.; Kobayashi, M.; Miyamoto, M.; Ishida, K.; Matsumoto, Y.; Kodama, M.; et al. Exosomal miR-99a-5p is elevated in sera of ovarian cancer patients and promotes cancer cell invasion by increasing fibronectin and vitronectin expression in neighboring peritoneal mesothelial cells. BMC Cancer 2018, 18, 1065. [Google Scholar] [CrossRef]
- Yang, S.J.; Wang, D.D.; Li, J.; Xu, H.Z.; Shen, H.Y.; Chen, X.; Zhou, S.Y.; Zhong, S.L.; Zhao, J.H.; Tang, J.H. Predictive role of GSTP1-containing exosomes in chemotherapy-resistant breast cancer. Gene 2017, 623, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.L.; Yan, W.; Liu, Y.W.; Wang, Z.; Hu, Q.; Nie, E.; Zhou, X.; Li, R.; Wang, X.F.; Jiang, T.; et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene 2017, 36, 5369–5381. [Google Scholar] [CrossRef] [PubMed]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Cesi, G.; Philippidou, D.; Kozar, I.; Kim, Y.J.; Bernardin, F.; Van Niel, G.; Wienecke-Baldacchino, A.; Felten, P.; Letellier, E.; Dengler, S.; et al. A new ALK isoform transported by extracellular vesicles confers drug resistance to melanoma cells. Mol. Cancer 2018, 17, 145. [Google Scholar] [CrossRef]
- Kalra, H.; Gangoda, L.; Fonseka, P.; Chitti, S.V.; Liem, M.; Keerthikumar, S.; Samuel, M.; Boukouris, S.; Al Saffar, H.; Collins, C.; et al. Extracellular vesicles containing oncogenic mutant beta-catenin activate Wnt signalling pathway in the recipient cells. J. Extracell. Vesicles 2019, 8, 1690217. [Google Scholar] [CrossRef]
- Crow, J.; Atay, S.; Banskota, S.; Artale, B.; Schmitt, S.; Godwin, A.K. Exosomes as mediators of platinum resistance in ovarian cancer. Oncotarget 2017, 8, 11917–11936. [Google Scholar] [CrossRef]
- Kanlikilicer, P.; Bayraktar, R.; Denizli, M.; Rashed, M.H.; Ivan, C.; Aslan, B.; Mitra, R.; Karagoz, K.; Bayraktar, E.; Zhang, X.; et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine 2018, 38, 100–112. [Google Scholar] [CrossRef]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef]
- Muturi, H.T.; Dreesen, J.D.; Nilewski, E.; Jastrow, H.; Giebel, B.; Ergun, S.; Singer, B.B. Tumor and endothelial cell-derived microvesicles carry distinct CEACAMs and influence T-cell behavior. PLoS ONE 2013, 8, e74654. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Yao, X.; Zhang, Y.; Gu, J.; Geng, Y.; Xue, F.; Zhang, J. Colorectal cancer cell-derived exosomes containing miR-10b regulate fibroblast cells via the PI3K/Akt pathway. Bull. Cancer 2018, 105, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef] [PubMed]
- van der Vos, K.E.; Abels, E.R.; Zhang, X.; Lai, C.; Carrizosa, E.; Oakley, D.; Prabhakar, S.; Mardini, O.; Crommentuijn, M.H.; Skog, J.; et al. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro. Oncol. 2015, 18, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, Y.; Wu, M.; Li, N. Regulation of Macrophage Activation and Polarization by HCC-Derived Exosomal lncRNA TUC339. Int. J. Mol. Sci. 2018, 19, 2958. [Google Scholar] [CrossRef]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef]
- Ye, S.B.; Zhang, H.; Cai, T.T.; Liu, Y.N.; Ni, J.J.; He, J.; Peng, J.Y.; Chen, Q.Y.; Mo, H.Y.; Jun, C.; et al. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.; Wang, D.; Luo, C.; Chen, L. Renal carcinoma cell-derived exosomes induce human immortalized line of Jurkat T lymphocyte apoptosis in vitro. Urol. Int. 2013, 91, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Bao, Q.; Hu, C.; Wang, J.; Zhou, Q.; Wei, L.; Tong, L.; Zhang, W.; Shen, Y. Exosomal miR-675 from metastatic osteosarcoma promotes cell migration and invasion by targeting CALN1. Biochem. Biophys. Res. Commun. 2018, 500, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guan, X.; Zhang, Y.; Ge, S.; Zhang, L.; Li, H.; Wang, X.; Liu, R.; Ning, T.; Deng, T.; et al. Exosomal miR-27a Derived from Gastric Cancer Cells Regulates the Transformation of Fibroblasts into Cancer-Associated Fibroblasts. Cell Physiol. Biochem. 2018, 49, 869–883. [Google Scholar] [CrossRef]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.; Dickman, C.; Towle, R.; Jabalee, J.; Javer, A.; Garnis, C. Extracellular vesicle secretion of miR-142-3p from lung adenocarcinoma cells induces tumor promoting changes in the stroma through cell-cell communication. Mol. Carcinog. 2019, 58, 376–387. [Google Scholar] [CrossRef]
- Yang, L.; Wu, X.H.; Wang, D.; Luo, C.L.; Chen, L.X. Bladder cancer cell-derived exosomes inhibit tumor cell apoptosis and induce cell proliferation in vitro. Mol. Med. Rep. 2013, 8, 1272–1278. [Google Scholar] [CrossRef]
- Qu, J.L.; Qu, X.J.; Zhao, M.F.; Teng, Y.E.; Zhang, Y.; Hou, K.Z.; Jiang, Y.H.; Yang, X.H.; Liu, Y.P. Gastric cancer exosomes promote tumour cell proliferation through PI3K/Akt and MAPK/ERK activation. Dig. Liver Dis. 2009, 41, 875–880. [Google Scholar] [CrossRef]
- Li, Z.; Tao, Y.; Wang, X.; Jiang, P.; Li, J.; Peng, M.; Zhang, X.; Chen, K.; Liu, H.; Zhen, P.; et al. Tumor-Secreted Exosomal miR-222 Promotes Tumor Progression via Regulating P27 Expression and Re-Localization in Pancreatic Cancer. Cell Physiol. Biochem. 2018, 51, 610–629. [Google Scholar] [CrossRef]
- Li, C.; Liu, D.R.; Li, G.G.; Wang, H.H.; Li, X.W.; Zhang, W.; Wu, Y.L.; Chen, L. CD97 promotes gastric cancer cell proliferation and invasion through exosome-mediated MAPK signaling pathway. World J. Gastroenterol. 2015, 21, 6215–6228. [Google Scholar] [CrossRef]
- Ghorbanian, M.; Babashah, S.; Ataei, F. The effects of ovarian cancer cell-derived exosomes on vascular endothelial growth factor expression in endothelial cells. EXCLI J. 2019, 18, 899–907. [Google Scholar]
- Lu, J.; Liu, Q.H.; Wang, F.; Tan, J.J.; Deng, Y.Q.; Peng, X.H.; Liu, X.; Zhang, B.; Xu, X.; Li, X.P. Exosomal miR-9 inhibits angiogenesis by targeting MDK and regulating PDK/AKT pathway in nasopharyngeal carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 147. [Google Scholar] [CrossRef]
- de Andrade, A.; de Oliveira, C.E.; Dourado, M.R.; Macedo, C.; Winck, F.V.; Paes Leme, A.F.; Salo, T.; Coletta, R.D.; de Almeida Freitas, R.; Galvao, H.C. Extracellular vesicles from oral squamous carcinoma cells display pro- and anti-angiogenic properties. Oral. Dis. 2018, 24, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.G.; IB, B.S.; Campos-Fernandez, E.; Marangoni, K.; VA, F.B.; Alves, P.T.; Goulart, L.R.; Alonso-Goulart, V. Extracellular vesicles as drivers of epithelial-mesenchymal transition and carcinogenic characteristics in normal prostate cells. Mol. Carcinog. 2018, 57, 503–511. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, I.Y.; Daher, A.; Destouches, D.; Firlej, V.; Kostallari, E.; Maille, P.; Huet, E.; Haidar-Ahmad, N.; Jenster, G.; de la Taille, A.; et al. Extracellular vesicles released by mesenchymal-like prostate carcinoma cells modulate EMT state of recipient epithelial-like carcinoma cells through regulation of AR signaling. Cancer Lett. 2017, 410, 100–111. [Google Scholar] [CrossRef]
- Brzozowski, J.S.; Bond, D.R.; Jankowski, H.; Goldie, B.J.; Burchell, R.; Naudin, C.; Smith, N.D.; Scarlett, C.J.; Larsen, M.R.; Dun, M.D.; et al. Extracellular vesicles with altered tetraspanin CD9 and CD151 levels confer increased prostate cell motility and invasion. Sci. Rep. 2018, 8, 8822. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.L.; Zhang, Y.; Tan, B.; Luo, C.L.; Wu, X.H. Renal tumor-derived exosomes inhibit hepaCAM expression of renal carcinoma cells in a p-AKT-dependent manner. Neoplasma 2014, 61, 416–423. [Google Scholar] [CrossRef][Green Version]
- Chiba, M.; Watanabe, N.; Watanabe, M.; Sakamoto, M.; Sato, A.; Fujisaki, M.; Kubota, S.; Monzen, S.; Maruyama, A.; Nanashima, N.; et al. Exosomes derived from SW480 colorectal cancer cells promote cell migration in HepG2 hepatocellular cancer cells via the mitogen-activated protein kinase pathway. Int. J. Oncol. 2016, 48, 305–312. [Google Scholar] [CrossRef][Green Version]
- Lopes-Rodrigues, V.; Di Luca, A.; Mleczko, J.; Meleady, P.; Henry, M.; Pesic, M.; Cabrera, D.; van Liempd, S.; Lima, R.T.; O’Connor, R.; et al. Identification of the metabolic alterations associated with the multidrug resistant phenotype in cancer and their intercellular transfer mediated by extracellular vesicles. Sci. Rep. 2017, 7, 44541. [Google Scholar] [CrossRef]
- Kawamura, Y.; Yamamoto, Y.; Sato, T.A.; Ochiya, T. Extracellular vesicles as trans-genomic agents: Emerging roles in disease and evolution. Cancer Sci. 2017, 108, 824–830. [Google Scholar] [CrossRef]
- Guo, D.; Chen, Y.; Wang, S.; Yu, L.; Shen, Y.; Zhong, H.; Yang, Y. Exosomes from heat-stressed tumour cells inhibit tumour growth by converting regulatory T cells to Th17 cells via IL-6. Immunology 2018, 154, 132–143. [Google Scholar] [CrossRef]
- Bu, N.; Li, Q.L.; Feng, Q.; Sun, B.Z. Immune protection effect of exosomes against attack of L1210 tumor cells. Leuk. Lymphoma 2006, 47, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef]
- Marton, A.; Vizler, C.; Kusz, E.; Temesfoi, V.; Szathmary, Z.; Nagy, K.; Szegletes, Z.; Varo, G.; Siklos, L.; Katona, R.L.; et al. Melanoma cell-derived exosomes alter macrophage and dendritic cell functions in vitro. Immunol. Lett. 2012, 148, 34–38. [Google Scholar] [CrossRef]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-beta1-mediated suppression. Oncotarget 2016, 7, 27033–27043. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-beta in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M.; et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef]
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar]
- Li, Y.; An, J.; Huang, S.; He, J.; Zhang, J. Esophageal cancer-derived microvesicles induce regulatory B cells. Cell Biochem. Funct. 2015, 33, 308–313. [Google Scholar] [CrossRef]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Giusti, I.; Di Francesco, M.; D’Ascenzo, S.; Palmerini, M.G.; Macchiarelli, G.; Carta, G.; Dolo, V. Ovarian cancer-derived extracellular vesicles affect normal human fibroblast behavior. Cancer Biol. Ther. 2018, 19, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, B.; Bosl, T.; Reiners, K.S.; Barnert, S.; Schubert, R.; Shatnyeva, O.; Zigrino, P.; Engert, A.; Hansen, H.P.; von Strandmann, E.P. Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front. Immunol. 2018, 9, 1358. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes Released by Gastric Cancer Cells Induce Transition of Pericytes Into Cancer-Associated Fibroblasts. Med. Sci. Monit. 2018, 24, 2350–2359. [Google Scholar] [CrossRef]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogne, J.M. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef]
- Kharaziha, P.; Chioureas, D.; Rutishauser, D.; Baltatzis, G.; Lennartsson, L.; Fonseca, P.; Azimi, A.; Hultenby, K.; Zubarev, R.; Ullen, A.; et al. Molecular profiling of prostate cancer derived exosomes may reveal a predictive signature for response to docetaxel. Oncotarget 2015, 6, 21740–21754. [Google Scholar] [CrossRef]
- Lv, M.M.; Zhu, X.Y.; Chen, W.X.; Zhong, S.L.; Hu, Q.; Ma, T.F.; Zhang, J.; Chen, L.; Tang, J.H.; Zhao, J.H. Exosomes mediate drug resistance transfer in MCF-7 breast cancer cells and a probable mechanism is delivery of P-glycoprotein. Tumour. Biol. 2014, 35, 10773–10779. [Google Scholar] [CrossRef]
- Ning, K.; Wang, T.; Sun, X.; Zhang, P.; Chen, Y.; Jin, J.; Hua, D. UCH-L1-containing exosomes mediate chemotherapeutic resistance transfer in breast cancer. J. Surg. Oncol. 2017, 115, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Pan, Q.; Jiang, L.; Chen, Z.; Zhang, F.; Liu, Y.; Xing, H.; Shi, M.; Li, J.; Li, X.; et al. Tumor endothelial expression of P-glycoprotein upon microvesicular transfer of TrpC5 derived from adriamycin-resistant breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, C.; Hua, Y.; Sun, L.; Cheng, K.; Jia, Z.; Han, Y.; Dong, J.; Cui, Y.; Yang, Z. Exosomes play an important role in the process of psoralen reverse multidrug resistance of breast cancer. J. Exp. Clin. Cancer Res. 2016, 35, 186. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, D.; Padula, M.P.; Lu, J.F.; Jaiswal, R.; Djordjevic, S.P.; Bebawy, M. The Role of CD44 and ERM Proteins in Expression and Functionality of P-glycoprotein in Breast Cancer Cells. Molecules 2016, 21, 290. [Google Scholar] [CrossRef]
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal transfer of MDR by exosomes in human osteosarcoma. Int. J. Oncol. 2016, 49, 189–196. [Google Scholar] [CrossRef]
- Zhang, F.F.; Zhu, Y.F.; Zhao, Q.N.; Yang, D.T.; Dong, Y.P.; Jiang, L.; Xing, W.X.; Li, X.Y.; Xing, H.; Shi, M.; et al. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. Eur. J. Pharmacol. 2014, 738, 83–90. [Google Scholar] [CrossRef]
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 2009, 23, 1643–1649. [Google Scholar] [CrossRef]
- Levchenko, A.; Mehta, B.M.; Niu, X.; Kang, G.; Villafania, L.; Way, D.; Polycarpe, D.; Sadelain, M.; Larson, S.M. Intercellular transfer of P-glycoprotein mediates acquired multidrug resistance in tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1933–1938. [Google Scholar] [CrossRef]
- Wang, X.; Qiao, D.; Chen, L.; Xu, M.; Chen, S.; Huang, L.; Wang, F.; Chen, Z.; Cai, J.; Fu, L. Chemotherapeutic drugs stimulate the release and recycling of extracellular vesicles to assist cancer cells in developing an urgent chemoresistance. Mol. Cancer 2019, 18, 182. [Google Scholar] [CrossRef]
- Ifergan, I.; Scheffer, G.L.; Assaraf, Y.G. Novel extracellular vesicles mediate an ABCG2-dependent anticancer drug sequestration and resistance. Cancer Res. 2005, 65, 10952–10958. [Google Scholar] [CrossRef]
- Goler-Baron, V.; Assaraf, Y.G. Structure and function of ABCG2-rich extracellular vesicles mediating multidrug resistance. PLoS ONE 2011, 6, e16007. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers (Basel) 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- de Souza, P.S.; Cruz, A.L.; Viola, J.P.; Maia, R.C. Microparticles induce multifactorial resistance through oncogenic pathways independently of cancer cell type. Cancer Sci. 2015, 106, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Khoo, X.H.; Paterson, I.C.; Goh, B.H.; Lee, W.L. Cisplatin-Resistance in Oral Squamous Cell Carcinoma: Regulation by Tumor Cell-Derived Extracellular Vesicles. Cancers 2019, 11, 1166. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, R.X.; Chan, K.W.; Hu, J.; Zhang, J.; Wei, L.; Tan, H.; Yang, X.; Liu, H. Exosomal transfer of p-STAT3 promotes acquired 5-FU resistance in colorectal cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 320. [Google Scholar] [CrossRef]
- Zhao, K.; Wang, Z.; Li, X.; Liu, J.L.; Tian, L.; Chen, J.Q. Exosome-mediated transfer of CLIC1 contributes to the vincristine-resistance in gastric cancer. Mol. Cell Biochem. 2019, 462, 97–105. [Google Scholar] [CrossRef]
- Guerra, F.; Paiano, A.; Migoni, D.; Girolimetti, G.; Perrone, A.M.; De Iaco, P.; Fanizzi, F.P.; Gasparre, G.; Bucci, C. Modulation of RAB7A Protein Expression Determines Resistance to Cisplatin through Late Endocytic Pathway Impairment and Extracellular Vesicular Secretion. Cancers (Basel) 2019, 11, 52. [Google Scholar] [CrossRef]
- Wu, H.; Zhou, J.; Mei, S.; Wu, D.; Mu, Z.; Chen, B.; Xie, Y.; Ye, Y.; Liu, J. Circulating exosomal microRNA-96 promotes cell proliferation, migration and drug resistance by targeting LMO7. J. Cell Mol. Med. 2017, 21, 1228–1236. [Google Scholar] [CrossRef]
- Yu, D.D.; Wu, Y.; Zhang, X.H.; Lv, M.M.; Chen, W.X.; Chen, X.; Yang, S.J.; Shen, H.; Zhong, S.L.; Tang, J.H.; et al. Exosomes from adriamycin-resistant breast cancer cells transmit drug resistance partly by delivering miR-222. Tumour. Biol. 2016, 37, 3227–3235. [Google Scholar] [CrossRef]
- Ozawa, P.M.M.; Alkhilaiwi, F.; Cavalli, I.J.; Malheiros, D.; de Souza Fonseca Ribeiro, E.M.; Cavalli, L.R. Extracellular vesicles from triple-negative breast cancer cells promote proliferation and drug resistance in non-tumorigenic breast cells. Breast Cancer Res. Treat. 2018, 172, 713–723. [Google Scholar] [CrossRef]
- Feng, Y.; Hang, W.; Sang, Z.; Li, S.; Xu, W.; Miao, Y.; Xi, X.; Huang, Q. Identification of exosomal and nonexosomal microRNAs associated with the drug resistance of ovarian cancer. Mol. Med. Rep. 2019, 19, 3376–3392. [Google Scholar]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 Controls Exosome Synthesis and Promotes Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef]
- Gu, Y.Y.; Yu, J.; Zhang, J.F.; Wang, C. Suppressing the secretion of exosomal miR-19b by gw4869 could regulate oxaliplatin sensitivity in colorectal cancer. Neoplasma 2019, 66, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Akao, Y.; Khoo, F.; Kumazaki, M.; Shinohara, H.; Miki, K.; Yamada, N. Extracellular disposal of tumor-suppressor miRs-145 and -34a via microvesicles and 5-FU resistance of human colon cancer cells. Int. J. Mol. Sci. 2014, 15, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- He, J.; He, J.; Min, L.; He, Y.; Guan, H.; Wang, J.; Peng, X. Extracellular vesicles transmitted miR-31-5p promotes sorafenib resistance by targeting MLH1 in renal cell carcinoma. Int. J. Cancer 2020, 146, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, K.; Shuting, J.; Yoshioka, Y.; Ochiya, T.; Kondo, T. Extracellular vesicle-encapsulated microRNA-761 enhances pazopanib resistance in synovial sarcoma. Biochem. Biophys. Res. Commun. 2018, 495, 1322–1327. [Google Scholar] [CrossRef]
- Yin, J.; Zeng, A.; Zhang, Z.; Shi, Z.; Yan, W.; You, Y. Exosomal transfer of miR-1238 contributes to temozolomide-resistance in glioblastoma. EBioMedicine 2019, 42, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Yuwen, D.; Ma, Y.; Wang, D.; Gao, J.; Li, X.; Xue, W.; Fan, M.; Xu, Q.; Shen, Y.; Shu, Y. Prognostic Role of Circulating Exosomal miR-425-3p for the Response of NSCLC to Platinum-Based Chemotherapy. Cancer Epidemiol. Biomark. Prev. 2019, 28, 163–173. [Google Scholar] [CrossRef]
- Ma, Y.; Yuwen, D.; Chen, J.; Zheng, B.; Gao, J.; Fan, M.; Xue, W.; Wang, Y.; Li, W.; Shu, Y.; et al. Exosomal Transfer Of Cisplatin-Induced miR-425-3p Confers Cisplatin Resistance In NSCLC Through Activating Autophagy. Int. J. Nanomed. 2019, 14, 8121–8132. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, W.; Wang, H.; Qiu, G.; Jiang, Z.; Wei, G.; Li, X. Exosomal MiR-744 Inhibits Proliferation and Sorafenib Chemoresistance in Hepatocellular Carcinoma by Targeting PAX2. Med. Sci. Monit. 2019, 25, 7209–7217. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Yu, S.; Zhou, L.; Shi, M.; Hu, Y.; Xu, X.; Shen, B.; Liu, S.; Yan, D.; Feng, J. Cisplatin-resistant lung cancer cell-derived exosomes increase cisplatin resistance of recipient cells in exosomal miR-100-5p-dependent manner. Int. J. Nanomed. 2017, 12, 3721–3733. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Zhuang, T.; Xing, B.H.; Li, N.; Li, Q. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem. Funct. 2017, 35, 296–303. [Google Scholar] [CrossRef]
- Dong, H.; Wang, W.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; Zhang, F.; Han, J. Exosome-mediated transfer of lncRNASNHG14 promotes trastuzumab chemoresistance in breast cancer. Int. J. Oncol. 2018, 53, 1013–1026. [Google Scholar]
- Takahashi, K.; Yan, I.K.; Kogure, T.; Haga, H.; Patel, T. Extracellular vesicle-mediated transfer of long non-coding RNA ROR modulates chemosensitivity in human hepatocellular cancer. FEBS Open Biol. 2014, 4, 458–467. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.K.; Wood, J.; Haga, H.; Patel, T. Involvement of extracellular vesicle long noncoding RNA (linc-VLDLR) in tumor cell responses to chemotherapy. Mol. Cancer Res. 2014, 12, 1377–1387. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Li, J.; Du, Y.; Wang, J.; Liu, J. Effects of long noncoding RNA (linc-VLDLR) existing in extracellular vesicles on the occurrence and multidrug resistance of esophageal cancer cells. Pathol. Res. Pract. 2019, 215, 470–477. [Google Scholar] [CrossRef]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.F.; et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef]
- Wang, J.; Lv, B.; Su, Y.; Wang, X.; Bu, J.; Yao, L. Exosome-Mediated Transfer of lncRNA HOTTIP Promotes Cisplatin Resistance in Gastric Cancer Cells by Regulating HMGA1/miR-218 Axis. Oncol. Targets Ther. 2019, 12, 11325–11338. [Google Scholar] [CrossRef]
- Luo, X.; Wei, J.; Yang, F.L.; Pang, X.X.; Shi, F.; Wei, Y.X.; Liao, B.Y.; Wang, J.L. Exosomal lncRNA HNF1A-AS1 affects cisplatin resistance in cervical cancer cells through regulating microRNA-34b/TUFT1 axis. Cancer Cell Int. 2019, 19, 323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yin, J.; Lu, C.; Wei, Y.; Zeng, A.; You, Y. Exosomal transfer of long non-coding RNA SBF2-AS1 enhances chemoresistance to temozolomide in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Chen, M.; Xing, P.; Yan, X.; Xie, B. Increased Expression of Exosomal AGAP2-AS1 (AGAP2 Antisense RNA 1) In Breast Cancer Cells Inhibits Trastuzumab-Induced Cell Cytotoxicity. Med. Sci. Monit. 2019, 25, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.H.; Lee, M.Y.; Choi, D.Y.; Lee, J.W.; You, S.; Lee, K.Y.; Kim, J.; Kim, K.P. Phospholipids of tumor extracellular vesicles stratify gefitinib-resistant nonsmall cell lung cancer cells from gefitinib-sensitive cells. Proteomics 2015, 15, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Faict, S.; Oudaert, I.; D’Auria, L.; Dehairs, J.; Maes, K.; Vlummens, P.; de Veirman, K.; de Bruyne, E.; Fostier, K.; Vande Broek, I.; et al. The Transfer of Sphingomyelinase Contributes to Drug Resistance in Multiple Myeloma. Cancers (Basel) 2019, 11, 1823. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Update 2016, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Luk, F.; Jaiswal, R.; George, A.M.; Grau, G.E.; Bebawy, M. Microparticle drug sequestration provides a parallel pathway in the acquisition of cancer drug resistance. Eur. J. Pharmacol. 2013, 721, 116–125. [Google Scholar] [CrossRef]
- Lu, J.F.; Luk, F.; Gong, J.; Jaiswal, R.; Grau, G.E.; Bebawy, M. Microparticles mediate MRP1 intercellular transfer and the re-templating of intrinsic resistance pathways. Pharmacol. Res. 2013, 76, 77–83. [Google Scholar] [CrossRef]
- Lu, J.F.; Pokharel, D.; Bebawy, M. A novel mechanism governing the transcriptional regulation of ABC transporters in MDR cancer cells. Drug Deliv. Transl. Res. 2017, 7, 276–285. [Google Scholar] [CrossRef]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.; Bebawy, M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Sandoghchian Shotorbani, S.; Baradaran, B. RNA interference and its role in cancer therapy. Adv. Pharm. Bull. 2014, 4, 313–321. [Google Scholar] [PubMed]
- Zhang, H.; McCarty, N. Tampering with cancer chemoresistance by targeting the TGM2-IL6-autophagy regulatory network. Autophagy 2017, 13, 627–628. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 2016, 1863, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yu, S.; Li, S.; Wu, J.; Ma, R.; Cao, H.; Zhu, Y.; Feng, J. Exosomes: Decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS ONE 2014, 9, e89534. [Google Scholar] [CrossRef]
- Sousa, D.; Matthiesen, R.; Lima, R.T.; Vasconcelos, M.H. Deep Sequencing Analysis Reveals Distinctive Non-Coding RNAs When Comparing Tumor Multidrug-Resistant Cells and Extracellular Vesicles with Drug-Sensitive Counterparts. Cancers (Basel) 2020, 12, 200. [Google Scholar] [CrossRef]
- Janas, T.; Janas, M.M.; Sapon, K.; Janas, T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef]
- Kong, J.N.; He, Q.; Wang, G.; Dasgupta, S.; Dinkins, M.B.; Zhu, G.; Kim, A.; Spassieva, S.; Bieberich, E. Guggulsterone and bexarotene induce secretion of exosome-associated breast cancer resistance protein and reduce doxorubicin resistance in MDA-MB-231 cells. Int. J. Cancer 2015, 137, 1610–1620. [Google Scholar] [CrossRef]
- Soekmadji, C.; Nelson, C.C. The Emerging Role of Extracellular Vesicle-Mediated Drug Resistance in Cancers: Implications in Advanced Prostate Cancer. Biomed. Res. Int. 2015, 2015, 454837. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Ther. Anostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long noncoding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2020, 146, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Guo, H.; Wang, X.; Zhu, X.; Yan, M.; Wang, X.; Xu, Q.; Shi, J.; Lu, E.; Chen, W.; et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019, 20, 12. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.N.; Tsuruga, T.; Yeung, T.L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; de Bruyne, E.; van Valckenborgh, E.; Lahoutte, T.; de Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef]
- Lobb, R.J.; van Amerongen, R.; Wiegmans, A.; Ham, S.; Larsen, J.E.; Moller, A. Exosomes derived from mesenchymal non-small cell lung cancer cells promote chemoresistance. Int. J. Cancer 2017, 141, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Challagundla, K.B.; Wise, P.M.; Neviani, P.; Chava, H.; Murtadha, M.; Xu, T.; Kennedy, R.; Ivan, C.; Zhang, X.; Vannini, I.; et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef] [PubMed]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Shedden, K.; Xie, X.T.; Chandaroy, P.; Chang, Y.T.; Rosania, G.R. Expulsion of small molecules in vesicles shed by cancer cells: Association with gene expression and chemosensitivity profiles. Cancer Res. 2003, 63, 4331–4337. [Google Scholar] [PubMed]
- Aubertin, K.; Silva, A.K.; Luciani, N.; Espinosa, A.; Djemat, A.; Charue, D.; Gallet, F.; Blanc-Brude, O.; Wilhelm, C. Massive release of extracellular vesicles from cancer cells after photodynamic treatment or chemotherapy. Sci. Rep. 2016, 6, 35376. [Google Scholar] [CrossRef]
- Lopes-Rodrigues, V.; Di Luca, A.; Sousa, D.; Seca, H.; Meleady, P.; Henry, M.; Lima, R.T.; O’Connor, R.; Vasconcelos, M.H. Multidrug resistant tumour cells shed more microvesicle-like EVs and less exosomes than their drug-sensitive counterpart cells. Biochim. Biophys. Acta 2016, 1860, 618–627. [Google Scholar] [CrossRef]
- Goler-Baron, V.; Sladkevich, I.; Assaraf, Y.G. Inhibition of the PI3K-Akt signaling pathway disrupts ABCG2-rich extracellular vesicles and overcomes multidrug resistance in breast cancer cells. Biochem. Pharmacol. 2012, 83, 1340–1348. [Google Scholar] [CrossRef]
- Yin, J.; Yan, X.; Yao, X.; Zhang, Y.; Shan, Y.; Mao, N.; Yang, Y.; Pan, L. Secretion of annexin A3 from ovarian cancer cells and its association with platinum resistance in ovarian cancer patients. J. Cell Mol. Med. 2012, 16, 337–348. [Google Scholar] [CrossRef]
- Yan, X.; Yin, J.; Yao, H.; Mao, N.; Yang, Y.; Pan, L. Increased expression of annexin A3 is a mechanism of platinum resistance in ovarian cancer. Cancer Res. 2010, 70, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Ciravolo, V.; Huber, V.; Ghedini, G.C.; Venturelli, E.; Bianchi, F.; Campiglio, M.; Morelli, D.; Villa, A.; Della Mina, P.; Menard, S.; et al. Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J. Cell Physiol. 2012, 227, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Role of the RAB7 Protein in Tumor Progression and Cisplatin Chemoresistance. Cancers (Basel) 2019, 11, 1096. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cancer Type from Released EVs | EVs Cargo | Type of Study | Refs |
|---|---|---|---|
| Promoting cell proliferation and escape from apoptosis | |||
| Glioblastoma | Splicing factor RBM11; CLIC1 | In vitro, In vivo | [48,49] |
| Melanoma | PDGFR-β | In vitro | [50] |
| Pancreatic cancer | Zinc transporter ZIP4; miR-23b-3p; miR-222 | In vitro, In vivo, Patients samples | [51,52,53] |
| Breast cancer | miR-1246 | In vitro | [54] |
| Cholangiocarcinoma | miR-205 | In vitro | [55] |
| Colon cancer | DNp73 mRNA; miR-193a; mir-200b; lnRNA PVT1 | In vitro, In vivo | [56,57,58,59] |
| Thyroid cancer | miR-222; miR-146b | In vitro | [60] |
| Ovarian serous carcinoma | miR-21 | In vivo, Patients samples | [61] |
| Acute leukemia | miR-118, miR-116 | In vivo, Patients samples | [62] |
| Gastric cancer | lnRNA ZFAS1 | In vitro | [63] |
| Esophageal cancer | miR-93-5p; lncRNA ZEB1-AS1 | In vitro, Patients samples | [52,64] |
| Hepatocelular carcinoma | lncRNA TUC339 | In vitro | [65] |
| Sustaining angiogenesis | |||
| Glioblastoma | lnRNA CCAT2; lncRNA POU3F3; miR-21; CXCR4 receptor; VEGF | In vitro, In vivo | [66,67,68,69] |
| Head and neck squamous cell carcinoma | EPHB2 | In vitro, In vivo | [70] |
| Hepatocellular carcinoma | Vasorin; miR-103 | In vitro, In vivo | [71,72] |
| Multiple myeloma | piRNA-823 | In vivo | [73] |
| Colon cancer | miR-25-3p | In vitro, In vivo | [74] |
| Lung cancer | miR-14-3p; miR-145-5p; miR-23a | In vitro | [75,76] |
| Ovarian carcinoma | miRNA-141-3p | In vitro | [77] |
| Oral cancer | miR-142-3p | In vitro, In vivo | [78] |
| Nasopharyngeal carcinoma | miR-23a | In vitro, Patients samples | [79] |
| Contributing to cancer cell invasion and metastasis | |||
| Cholangiocarcinoma | miR-205-5p | In vitro | [55] |
| Breast cancer | Caveolin-1 | In vitro | [80] |
| Colon cancer | Wnt5b; AREG | In vitro | [81,82] |
| Hepatocarcinoma | CXCR4; SMAD3; miR-93; miR-103 | In Vitro; In vivo; Patient Samples | [72,83,84,85,86] |
| Gastric cancer | miR-423-5p | In vitro, In vivo; Patient samples | [87] |
| Prostate cancer | miR-1246 | In vitro, In vivo; Patient samples | [88] |
| Glioblastoma | miR-148a | In vitro; Patient Samples | [89] |
| Ovarian Cancer | miR-99a-5p | In vitro | [90] |
| Reprogramming energy metabolism | |||
| Breast Cancer | GSTP1; miR-122 | In vitro; In vivo; Patient samples | [91,92] |
| Transferring mutations | |||
| Glioblastoma | fusion genes PTPRZ1-Met, EGFRvIII | In vitro; In vivo | [93,94] |
| Melanoma | mRNA truncated Alk form | In vitro | [95] |
| Colon cancer | Mutant β-catenin | In vitro | [96] |
| Ovarian cancer | SMAD4 | In vitro | [97] |
| Modulating the Tumor Microenvironment: evading immune response and promoting inflammation | |||
| Ovarian Cancer | miR-1246, metabolic checkpoint molecular arginase-1 | In vitro | [98,99] |
| Colon cancer | miR-1246; miR-10b; CEACAM-family; Fas ligand | In vitro | [100,101,102] |
| Liposarcoma | miR-25-3p; miR-921-3p | In vitro, In vivo, Patients samples | [103] |
| Glioblastoma | miR-21, PD-L1 | In vitro | [104,105] |
| Hepatocellular carcinoma | lncRNA TUC339; miR-21 | In vitro | [106,107] |
| Nasopharyngeal carcinoma | miR-24-3p, galectin-9 | In vivo, Patients Samples | [108,109,110] |
| Renal cell carcinoma | Fas ligand | In vitro | [111] |
| Osteosarcoma | miR-675 | In vitro | [112] |
| Gastric Cancer | miR-27a | In vivo; Patient samples | [113] |
| Melanoma | miR-155-5p | In vitro, In vivo | [114] |
| Lung cancer | miR142-3p | In vitro | [115] |
| Intercellular Transfer | Specific EVs Cargo Transferred | Anti-Cancer Drugs | Cancer Type | Cell Lines/ Model/ Patients Studies | Refs |
|---|---|---|---|---|---|
| Drug-efflux pumps | MRP1/ABCC1 | Multi-drug | Leukemia, | HL-60 | [146] |
| ABCA3 | rituximab | B-cell lymphoma | Su-DHL-4; Balm3; OCI-Ly1 | [147] | |
| Pgp | Docetaxel Adriamycin Doxorubicin Paclitaxel Multi-drug | Prostate, Breast Breast Breast; Osteosarcoma Ovarian; Neuroblastoma; Leukemia | DU145 Tax-Sen/Tax-Res; 22Rv1 Doc-Res; MCF7 MCF7/ADM vs MCF7/WT MCF7;MG-63DXR30 A2780; BE (2)-C; VLB100; xenograft mouse models | [148,149,150] [151,152,153] [154,155] [156] [157,158] | |
| TrpC5 | Adriamycin | Breast | MCF7 | [152] | |
| ABCB1 | Vincristine, cisplatin, doxorubicin | Oral squamous carcinoma | KBv200 | [159] | |
| ABCG2 | Mitoxantrone Topotecan | Breast | MCF7/MR; MCF7/FLV1000 | [160] | |
| Imidazoacridinones methotrexate | Breast | MCF7/MR; MCF7/FLV1000 | [161] | ||
| Apoptotic modulators | Survivin | Paclitaxel | Triple-negative Breast | MDAMB231 | [162,163] |
| Inhibitors of apoptosis proteins XIAP and IAP | Multi-drug | Breast Chronic myeloid leukemia Lungs | MCF7 CML562 A529 | [163] | |
| Other Proteins | ATP1A1ATP1B3 | cisplatin | Squamous cell carcinoma | H314/H103 | [164] |
| TGM2 | cisplatin | Squamous cell carcinoma | H314/H103 | [164] | |
| GSTP1p-STAT3 | 5-fluoracil | Colorectal | RKO | [165] | |
| CLIC1 | vincristine | Gastric | SGC-7901 | [166] | |
| RAB7A | Cisplatin | Ovarian Cervical | HeLa; A431; 2008 (and cisplatin-resistant counterpart cell lines) | [167] | |
| miRNAs | miR-21 | Multi-drug | Chronic myeloid leukemia Breast | CMLK562 MCF7 | [163] |
| miR-96 | Cisplatin | Lung | A549, H1299, MCF-7 | [168] | |
| miR-222 | Adriamycin | Breast | [169] | ||
| miR-155-5p, miR-542-3p, let-7 and miR-28 | Docetaxel Doxorubicin | Triple-negative Breast | MCF10A | [170] | |
| miR-183-5p | Taxol Cisplatin Multidrug | Ovarian | SKOV3, A2780, HEYA8 | [171] | |
| miR-155 | Gemcitabine | Pancreatic ductal adenocarcinoma | Panc1, MiaPaCa2, Colo-357 and PSN1 cell lines Patients samples, Murine xenograft model | [172,173] | |
| miR-221/222 | Tamoxifen | Breast | MCF7 | [174] | |
| miR-19b | Oxaliplatin | Colorectal | SW480 | [175] | |
| miR-145 miR-34a | 5-fluoracil | Colon cancer | DLD-1 | [176] | |
| miR-31-5p | Sorafenib | Renal Cell Carcinoma | 786-0, ACHN, Patients samples | [177] | |
| miR-761 | Pazopanib | Synovial Sarcoma | SYO-1, HS-SYII, 1273/99, YaFuSS | [178] | |
| miR-1238 | Temozolomide | Glioblastoma | U251 cell line, patient samples | [179] | |
| miR-425-3p | Cisplatin | Non-small cell lung cancer | A549 cell line, Patients samples | [180,181] | |
| miR-744 | Sorafenin | Hepatocellular carcinoma | HepG2 cell line, Patients samples | [182] | |
| miR-100-5p | Cisplatin | Lung | A549 | [183] | |
| mRNAs | DNMT1 mRNA | Cisplatin | Ovarian cancer | Xenograft mouse model | [184] |
| GSTP1 mRNA | Adriamycin | Breast | MCF7 | [91] | |
| lncRNAs | lncSNHG14 | Trastruzumab | HER2-positive Breast | HER2-positive SKBR-3, HER2-positive BT474 | [185] |
| linc-ROR linc-VLDLR | Sorafenib Doxorubicin | Hepatocellular carcinoma | HepG2, Hep3B, PLC/PRF-5 and Huh-7 | [186,187] | |
| linc-VLDLR-ABCG2 | Multidrug | Esophageal cancer | Eca109 cell line, Patients samples | [188] | |
| lncARSR | Sunitinib | Renal Cell carcinoma | 786-O, ACHN, xenograft mouse models | [189] | |
| lncHOTTIP | Cisplatin | Gastric | Xenograft mouse models, Patients samples | [190] | |
| lncHNF1A-AS1 | Cisplatin | Cervical cancer | HeLa | [191] | |
| linc-SBF2-AS1 | Temozolomide | Glioblastoma | U87, LN229, A172, T98, U251, Patients samples | [192] | |
| linc-AGAP2-AS1 | Trastuzumab | HER2-positive Breast | HER2-positive SKBR-3, HER2-positive BT474 | [193] | |
| Lipids | Multiple phospholipids | Gefitinib | Lung | PC9R | [194] |
| Acid Sphingomyelinase | Melphalan Bortezomib | Multiple myeloma | JJN3, LP1, OPM2, U266 | [195] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimarães, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. https://doi.org/10.3390/cells9051141
Xavier CPR, Caires HR, Barbosa MAG, Bergantim R, Guimarães JE, Vasconcelos MH. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells. 2020; 9(5):1141. https://doi.org/10.3390/cells9051141
Chicago/Turabian StyleXavier, Cristina P. R., Hugo R. Caires, Mélanie A. G. Barbosa, Rui Bergantim, José E. Guimarães, and M. Helena Vasconcelos. 2020. "The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance" Cells 9, no. 5: 1141. https://doi.org/10.3390/cells9051141
APA StyleXavier, C. P. R., Caires, H. R., Barbosa, M. A. G., Bergantim, R., Guimarães, J. E., & Vasconcelos, M. H. (2020). The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells, 9(5), 1141. https://doi.org/10.3390/cells9051141